
Abstract
Molecular alterations preceding endometrial and ovarian cancer and the sequence of events are unknown. Consecutive specimens from lifelong surveillance for Lynch syndrome provides a natural setting to address such questions. To molecularly define the multistep gynecological tumorigenesis, DNA mismatch repair gene mutation carriers with endometrial or ovarian carcinoma or endometrial hyperplasia were identified from a nation-wide registry and endometrial biopsy specimens taken from these individuals during 20 years of screening were collected. A total of 213 endometrial and ovarian specimens from Lynch syndrome individuals and 197 histology-matched (non-serous) samples from sporadic cases were available for this investigation. The specimens were profiled for markers linked to endometrial and ovarian tumorigenesis, including ARID1A protein expression, mismatch repair status, and tumor suppressor gene promoter methylation. In Lynch syndrome-associated endometrial and ovarian carcinomas, ARID1A protein was lost in 61–100% and mismatch repair was deficient in 97–100%, compared to 0–17% and 14–44% in sporadic cases (P = 0.000). ARID1A loss appeared in complex hyperplasia and deficient mismatch repair and tumor suppressor gene promoter methylation in histologically normal endometrium. Despite quantitative differences between Lynch syndrome and sporadic cases, ARID1A expression, mismatch repair, and tumor suppressor gene promoter methylation divided endometrial samples from both patient groups into three categories of increasing abnormality, comprising normal endometrium and simple hyperplasia (I), complex hyperplasia with or without atypia (II), and endometrial cancer (III). Complex hyperplasias without vs. with atypia were molecularly indistinguishable. In conclusion, surveillance specimens from Lynch syndrome identify mismatch repair deficiency, tumor suppressor gene promoter methylation, and ARID1A loss as early changes in tumor development. Our findings are clinically relevant for the classification of endometrial hyperplasias and have potential implications in cancer prevention in Lynch syndrome and beyond.
Similar content being viewed by others

Introduction
Endometrial and ovarian carcinomas are among the most common cancers in females from industrialized countries, ranking in the fourth and eighth place, respectively, in the United States [1, 2]. Epidemiological and histopathological features combined with clinical behavior divide endometrial carcinoma into type I (endometrioid, estrogen-related) and type II (non-endometrioid, non-estrogen-related) [3]. Analogous categorization applies to epithelial ovarian cancer, with type I (non-serous, low-grade) consisting of low-grade serous carcinoma, mucinous, endometrioid, and clear cell carcinomas, and type II of high-grade serous carcinoma [4]. Among endometrial carcinomas, type I comprises 80% of cases and likely evolves via endometrial hyperplasia [3]. Mutations in PTEN, CTNNB1, PIK3CA, ARID1A, and KRAS are common in type I endometrial carcinomas, and microsatellite instability is present in one third [5]. Type I ovarian carcinoma reveals the involvement of many of the same genes [6]. The relative proportions of type I vs. type II, however, are opposite to those seen in endometrial carcinomas: high-grade serous type accounts for 70% of all epithelial ovarian carcinomas, compared to 10% for endometrioid and clear cell carcinomas each [7]. Frequent TP53 mutations are a feature of type II ovarian carcinomas [6].
The cellular origins of epithelial ovarian cancers are the subject of an ongoing research. Recent findings suggest that the three most common types (high-grade serous, endometrioid, and clear cell carcinoma) all arise from cells not normally located in the ovary [8]. Endometrioid and clear cell ovarian carcinomas have been suggested to arise from endometrial epithelial cells via atypical endometriosis and borderline tumors, whereas variable origins (mainly the fallopian tube and endosalpingiosis) have been proposed for high-grade serous carcinoma [8, 9].
In Lynch syndrome, a prevalent cancer predisposition syndrome associated with hereditary defects in DNA mismatch repair, up to 54% and 24% of female mutation carriers develop endometrial and ovarian cancer, respectively, during their lifetime [10, 11]. Importantly, mutation carriers are enrolled in lifelong surveillance against gynecological cancer, providing specimens from endometrial aspiration biopsies taken every 2 to 3 years [12]. We here profiled surveillance specimens from Lynch syndrome mutation carriers and corresponding tissues from sporadic cases for genetic and epigenetic changes. Our results define the molecular trajectories of endometrial and ovarian cancer and guide patient management.
Materials and methods
Patients and samples
Mismatch repair gene mutation carriers diagnosed with endometrial or ovarian carcinoma or endometrial hyperplasia were ascertained from the nation-wide Hereditary Colorectal Cancer Registry of Finland. A total of 213 endometrial and ovarian specimens from 66 Lynch syndrome mutation carriers (52 cases with MLH1, 10 with MSH2, and 4 with MSH6) were retrieved and compared to 197 samples from sporadic cases (Table 1). For endometrial hyperplasia samples, classification into four categories (simple hyperplasia, simple atypical hyperplasia, complex hyperplasia without atypia, and complex atypical hyperplasia) according to the WHO classification of 1994/2003 [13, 14] was maintained as it had originally been used to diagnose the cases. A simple atypical hyperplasia category was not included because we only had a single simple atypical hyperplasia sample (case ID LCAH7 in Supplementary Figure 1). Sporadic sets of endometrial carcinoma [15], ovarian carcinoma [16], and endometrial hyperplasias [17] were derived from larger consecutive series to represent histological types common in Lynch syndrome. Endometrial carcinoma in Lynch syndrome is mainly of endometrioid histology and ovarian carcinomas are non-serous [18, 19], which was taken into account when selecting the sporadic reference series (Table 1). Regarding other clinicopathological features, such as clinical stage or age at onset, the Lynch syndrome and sporadic series reflected the inherent characteristics of disease in the respective groups [18, 19]. Samples of normal endometrium from 18 unrelated individuals undergoing biopsy or surgery for non-malignant reasons [17] were used for normal endometrial tissue references for sporadic cases. For microsatellite instability analyses, an additional 20 specimens of histologically normal endometrium from patients diagnosed with non-Lynch endometrial cancer [20] were available.
The histology of carcinomas and hyperplasias had originally been determined by a gynecological pathologist, and the accuracy of the diagnoses was confirmed by one of the authors (RB). Archival formalin-fixed paraffin-embedded tissue sections were stained with hematoxylin and eosin and areas containing over 60% of tumor cells were selected for DNA extraction. Normal, hyperplasia, and tumor tissues were separated by manual microdissection and DNA extracted by a customized protocol [21]. The Institutional Review Boards of the Departments of Surgery (466/E6/01) and the Obstetrics and Gynecology (040/95) of the Helsinki University Central Hospital (Helsinki, Finland) and that of the Jyväskylä Central Hospital (Jyväskylä, Finland) (Dnro 5/2007) approved this study. The collection of archival specimens was approved by the National Authority for Medicolegal Affairs (Dnro 1272/04/044/07) and the National Supervisory Authority for Welfare and Health (Valvira/Dnro 10741/06.01.03.01/2015).
Immunohistochemistry for ARID1A protein expression
The 4 μm slides were deparaffinized with xylene and dehydrated with graded alcohols. Antigen retrieval was performed by PT-Module (Lab Vision, CA, USA) at 98 °C for 20 min in Envision TM Flex Target Retrieval solution, pH 6.1. Anti-ARID1A antibody produced in rabbit (1:200 for 20 min, HPA005456, polyclonal, Lot D104841, Sigma-Aldrich, USA) was used. Staining was conducted on Autostainer 480 automated immunostainer (Lab Vision, CA, USA). Tissue sections were counterstained with hematoxylin (Mayers HTX, Histolab), dehydrated, cleared in xylene, and mounted.
Slides were scored by two authors (RB and AP). ARID1A expression was regarded as negative/abnormal when there was no nuclear staining of the tumor cells but stromal cells showed positive expression functioning as internal control.
Microsatellite instability and mismatch repair protein expression to determine mismatch repair status
DNA was analyzed for microsatellite instability by polymerase chain reaction (PCR) using sensitive mononucleotide repeat markers BAT25 and BAT26. Fluorescently labeled PCR products were run on ABI 3730 Automatic DNA Sequencer and visual inspection of electropherograms was performed by GeneMapper 5.0 (Applied Biosystems). Samples with at least one unstable repeat marker were considered as microsatellite instability-high, otherwise the sample was microsatellite stable [22]. Mismatch repair protein expression was investigated by immunohistochemistry as described [23]. Mismatch repair deficiency was defined as microsatellite instability or absent mismatch repair protein or both.
Methylation-specific multiplex ligation-dependent probe amplification for methylation analysis
The methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) test SALSA MLPA ME001-C2 (MRC-Holland, Amsterdam, The Netherlands) was used to detect aberrant methylation in 24 general tumor suppressor genes (specified at http://www.mrc-holland.com). In addition, Salsa MLPA kit P-300-B1 human DNA reference-2 with custom designed probe mix was applied to detect abnormal methylation of 7 endometrial and ovarian cancer-related genes [24]. The test is based on probes that contain a restriction site (GCGC) for the methylation-sensitive endonuclease HhaI (Promega, USA), which binds to the first unmethylated CpG dinucleotide of a GCGC site and subsequently digests the site. If the GCGC site is methylated, the site stays undigested and will generate a signal peak in PCR. The reactions were carried out following the manufacturer’s instructions (http://www.mrc-holland.com) using 100–250 ng of formalin-fixed paraffin-embedded tissue DNA. PCR products were separated by capillary electrophoresis (ABI 3730 Automatic DNA Sequencer, Applied Biosystems, USA) and results analyzed by GeneMapper 5.0 genotyping software (Applied Biosystems, USA). The methylation dosage ratio varies between 0 and 1.0 and compares to the percentage of methylated DNA. The methylation dosage ratio was calculated individually for each tumor and normal sample as described [25]. The methylation dosage ratio value of ≥0.15 (corresponding to 15% of methylated DNA) was considered as the cut-off for tumor-specific hypermethylation for all genes included in the commercial 24 general tumor suppressor gene MS-MLPA test [25], except for CDKN2B. The thresholds for hypermethylation for CDKN2B and all 7 endometrial and ovarian carcinoma-related genes included in the custom assay were determined using the normal endometrial samples of Lynch (n = 99) and sporadic cases (n = 18) and specified as the mean methylation dosage ratio in normal endometria plus 1 standard deviation (the thresholds were separate for Lynch and sporadic cases).
Statistical analyses
Statistical evaluations were performed using SPSS software, version 22.0 (IBM® SPSS® Statistics, Inc. Chicago, IL, USA). Fisher’s exact test (two-tailed P values adjusted for multiple comparisons by Bonferroni correction when appropriate) was applied to evaluate frequency data. For two-group comparisons involving numbers of methylated genes or the methylation dosage ratio values, Shapiro–Wilk test was first applied to see if the data were normally distributed, followed by the Student’s t-test (for normally distributed samples) or nonparametric Mann–Whitney U test (for samples not normally distributed). Statistical significance of methylation changes between multiple groups was studied by the nonparametric Kruskal–Wallis one-way ANOVA test with pairwise comparisons because all series were either not normally distributed or did not reach the homogeneity of variances by Levene’s test. P values < 0.05 (two-tailed) were regarded significant.
Results
Study design
This investigation was designed to identify molecular changes in non-malignant endometrial specimens that might signal endometrial and/or ovarian carcinoma (Fig. 1). We took advantage of a surveillance program offered for Lynch syndrome mutation carriers in Finland since 1996 [26], supplemented with samples from sporadic cohorts (Table 1). The following key parameters of endometrial and ovarian tumorigenesis were investigated: (i) ARID1A, a chromatin remodeler and tumor suppressor often mutant in endometrial and non-serous ovarian tumors, including mismatch repair-deficient and proficient cases [5, 6, 27], (ii) mismatch repair status, a key feature of Lynch syndrome and type I endometrial and ovarian cancers in general [5, 6], and (iii) tumor suppressor gene methylation to represent epigenetic changes, fingerprints of cancer cell origins [28] that are histology-specific [5, 24].

Flowchart of this investigation. MS-MLPA methylation-specific multiplex ligation-dependent probe amplification. All endometrial carcinomas included in our molecular studies were of endometrioid histology. Grade and stage information of the endometrial and ovarian tumors is available in Table 1
Loss of ARID1A protein expression and mismatch repair deficiency characterize Lynch-associated endometrial and ovarian carcinomas
Figure 2a and b displays the frequencies of ARID1A and mismatch repair alterations in endometrial and ovarian carcinomas from Lynch syndrome and sporadic cases. Of Lynch syndrome-associated carcinomas, 61–100% showed loss of ARID1A protein expression (Fig. 2a) and 97–100% were mismatch repair-deficient (Fig. 2b). In sporadic carcinomas, ARID1A protein was absent in 0–17% and mismatch repair defective in 14–44% of tumors. The differences between Lynch syndrome and sporadic tumors were significant (P = 0.000).

Occurrence of ARID1A (a), mismatch repair (b), and tumor suppressor gene methylation aberrations (c, d) in Lynch syndrome-associated and sporadic endometrial and ovarian carcinomas. The number of tumors studied in each category is given below the bar graphs. Significance values by Fisher’s exact test (a, b) and by Mann–Whitney U test (c, d) for Lynch syndrome vs. sporadic comparisons are indicated on the right. EnCa endometrioid endometrial carcinoma, OvCC clear cell ovarian carcinoma, OvE endometrioid ovarian carcinoma, ns non-significant, N/A result not available
Figure 2c and d depicts the average numbers of tumor suppressor genes with promoter hypermethylation (i.e., showing increased methylation when compared to the corresponding normal tissues) in the different carcinoma groups. Gene-specific values for the most informative methylation markers (RSK4, SPARC, HOXA10, and HOXA9 among 7 endometrial and ovarian carcinoma-related genes and RASSF1A and CDH13 among 24 general tumor suppressor genes) are given in Supplementary Table 1. Differences between Lynch syndrome and sporadic cases were not striking and their direction varied depending on tumor type and markers. In sporadic tumors, the CpG Island Methylator Phenotype is known to be linked to deficient mismatch repair via MLH1 promoter methylation [29]. In our sporadic series, mismatch repair-deficient cases comprised 44% (16/36) of endometrial carcinomas and 15% (10/67) of ovarian carcinomas (Fig. 2b), corresponding well to reported frequencies of deficient mismatch repair in endometrial [5] and ovarian carcinomas [7]. Endometrial carcinomas with a notable mismatch repair-deficient subgroup were stratified by mismatch repair status (Supplementary Table 1). The average numbers of hypermethylated tumor suppressor genes were significantly higher in sporadic microsatellite instability-high endometrial carcinomas compared to sporadic microsatellite stable endometrial carcinomas (P = 0.016 for 7 endometrial and ovarian cancer-related genes and P = 0.001 for 24 general tumor suppressor genes) or Lynch syndrome endometrial carcinomas (P = 0.000 and P = 0.001 for the two marker panels, respectively).
ARID1A, mismatch repair deficiency, and tumor suppressor gene methylation are early markers of endometrial and ovarian tumorigenesis
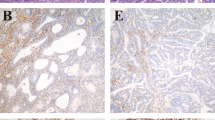
Having established the frequencies of ARID1A, deficient mismatch repair and tumor suppressor gene promoter methylation in endpoint lesions (carcinomas), we set out to examine non-malignant endometrial samples. Loss of ARID1A protein occurred at endometrial hyperplasia stage (Lynch syndrome and sporadic, Fig. 3). While ARID1A protein was expressed in normal endometrium and simple hyperplasia, expression was lost in 25% (1/4) of complex hyperplasias without atypia and 20% (6/30) of complex atypical hyperplasias from Lynch syndrome patients and 20% (4/20) of complex atypical hyperplasia samples from sporadic cases (Fig. 3a). Examples of immunohistochemical staining patterns depicting loss or retention of ARID1A protein in precursor lesions from Lynch syndrome patients whose endometrial carcinoma lacked ARID1A protein are given in Fig. 3b.

a Frequencies of absent ARID1A protein in endometrial hyperplasia as part of progressive endometrial tumorigenesis. The number of specimens studied in each category is given below the bar graphs. Categories are normal endometrium (NE), simple hyperplasia (SH), complex hyperplasia without atypia (CH), complex atypical hyperplasia (CAH), and endometrioid endometrial carcinoma (EnCa). Significance values by Fisher’s exact test for Lynch syndrome vs. sporadic comparisons are shown on the right. b Examples of normal and abnormal immunohistochemistry results of ARID1A in multiple endometrial samples taken from the same Lynch syndrome individuals. Representative histological areas with immunohistochemical changes are marked with solid arrows whereas reference normal cells expressing ARID1A protein are indicated with open arrows. Case LEC15 (left) shows ARID1A expression in normal endometrium whereas complex hyperplasia without atypia and concurrent endometrial carcinoma lack ARID1A protein. In case LEC1 (right), ARID1A is expressed in specimens of normal endometrium and complex hyperplasia without atypia taken two years before endometrial carcinoma, in which ARID1A protein is lost. Complex atypical hyperplasia concurrent with endometrial carcinoma still expresses ARID1A. Original magnification, ×20 (all images). ns non-significant, N/A result not available
As shown in Fig. 3a, the earliest endometrial lesions exhibiting loss of ARID1A protein expression were Lynch-associated complex hyperplasias without atypia. Mismatch repair defects (Fig. 4a) appeared even earlier than ARID1A loss, in histologically normal endometrium from Lynch syndrome patients. The proportion of mismatch repair-deficient samples increased from normal endometrium to simple hyperplasia to complex hyperplasia without atypia to complex atypical hyperplasia to endometrial carcinoma and was significantly higher in Lynch syndrome than sporadic cases in all histological groups (Fig. 4a). Mismatch repair defects (microsatellite instability-high) were observed in 12% (12/99) of normal endometrial specimens as a remarkable feature of Lynch syndrome. Analysis of consecutive specimens showed that microsatellite instability was in some cases detectable several years before endometrial or ovarian cancer (Fig. 4b, Supplementary Figure 1). For example, in case LEC1, microsatellite instability occurred in normal endometrium 4 years before endometrial carcinoma (Fig. 4b, left) and in case LOC20, 6 years before endometrioid ovarian cancer (Supplementary Figure 1). The endometrial areas with microsatellite instability were apparently focal since microsatellite instability was variably present in consecutive specimens of normal endometrium as illustrated by case LCAH5 in Fig. 4b (right). Normal endometrium (n = 18) from our basic reference series (diagnostic specimens taken for non-malignant reasons) and normal endometrium (n = 20) from cases with non-Lynch endometrial carcinoma (see Materials and Methods) showed no microsatellite instability; the difference relative to Lynch syndrome was significant (P = 0.037). All 38 non-Lynch normal endometrial samples were unrelated to the sporadic endometrial or ovarian carcinomas belonging to our study series (paired normal endometrium from our sporadic cases of endometrial and ovarian carcinomas was unavailable).

a Frequencies of mismatch repair aberrations in progressive endometrial lesions from Lynch syndrome and sporadic cases. The number of specimens studied in each category is given below the bar graphs. Categories are normal endometrium (NE), simple hyperplasia (SH), complex hyperplasia without atypia (CH), complex atypical hyperplasia (CAH), and endometrioid endometrial carcinoma (EnCa). Significance values by Fisher’s exact test for Lynch syndrome vs. sporadic comparisons are indicated on the right. b Examples of microsatellite instability (MSI) tracings in cases where normal endometrium is unstable. Results from consecutive specimens are shown with the endpoint diagnosis at the bottom. Reference normal DNA (top) illustrates the microsatellite stable (MSS) pattern. The wild type size for BAT26 is 116 bp (vertical lines) and arrows pointing to the left indicate contractions of the microsatellite repeat. Electropherogram on the left demonstrates microsatellite instability in normal endometrium prior to endometrial carcinoma (case LEC1). Intermittent or focal microsatellite instability in normal endometrium prior to or concurrent with complex atypical hyperplasia is shown on the right (case LCAH5)
Consecutive biopsy specimens from Lynch syndrome patients also demonstrated the presence of tumor suppressor promoter methylation in normal endometrium before endometrial or ovarian carcinoma with hypermethylation of the same loci. Please see cases LEC1, LEC3, LEC5, LOC13, LOC15, and LOC20 in Supplementary Figure 1 as examples.
Molecular changes divide endometrial specimens into three categories
In Lynch syndrome and sporadic cases, loss of ARID1A protein (Fig. 3a) and deficient mismatch repair (Fig. 4a) showed an increasing trend of molecular alterations along with increasing histological abnormality of endometrial lesions. Tumor suppressor gene promoter hypermethylation likewise revealed a progressive trend (Fig. 5). By pairwise comparisons detailed in Fig. 5, no significant differences were seen in normal endometrium vs. simple hyperplasia and in complex hyperplasia without atypia vs. complex atypical hyperplasia comparisons, whereas complex hyperplasia without atypia/complex atypical hyperplasia vs. normal endometrium and complex hyperplasia without atypia/complex atypical hyperplasia vs. endometrial carcinoma often and normal endometrium vs. endometrial carcinoma almost always showed significant differences. This suggested division of endometrial specimens into three groups: (I) normal endometrium plus simple hyperplasia, (II) complex hyperplasia without atypia plus complex atypical hyperplasia, and (III) endometrial carcinoma. On the basis of the observations described above, the data shown individually for normal endometrium, simple hyperplasia, complex hyperplasia without atypia, complex atypical hyperplasia, and endometrial carcinoma in Figs 3, 4, and 5 were reorganized into combined categories I–III (Fig. 6). The significant differences between the three groups illustrate the validity of the proposed categorization.

Distributions of the numbers of hypermethylated endometrial and ovarian cancer-related tumor suppressor genes (among 7 genes evaluated) and general tumor suppressor genes (among 24) in Lynch-associated and sporadic samples of normal endometrium (NE), simple hyperplasia (SH), complex hyperplasia without atypia (CH), complex atypical hyperplasia (CAH), and endometrioid endometrial carcinoma (EnCa). The horizontal line within the box denotes the median. Significance values by Kruskal–Wallis pairwise comparisons are shown

Occurrence of ARID1A, mismatch repair, and methylation aberrations in progressive endometrial lesions, categorized I (normal endometrium + simple hyperplasia), II (complex hyperplasia without atypia + complex atypical hyperplasia), and III (endometrioid endometrial carcinoma). Statistically significant differences by Fisher’s exact test (for ARID1A and mismatch repair) and Kruskal–Wallis test (for hypermethylation data) are pointed out when present
When the molecular characteristics of endometrial hyperplasias were compared with endometrial or ovarian carcinomas concurrently or later diagnosed in the same Lynch syndrome individuals, complex hyperplasia without atypia, and complex atypical hyperplasia showed a high degree of similarity to the carcinomas (Supplementary Figure 1). In contrast, simple hyperplasia revealed discordant patterns. This implies equal pre-malignant potential for complex hyperplasia without atypia and complex atypical hyperplasia and argues against pre-malignant potential of simple hyperplasia, observations consistent with our 3-tiered classification of Fig. 5.
Discussion
We used Lynch syndrome as a longitudinal model to define the sequence of events leading to endometrial and ovarian cancer development. In our Lynch syndrome series, 12% (12/99) of histologically normal endometria were mismatch repair-deficient compared to 0/38 samples from non-Lynch syndrome individuals (P = 0.037). Microsatellite instability could be detected several years before endometrial cancer and was apparently focal (Fig. 4b and Supplementary Figure 1). Our endometrial findings resemble those of Kloor et al. [30] who identified frequent lesions (termed mismatch repair-deficient crypt foci) in non-tumorous intestinal mucosa from mismatch repair gene mutation carriers. While the possible neoplastic nature of mismatch repair deficiency observed in colorectal or endometrial mucosae is unsettled, so far, the findings do indicate a high frequency of biallelic mismatch repair gene inactivation in seemingly normal tissues from Lynch syndrome individuals. Since the abundance of mismatch repair-deficient crypt foci greatly exceeded the number of colorectal tumors, Kloor et al. [30] concluded that other tumor-promoting events are required in addition to mismatch repair deficiency. In our investigation, hypermethylation of tumor suppressor gene promoters could constitute such accessory events. For example, normal endometrium from LOC20 displayed hypermethylation at SPARC, PROM1, HOXA10, HOXA9, APC, and CDH13 six years before endometrioid ovarian carcinoma in which the same loci were hypermethylated (Supplementary Figure 1).
Follow-up studies of sporadic cases indicate an up to 30% risk of malignant progression for atypical endometrial hyperplasias, which is why these are traditionally recognized as precursors of endometrial carcinoma [31, 32]. In agreement, WHO 2014 classification [33] divides hyperplasias into two categories, hyperplasia without atypia and atypical hyperplasia. These classifications reflect the fact that the role of non-atypical hyperplasias in endometrial tumorigenesis is unclear. In our investigation, loss of ARID1A protein, mismatch repair deficiency, and tumor suppressor gene promoter methylation divided endometrial specimens into three groups: normal endometrium plus simple hyperplasia, complex hyperplasia without atypia plus complex atypical hyperplasia, and endometrial carcinoma (Fig. 6) and suggested that complex hyperplasia without atypia should be regarded as a precursor for endometrial carcinoma just as complex atypical hyperplasia. Our study is not the only one emphasizing the significance of complex hyperplasia without atypia. Van der Putten et al. [32] used a sensitive sequencing technique to screen endometrial hyperplasias for known mutations in established cancer genes and found that complex hyperplasia without atypia and complex atypical hyperplasia were mutated in 22 and 33%, respectively, compared to 5% of simple hyperplasia and 0% of simple atypical hyperplasia. The findings suggested that complex hyperplasia regardless of atypia is important in endometrioid endometrial carcinogenesis. Histological assessment of endometrial hyperplasias for the presence vs. absence of atypia is difficult and diagnostic reproducibility poor, as has been demonstrated repeatedly [34,35,36]. Therefore, our endometrial hyperplasia specimens underwent two independent evaluations, a primary diagnostic assessment followed by re-evaluation as part of research (see Materials and methods). Accurate knowledge of carcinoma precursors is clinically relevant since it guides treatment decisions: atypical hyperplasia is usually treated with total hysterectomy whereas non-atypical hyperplasias are treated with medication and preventive hysterectomy is considered only in exceptional cases [37].
Although ARID1A protein expression, tumor suppressor gene promoter methylation, and mismatch repair status classified endometrial hyperplasias into similar categories in Lynch syndrome vs. sporadic cases, quantitative differences did exist between Lynch syndrome and sporadic cases and were remarkable in carcinomas. The frequencies of ARID1A protein loss and defective mismatch repair were significantly (P = 0.000) higher in Lynch syndrome-associated endometrial and ovarian carcinomas compared to their sporadic counterparts (Figs. 3 and 4). Our results differ from those of Bosse et al. [38] who detected loss of ARID1A protein in 75% (24/32) among sporadic microsatellite instability-high endometrial carcinomas vs. 14% (5/36) among Lynch syndrome-associated cases collected at the Leiden University Medical Center (P = 0.000). They interpreted their results to suggest that ARID1A, a chromatin remodeler protein, might contribute to microsatellite instability (through epigenetic silencing of MLH1), instead of being a target gene for microsatellite instability. In our endometrial carcinomas, ARID1A was lost in 25% (4/16) and 11% (2/19) of sporadic microsatellite instability-high and microsatellite stable endometrial carcinomas, respectively (statistically non-significant), whereas 61% (14/23) of Lynch syndrome endometrial carcinomas (97% of which were mismatch repair-deficient) lacked ARID1A protein (the difference between sporadic microsatellite instability-high and Lynch syndrome endometrial carcinomas was significant, P = 0.049). Our results comply with Bosse et al. [38] in that mismatch repair status per se does not explain the difference between Lynch syndrome and sporadic cases. ARID1A is known to be regulated by somatic mutations, copy number changes, and promoter methylation [39]. In agreement with our finding of common inactivation of ARID1A in Lynch syndrome endometrial and ovarian carcinomas, our recent panel sequencing of cancer-relevant genes identified ARID1A as the top somatically mutant gene (with 50% showing high-frequency mutations) in Lynch syndrome ovarian carcinomas [40]. Although we have not sequenced sporadic tumors, it is possible that the mechanisms of ARID1A inactivation and/or selection of clones with inactive ARID1A differ in Lynch syndrome and sporadic tumors. Furthermore, results from our registry-based Lynch syndrome cohort cannot be directly extrapolated to any possible population-based cohorts of Lynch syndrome.
In summary, three main discoveries emanate from our investigation. First, we identify early tumorigenic changes, including ARID1A loss, which appears in endometrial hyperplasia (Lynch syndrome and sporadic), whereas defective mismatch repair (Lynch syndrome) and tumor suppressor gene promoter hypermethylation (Lynch syndrome and sporadic) are detectable even in histologically normal endometrium. Second, in Lynch syndrome and sporadic cases, molecular alterations classify endometrial samples into three groups of increasing abnormality, and endometrial hyperplasia with and without atypia closely resemble each other. Third, there are quantitative differences between Lynch syndrome and sporadic cases; notably, loss of ARID1A protein and deficient mismatch repair are characteristic of Lynch syndrome-associated endometrial and ovarian carcinomas (the frequencies of mismatch repair defects differ between Lynch syndrome and sporadic cases in normal endometrium and endometrial hyperplasia already). These observations have important clinical implications. As discussed above, information of cancer precursor lesions may affect treatment decisions. In regard to Lynch syndrome specifically, no consensus guidelines for gynecological cancer screening currently exist although regular surveillance of the endometrium, starting at age 30–40 years, is recommended [12, 41]. Definitive evidence of efficacy for gynecological cancer screening is lacking, so far [12, 41], and surveillance biopsies could best be used during the interval between Lynch syndrome diagnosis and eventual prophylactic surgery when other forms of intervention are less useful or more toxic. Molecular alterations in endometrial biopsy specimens could identify mutation carriers who would need intensive surveillance and active cancer prevention, including aspirin-based chemoprevention that may be efficient against non-colonic Lynch syndrome cancers in addition to colorectal cancer [42]. Molecular changes could also guide optimal timing for prophylactic surgery. Of relevance for treatment considerations, the interval between a detectable molecular abnormality and developing cancer can be several years (Figs. 3b and 4b and Supplementary Figure 1).
References
Sorosky JI. Endometrial cancer. Obstet Gynecol. 2012;120:383–97.
Baldwin LA, Huang B, Miller RW, et al. Ten-year relative survival for epithelial ovarian cancer. Obstet Gynecol. 2012;120:612–8.
Prat J, Gallardo A, Cuatrecasas M, Catasus L. Endometrial carcinoma: pathology and genetics. Pathology. 2007;39:72–87.
Kurman RJ, Shih LEM. Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int J Gynecol Pathol. 2008;27:151–60.
Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15.
Prat J. Ovarian carcinomas: five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012;460:237–49.
Karnezis AN, Cho KR, Gilks CB, Pearce CL, Huntsman DG. The disparate origins of ovarian cancers: pathogenesis and prevention strategies. Nat Rev Cancer. 2017;17:65–74.
Ng A, Barker N. Ovary and fimbrial stem cells: biology, niche and cancer origins. Nat Rev Mol Cell Biol. 2015;16:625–38.
Bonadona V, Bonaiti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305:2304–10.
Moller P, Seppala T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66:464–72.
Lu KH, Daniels M. Endometrial and ovarian cancer in women with Lynch syndrome: update in screening and prevention. Fam Cancer. 2013;12:273–7.
Scully RE, Poulsen HE. Histological typing of female genital tract tumours. 2nd ed. Berlin; New York: Springer-Verlag; 1994. 189 p.
Silverberg SGKR, Nogales F, Mutter GL, Kubik-Huch RA, Tavassoli FA. Epithelial tumours and related lesions. In: Tavassoli FADP, editors. Pathology and genetics of tumours of the breast and female genital organs. World Health Organization classification of tumours. Lyon (France): IARC Press; 2003. pp. 221–32.
Pasanen A, Tuomi T, Isola J, et al. L1 cell adhesion molecule as a predictor of disease-specific survival and patterns of relapse in endometrial cancer. Int J Gynecol Cancer. 2016;26:1465–71.
Soovares P, Pasanen A, Butzow R, Lassus H. L1CAM expression associates with poor outcome in endometrioid, but not in clear cell ovarian carcinoma. Gynecol Oncol. 2017;146:615–22.
Nieminen TT, Gylling A, Abdel-Rahman WM, et al. Molecular analysis of endometrial tumorigenesis: importance of complex hyperplasia regardless of atypia. Clin Cancer Res. 2009;15:5772–83.
Rossi L, Le Frere-Belda MA, Laurent-Puig P, et al. Clinicopathologic characteristics of endometrial cancer in lynch syndrome: a french multicenter study. Int J Gynecol Cancer. 2017;27:953–60.
Grindedal EM, Renkonen-Sinisalo L, Vasen H, et al. Survival in women with MMR mutations and ovarian cancer: a multicentre study in Lynch syndrome kindreds. J Med Genet. 2010;47:99–102.
Ollikainen M, Abdel-Rahman WM, Moisio AL, et al. Molecular analysis of familial endometrial carcinoma: a manifestation of hereditary nonpolyposis colorectal cancer or a separate syndrome? J Clin Oncol. 2005;23:4609–16.
Isola J, DeVries S, Chu L, Ghazvini S, Waldman F. Analysis of changes in DNA sequence copy number by comparative genomic hybridization in archival paraffin-embedded tumor samples. Am J Pathol. 1994;145:1301–8.
Loukola A, Eklin K, Laiho P, et al. Microsatellite marker analysis in screening for hereditary nonpolyposis colorectal cancer (HNPCC). Cancer Res. 2001;61:4545–9.
Niskakoski A, Kaur S, Renkonen-Sinisalo L, et al. Distinct molecular profiles in Lynch syndrome-associated and sporadic ovarian carcinomas. Int J Cancer. 2013;133:2596–608.
Niskakoski A, Kaur S, Staff S, et al. Epigenetic analysis of sporadic and Lynch-associated ovarian cancers reveals histology-specific patterns of DNA methylation. Epigenetics. 2014;9:1577–87.
Gylling AH, Nieminen TT, Abdel-Rahman WM, et al. Differential cancer predisposition in Lynch syndrome: insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis. 2008;29:1351–9.
Renkonen-Sinisalo L, Butzow R, Leminen A, et al. Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int J Cancer. 2007;120:821–4.
Takeda T, Banno K, Okawa R, et al. ARID1A gene mutation in ovarian and endometrial cancers (review). Oncol Rep. 2016;35:607–13.
Kundaje A, Meuleman W, Ernst J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30.
Joensuu EI, Abdel-Rahman WM, Ollikainen M, et al. Epigenetic signatures of familial cancer are characteristic of tumor type and family category. Cancer Res. 2008;68:4597–605.
Kloor M, Huth C, Voigt AY, et al. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: a pathological study. Lancet Oncol. 2012;13:598–606.
Kurman RJ, Kaminski PF, Norris HJ. The behavior of endometrial hyperplasia. A long-term study of “untreated” hyperplasia in 170 patients. Cancer. 1985;56:403–12.
van der Putten LJM, van Hoof R, Tops BBJ, et al. Molecular profiles of benign and (pre)malignant endometrial lesions. Carcinogenesis. 2017;38:329–35.
Zaino RCSG, Ellenson LH. WHO classification of tumours of female reproductive organs. 4th ed. In: Kurman RJ, Carcangiu, ML, Herrington, CS, Young, RH, editors. WHO Classification of Tumours, Vol. 6. Lyon, WHO Press; 2014. pp. 125–6.
Bergeron C, Nogales FF, Masseroli M, et al. A multicentric European study testing the reproducibility of the WHO classification of endometrial hyperplasia with a proposal of a simplified working classification for biopsy and curettage specimens. Am J Surg Pathol. 1999;23:1102–8.
Kendall BS, Ronnett BM, Isacson C, et al. Reproducibility of the diagnosis of endometrial hyperplasia, atypical hyperplasia, and well-differentiated carcinoma. Am J Surg Pathol. 1998;22:1012–9.
Zaino RJ, Kauderer J, Trimble CL, et al. Reproducibility of the diagnosis of atypical endometrial hyperplasia: a Gynecologic Oncology Group study. Cancer. 2006;106:804–11.
Trimble CL, Method M, Leitao M, et al. Management of endometrial precancers. Obstet Gynecol. 2012;120:1160–75.
Bosse T, ter Haar NT, Seeber LM, et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol. 2013;26:1525–35.
Zhang X, Sun Q, Shan M, et al. Promoter hypermethylation of ARID1A gene is responsible for its low mRNA expression in many invasive breast cancers. PLoS ONE. 2013;8:e53931.
Porkka N, Valo S, Nieminen TT, et al. Sequencing of Lynch syndrome tumors reveals the importance of epigenetic alterations. Oncotarget. 2017;8:108020–30.
Vasen HF, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62:812–23.
Burn J, Mathers JC, Bishop DT. Chemoprevention in Lynch syndrome. Fam Cancer. 2013;12:707–18.
Acknowledgements
We thank the patients and responsible clinical experts for participation. Saila Saarinen is thanked for expert technical assistance. This work was supported by Jane and Aatos Erkko Foundation (to P.P. and J.-P.M.), the Academy of Finland (Grant No. 294643, to P.P.), the Finnish Cancer Organizations (to P.P. and J.-P.M), the Sigrid Juselius Foundation (to P.P.), the HiLIFE Fellows 2017–2020 (to P.P.), K Albin Johanssons Stiftelse (to A.N.), and the Integrative Life Science Doctoral Program ILS (to A.N.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Niskakoski, A., Pasanen, A., Lassus, H. et al. Molecular changes preceding endometrial and ovarian cancer: a study of consecutive endometrial specimens from Lynch syndrome surveillance. Mod Pathol 31, 1291–1301 (2018). https://doi.org/10.1038/s41379-018-0044-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0044-4
