
Abstract
Recurrent mutations in IDH1 or IDH2 in acute myeloid leukemia (AML) are associated with increased DNA methylation, but the genome-wide patterns of this hypermethylation phenotype have not been comprehensively studied in AML samples. We analyzed whole-genome bisulfite sequencing data from 15 primary AML samples with IDH1 or IDH2 mutations, which identified ~4000 focal regions that were uniquely hypermethylated in IDHmut samples vs. normal CD34+ cells and other AMLs. These regions had modest hypermethylation in AMLs with biallelic TET2 mutations, and levels of 5-hydroxymethylation that were diminished in IDH and TET-mutant samples, indicating that this hypermethylation results from inhibition of TET-mediated demethylation. Focal hypermethylation in IDHmut AMLs occurred at regions with low methylation in CD34+ cells, implying that DNA methylation and demethylation are active at these loci. AML samples containing IDH and DNMT3AR882 mutations were significantly less hypermethylated, suggesting that IDHmut-associated hypermethylation is mediated by DNMT3A. IDHmut-specific hypermethylation was highly enriched for enhancers that form direct interactions with genes involved in normal hematopoiesis and AML, including MYC and ETV6. These results suggest that focal hypermethylation in IDH-mutant AML occurs by altering the balance between DNA methylation and demethylation, and that disruption of these pathways at enhancers may contribute to AML pathogenesis.
Similar content being viewed by others


Introduction
DNA methylation changes in acute myeloid leukemia (AML) are caused by disruptions in the processes that add or remove 5-methyl groups to cytosines (5mC) [1, 2]. In normal and malignant hematopoietic cells, de novo DNA methylation is catalyzed primarily by the DNA methyltransferase DNMT3A [3, 4], which methylates unmethylated DNA substrates. Demethylation occurs passively after DNA synthesis in the absence of DNMT1-mediated propagation of hemi-methylated DNA, and actively via hydroxylation of 5mC by the TET family of hydroxylases. Alterations in these opposing forces result in either increased or decreased DNA methylation in AML cells. These changes include diffuse hypomethylation across large genomic regions and focal hypermethylation in CpG islands (CGIs). We recently showed that CGI hypermethylation in AML is mediated by DNMT3A and is present in nearly all AML subtypes [5]. In addition to these changes, specific DNA methylation patterns correlate with AML mutations that influence DNA methylation. This includes the DNMT3AR882 mutation, which impairs DNA methylation activity and results in a focal, canonical hypomethylation phenotype [5].
Mutations in IDH1 and IDH2 are also associated with altered DNA methylation patterns [6, 7] that are thought to occur by disrupting active DNA demethylation. IDH1 and IDH2 encode metabolic enzymes not normally involved in DNA methylation, but when mutated produce 2-hydroxyglutarate (2HG) [8] that inhibits the TET family of enzymes [9], thereby reducing active demethylation. Analysis of DNA methylation in primary AML samples using array-based technologies and enhanced reduced-representation bisulfite sequencing has demonstrated that DNA methylation is increased in samples with IDH mutations [6, 10]. While the direct effects of these changes on gene regulation have been challenging to identify, the contribution of IDH mutations to leukemogenesis has been established in mouse models. Expression of either IDH1R132H or IDH2R140Q blocks normal hematopoietic differentiation, promotes myeloproliferation [11,12,13], and can result in AML transformation in the presence of cooperating mutations [13, 14]. These studies establish the contribution of IDH mutations to AML development and suggest this may occur by disrupting the balance between DNA methylation and demethylation.
Although previous studies using targeted DNA methylation approaches have reported the general effects of IDH1 and IDH2 mutations on DNA methylation [6, 7, 10, 15], genome-wide methylation analysis in primary AML samples has not yet been described. It is therefore unclear whether IDH1 vs. IDH2 mutations cause hypermethylation at the same or different genomic loci, and whether these methylation changes are distinct from DNMT3A-mediated CGI hypermethylation. In addition, although IDH mutations are thought to cause hypermethylation via inhibition of TET enzymes, the overlap in methylation phenotypes between AML samples with these mutations is unclear. Here, we performed a genome-wide analysis of DNA methylation in primary AML samples with recurrent mutations in IDH1, IDH2, or TET2 using whole-genome bisulfite sequencing (WGBS). WGBS data from normal hematopoietic cells and AML samples with other mutational profiles were included to define the methylation phenotypes specific to IDH mutations and to determine their relationship to “generic” AML-associated methylation changes. We integrated these data with epigenetic modifications and three-dimensional (3D) genome architecture from primary AML samples to characterize the functional genomic elements that may be affected by disruption of the balance between DNA methylation and demethylation in AML.
Materials and methods
Patient samples
Primary AML samples and normal hematopoietic cells for epigenetic studies were obtained from presentation AML and normal bone marrow aspirates, following informed consent using the protocol (201011766) approved by the Human Research Protection Office at Washington University as described previously [5] (Table S1). All experiments with AML samples used total bone marrow cells for DNA preparation.
Whole-genome bisulfite and oxidative bisulfite sequencing and data analysis
WGBS data for 38 samples were described previously [5]. Data for 13 additional samples were generated using 50 ng of DNA with the Swift Accel-NGS Methyl-Seq library preparation kit. Oxidative bisulfite sequencing libraries were prepared following treatment of 200 ng of DNA with the TrueMethyl oxBS module (Cambridge Epigenetix) prior to bisulfite conversion and Swift library construction and sequencing on NovaSeq 6000 instruments (Table S1). Data were aligned to the GRCh38 reference and processed into methylated read counts using biscuit [16] with default parameters. Differentially methylated CpGs (DMCs) were identified between AML groups and CD34+ cells using read count data via DSS [17] and required a minimum methylation difference of 0.2. DMCs were then used to identify differentially methylated regions (DMRs) with >10 CpGs and a difference in mean methylation of 0.2. IDHmut- and TET2mut-specific DMCs and DMRs were subsequently identified by comparing these samples to all other AML samples via the DSS beta-binomial test in the methylkit Bioconductor package [18]. 5hmC values were obtained by subtracting the methylation ratios from OxWGBS data from WGBS data at all CpGs with coverage >10×.
ChIP-seq for histone modifications
ChIP-seq was performed using ChIPmentation [19] with the following antibodies: H3K27me3 (9733 S), and H3K27ac (8173 S) from Cell Signaling Technology, and H3K4me1 (ab1012) from Abcam. Sequencing was performed on a NovaSeq 6000 (Illumina, San Diego, CA) to obtain ~50 million 2 × 150 bp reads. Data were analyzed via adapter trimming with trimgalore and alignment to GRCh38 using bwamem [20]. Normalized coverage for visualization and analysis used the deeptools “bamCoverage” tool [21], and peaks were called with MACS2 [22]. Statistical comparisons with DESeq2 [23] used raw fragment counts at peak summits, and visualizations were prepared with Gviz [24]. Superenhancer analysis was conducted using ROSE software [25, 26] with default parameters.
RNA-seq analysis
RNA-seq data from AML samples were obtained from the AML TCGA study [15]. TPM values were obtained using kallisto [27] and gene counts were generated using the tximport Bioconductor package [28] in R with the tx2gene option set to accomplish gene-level summarization. Previously published RNA-seq data for normal CD34+ cells generated using the same procedures that were used for the AML samples [29, 30] were obtained as raw sequencing reads from the short-read archive (GSE48846) and processed as described above.
HiC data analysis
HiC data were obtained from previous studies of 3D genome interactions in primary AML samples [31] and normal hematopoietic stem/progenitors [32]. All libraries were generated using MboI digestion prior to proximity ligation and data were analyzed using the juicer pipeline [33]. Loops were identified with HICCUPs and were analyzed using bedtools [34] to identify overlap with genes and putative enhancers. Visualizations used the GenomicInteractions and Gviz R packages [24].
Results
Primary AML samples with IDH1 or IDH2 mutations are focally hypermethylated at regions with low methylation in normal hematopoietic cells
We performed WBGS using 15 primary bone marrow aspirate samples from AML patients with canonical IDH mutations, including seven with IDH1R132C/G, seven with IDH2R140Q, and one with an IDH2R172K allele (referred to hereafter as IDHmut). These data were analyzed with WGBS data from 36 other primary AML samples representing nine mutational categories, including five with biallelic loss-of-function mutations in TET2, and primary CD34+ cells from six healthy adults bone marrow donors [5]. All AML samples were previously sequenced using whole-genome and/or whole-exome sequencing [15, 35] that confirmed the mutations affecting DNA methylation were present in the dominant leukemic clone (Fig. 1A). Importantly, the 15 AML samples with IDH mutations were wild type for DNMT3A and TET2 to minimize the effects of other mutations on DNA methylation patterns. We first performed an unsupervised analysis of genome-wide methylation in 1 kb bins using principal component analysis. This demonstrated that most AML samples formed a diffuse cluster separate from CD34+ cells (Fig. 1B). AML samples with either DNMT3AR882 or IDH mutations (and some with TET mutations) formed sub-clusters on opposite sides of the main AML group, which is consistent with the hypomethylation phenotype of AML cells with the DNMT3AR882 mutation [5] and suggests IDHmut samples may also have unique methylation features compared to other AMLs.

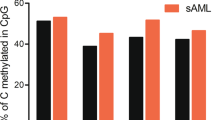
A Summary of the mutations in 51 primary AML patients analyzed using whole-genome bisulfite sequencing. B Principal component analysis of genome-wide methylation in AML samples and CD34+ cells. Points show the values of the first and second principal components by variance explained from an analysis of genome-wide methylation summarized in mean methylation in 1 kb bins, with colors representing the defining mutation for each sample. Stratification of IDHmut samples from CD34+ cells and AML samples with DNMT3AR882 mutations are highlighted by colored ellipses. C Two-way hierarchical clustering of relative (difference from CD34+ cells) mean methylation levels in genomic regions defined by 15 chromatin states [36] in CD34+ cells, where rows are AML samples and columns are chromatin states. Blue is less methylated than CD34+ cells and red is more methylated. IDH1 and IDH2 mutation status are indicated in the colored bar on the left, and selected chromatin states are shown underneath the panel. D Mean methylation levels in the enhancer chromHMM state (derived from publicly available CD34+ epigenetic data) from WGBS for CD34+ cells (N = 6) and AML subtypes (IDH1mut or IDH2mut, n = 15; TET2mut, n = 5; DNMT3AR882, n = 6; DNMT3AR882/IDHmut, n = 7; normal karyotype with NPM1c and wild-type IDH1, IDH2, TET2, and DNMT3A, n = 4; Normal karyotype with wild-type NPM1, IDH1, IDH2, TET2, and DNMT3A, n = 4; CBFB-MYH11, n = 3; MLL-ELL, n = 3; RUNX1-RUNX1T1, n = 3). E Mean methylation levels for ~28 million genome-wide CpGs in CD34+ cells and AML subtypes. F Mean methylation at CpG islands in CD34+ cells and AML subtypes. G Number of differentially methylated regions (DMRs) identified for each AML subtype compared with normal CD34+ cells. Teal and orange bars represent hypomethylated and hypermethylated DMRs with respect to normal CD34+ cells, respectively. Mean number of CpGs per DMR (top panel) and DMR length (bottom panel) are shown for each AML subtype. H Mean methylation in IDHmut DMRs in IDHmut samples versus CD34+ cells. The red line indicates the percent of all DMRs with mean methylation in CD34+ cells <0.3.
We next determined whether IDH mutations have global or context-dependent effects on DNA methylation by analyzing methylation levels in regions defined by chromatin states in hematopoietic stem/progenitors [36]. This demonstrated that quiescent and repressed chromatin states had lower methylation in most AMLs compared to CD34 cells, whereas bivalent regions (which are enriched for CGIs) were hypermethylated in nearly all samples (Fig. 1C). Enhancers and regions flanking transcriptional start sites (TSS) supervised a cluster of hypermethylated AMLs containing 14 of the 15 IDHmut samples. Mean methylation in IDHmut AMLs at enhancer regions was significantly higher vs. both CD34+ cells and AMLs without IDH mutations (Fig. 1D, P = 0.009 and P = 0.0002, respectively). IDHmut AMLs also tended to have higher mean methylation vs. other AML groups both genome-wide (Fig. 1E, adjusted P = 0.02) and in regions with other chromatin states (Figs. S1A, B), but not in CGIs (Fig. 1F; P = 0.14), indicating that IDH mutations do not result in an exaggerated CGI hypermethylation phenotype.
We next determined the extent to which IDH mutations result in focal methylation changes by performing differential methylation analysis [37] between AMLs with IDH1 or IDH2 mutations and CD34 cells. There were 6309 differentially methylated regions (DMRs) in IDH1mut AMLs, of which 99% were hypermethylated relative to CD34+ cells (methylation difference >0.2, FDR < 0.05 with >10 CpGs; Fig. 1G); this was more than any mutation-defined AML group. IDH2mut AMLs had fewer DMRs (N = 4195), although most were also hypermethylated (85%). AMLs with IDH1 or IDH2 mutations also had the highest fraction of hypermethylated CpGs (DMCs) (85% and 87%, respectively; see Fig. S1C), most of which were contained in DMRs (Fig. S1D, E). Interestingly, although IDH mutations are thought to inhibit active demethylation, most IDHmut DMRs had low methylation in normal hematopoietic cells. For example, 60% of the IDHmut DMRs had mean methylation <0.3 in both CD34+ cells (Fig. 1H) and more mature myeloid cell populations (Fig. S1F), suggesting that DNA methylation pathways must be active in these regions despite the low methylation levels at these loci in normal cells.
IDH mut-specific methylation changes are distinct from AML-associated CGI hypermethylation and are influenced by IDH mutation type
We next performed a second statistical comparison of the DMRs (and DMCs) identified in AMLs with IDH mutations vs. CD34+ cells to identify loci with methylation levels in the IDHmut samples that were significantly different from all other AML samples. AMLs with mutations in DNMT3A or TET2 were excluded from this analysis given their established hypomethylation phenotype (DNMT3A) and potential to phenocopy IDH mutations (TET2). This resulted in 4388 and 2552 IDH1mut and IDH2mut-specific DMRs, respectively, nearly all of which were hypermethylated relative to the other AML samples (Fig. 2A–C, Tables S2, 3). Similar results were observed at the DMC level (Fig. S2A, B). Most of these DMRs displayed low methylation in normal cells, with 60% of IDH1mut-specific and 58% of IDH2mut-specific loci having a methylation level <0.3 in CD34+ and mature myeloid cells (Fig. S2C, D). There was extensive overlap between the IDH mutation-specific DMRs (94% [2399/2552] of IDH2mut-specific DMRs overlapped an IDH1mut-specific DMR), and AML samples with either mutation were hypermethylated at both DMR sets (Fig. 2D). However, hierarchical clustering demonstrated considerable variability in methylation between the IDH1mut and IDH2mut samples (Fig. 2E). Notably, three IDH2mut AMLs had lower methylation across the union of IDHmut-specific DMRs (N = 4541). IDH2mut AML samples were also less methylated than IDH1mut samples at the combined set of IDHmut DMRs (0.54 vs. 0.70, respectively; P = 0.04), but were hypermethylated relative to CD34+ cells (Fig. 2E). This was not related to mutant IDH allele abundance (all samples had VAFs >30%, Table S1), and did not correlate with other recurrent mutations, including NPM1c (four in IDH1mut and 3 in IDH2mut samples, Fig. 2D; all samples were wild type for DNMT3A and TET2). Comparable differences in methylation were observed at the DMC level (Fig. S2E, F), suggesting this phenomenon was not an artifact of DMR identification.

A Mean methylation in all IDH1mut associated DMRs vs. CD34+ cells (n = 6309; black points) and in IDH1mut-specific DMRs that are unique compared with all other AMLs (n = 4388; red points). B Mean methylation in all IDH2mut associated DMRs vs. CD34+ cells (n = 4195; black points) and in IDH2mut-specific DMRs that are unique compared to all other AMLs (n = 2552; red points). C Locus heatmap showing mean methylation values by mutation group for the union of IDH1mut and IDH2mut-specific DMRs, where each column is centered over the DMR with the window extending 5 kb upstream and downstream of the DMR center. D Mean methylation across all IDH1/2mut-specific DMRs (rows) in 15 individual IDHmut cases (columns). The mutation status of IDH1, IDH2, and NPM1 are indicated by the colored bars above the heatmap. Note that all AML samples in this panel are wild type for DNMT3A and TET2. E Aggregate DMR methylation across 4388 IDH1mut specific DMRs and 2552 IDH2mut specific DMRs respectively. F Fraction of generically hypermethylated DMRs, IDH1mut-specific DMRs, and IDH2mut-specific DMRs overlapping functional genomic elements. G Distribution of CpG densities across generically hypermethylated regions in primary AML and IDH1mut-and IDH2mut-specific DMRs.
Interestingly, the IDHmut-specific DMRs demonstrated markedly different CpG density and overlap with genomic annotations compared to hypermethylated regions in other AML samples. For example, both IDH1mut-specific and IDH2mut-specific DMRs displayed significantly less overlap with annotated CGIs compared to 4573 hypermethylated regions identified in at least two other AML mutation categories (20% and 18% of IDH1mut and IDH2mut DMRs overlapped a CGI, respectively, compared to 54% of commonly hypermethylated regions; see Fig. 2F), and had lower CpG density (mean CpG density of 0.81 and 0.79 vs. 1.26, respectively; P values < 0.0001; Fig. 2G). Promoters were also underrepresented in IDH1mut-specific and IDH2mut-specific DMRs (21% and 20% of IDH1mut and IDH2mut DMRs overlapped a promoter, vs 31% of commonly hypermethylated regions; Fig. 2G). IDHmut-specific DMCs showed similar levels of overlap with annotated regions as DMRs (Fig. S2G), further suggesting that IDH-associated hypermethylation is distinct from AML-associated CGI hypermethylation.
Hypermethylation in TET2 mut AMLs overlaps with IDH mut-specific hypermethylation, but does not phenocopy the extent of methylation changes
We next determined whether AML samples with biallelic loss-of-function mutations in TET2 shared similar genome-wide patterns of hypermethylation with IDHmut AMLs. Initial comparison of the TET2mut AMLs vs. normal CD34+ cells yielded fewer DMRs (and DMCs) and a lower proportion of hypermethylated regions compared to the combined set of DMRs in IDHmut samples (1879 vs. 7569 DMRs, and 75% vs 99% hypermethylated regions, respectively; see Fig. 1G and S1A), consistent with previous reports [6, 10]. Hierarchical clustering of TET2mut samples with the set of IDHwt/TET2wt/DNMT3Awt AMLs at these regions did not reveal striking methylation differences between the two groups (Fig. S3A). Consistent with this result, only 188 TET2mut-specific DMRs were identified using the approach described above (with IDHmut and DNMT3AR882 AMLs excluded from the analysis) (Fig. 3A). Although most TET2mut-specific DMRs were hypermethylated relative to CD34+ cells and other AMLs (171 of 188), the fraction was less than in either IDH1mut or IDH2mut AMLs (89% vs 99% and 99%, respectively). Similarly, TET2mut-specific DMCs showed subtle hypermethylation (Fig. S3B). TET2mut-specific DMRs were also not enriched for CGIs and promoters compared to a set of regions commonly hypermethylated in AML (12% of TET2 DMRs overlapped a CGI vs. 54% of common hypermethylated DMRs; 17% of TET2 DMRs overlapped a promoter vs. 31% of hypermethylated DMRs; see Fig. S3C, D), suggesting these regions do not reflect CGI hypermethylation.

A Mean DMR methylation across 1879 TET2mut DMRs called vs. CD34+ cells (black points) and 188 in TET2mut-specific DMRs (red points) in TET2 mutant samples versus all other AMLs that are wild type for IDH, TET2, and DNMT3A. B Intersection of TET2mut-specific and IDHmut-specific DMRs. C Aggregate methylation over IDHmut-specific DMRs in IDHmut and in TET2mut AML and CD34+ cells. D Locus heatmap of mean methylation values for all IDHmut-specific DMRs (rows), where each column is centered over the DMR with the window extending 5 kb up- and downstream the DMR center point. E Mean 5hmC (WGBS minus oxWGBS) levels in IDHmut, TET2mut, and IDHwt/TET2wt AML samples at 105,519 ChromHMM heterochromatic regions, 4586 generically hypermethylated regions (i.e., regions that were hypermethylated vs. CD34+ cells in at least two AMLs without IDH1, IDH2, or TET2 mutations), and 4541 IDHmut-specific hypermethylated DMRs.
To investigate the interaction between IDHmut and TET2-mediated demethylation, we compared TET2mut-specific and IDHmut-specific DMRs and performed oxidative bisulfite sequencing [38] to measure 5-hydroxymethylation (5hmC) in TET2mut, IDHmut, and TET2wt/IDHwt samples. This analysis showed that 68% (127 of 188) of the TET2mut-specific DMRs overlapped an IDHmut-specific hypermethylated region (Fig. 3B; P < 0.0001 using a permutation test for overlaps using all DMRs identified in any AML group). TET2mut AMLs also displayed higher methylation levels at the combined set of 4541 IDHmut-specific DMRs compared to CD34+ cells (mean methylation of 0.35 vs. 0.26; 40% of DMRs with increased methylation via beta-binomial hypothesis testing with adjusted P < 0.05; Fig. 3C, D). Analysis of 5hmC using paired oxidative and standard whole-genome bisulfite sequencing (oxWGBS and WGBS with conversion rates ranging from 73–83%; see Fig. S3E) demonstrated low calculated levels of 5hmC across the genomes of all samples (0.44–0.66% in TET2mut, 0.52–1.22% in TET2wt, 0.17–0.25% in IDHmut; Fig. S3F), with higher levels in enhancer regions (Fig. S3G) and identifiable peaks at selected loci (Fig. S3H). Calculated 5hmC was statistically higher in IDHmut DMRs compared to regions that were hypermethylated in other AML samples or in constitutively methylated heterochromatic regions (adjusted P = 0.0009 and P = 4 × 10−7 for a difference in mean 5hmC in all samples at IDHmut DMRs vs. 4586 commonly hypermethylated DMRs and 105,519 heterochromatin regions, respectively; see Fig. 3E, S2I, J). AML samples with TET2, IDH1, or IDH2 mutations had lower calculated 5hmC levels at IDHmut DMRs compared to AMLs that were wild type for these genes (Fig. 3E, S2I, J), providing evidence that these mutations influence methylation turnover at these loci.
DNA hypermethylation in IDH mut AML cells requires DNMT3A
To assess whether de novo DNA methylation by DNMT3A contributes to IDHmut-associated hypermethylation, we analyzed methylation levels at IDHmut-specific DMRs in seven AML samples with co-occurring IDH1 (N = 5) or IDH2 (N = 2) and DNMT3AR882 mutations (R882 mutations have a more severe hypomethylation phenotype than other DNMT3A mutations [4, 39]). Interestingly, although DNMT3AR882/IDHmut AMLs were still hypermethylated at IDHmut-specific DMRs, the degree of hypermethylation was diminished, with 67% of these regions having significantly lower DNA methylation levels than samples with IDH mutations alone (3024 of 4541 regions having a beta-binomial adjusted P < 0.05; see Fig. 4A–C, S3A). Similar findings were observed in seven additional DNMT3AR882/IDHmut AML samples using methylation array data from the TCGA AML study [15] (Fig. S4B). To further characterize the extent of this interaction, we analyzed DNA methylation levels in DNMT3AR882/IDHmut AML samples at hypomethylated DMRs in AMLs with the DNMT3AR882 allele [5]. Surprisingly, these regions remained nearly fully methylated in the DNMT3AR882/IDHmut double mutant samples, with 93% of the regions having significantly higher methylation than AMLs with DNMT3AR882 alone (4209 of 4541 regions having a beta-binomial adjust P < 0.05; see Fig. 4D–F, Fig. S4C). Similar findings were observed in the AML TCGA data [15] (Fig. S4D), strongly suggesting that DNMT3A-mediated methylation and TET-mediated demethylation occur at the same places in the genome.

A Locus heatmap of mean methylation at IDHmut DMRs (rows) in IDH1 or IDH2 mutant, DNMT3AR882/IDHmut double mutant, and DNMT3AR882 AMLs, and CD34+ cells. B Distribution of IDHmut-specific DMR methylation levels by AML subtype. C Example IDHmut-specific DMR locus within the ETV6 gene demonstrating an intermediate methylation phenotype of double mutant samples with respect to IDHmut and DNMT3AR882 mutant AMLs. D Methylation locus heatmap of average subtype methylation across DNMT3AR882 DMRs called vs. CD34+ cells in IDHmut, DNMT3AR882/IDHmut double mutant, and DNMT3AR882 AMLs, and CD34+ cells. E Distribution of DNMT3AR882 DMR methylation levels by AML subtype. F Example DNMT3AR882 DMR locus within the MLLT1 gene, demonstrating the hypomethylation phenotype of DNMT3AR882 mutant samples with respect to IDHmut and DNMT3AR882/IDHmut double mutant AML samples.
IDH mut-specific hypermethylated DMRs are enriched for enhancers
We next asked whether IDHmut-specific DMRs were associated with certain chromatin states. Annotation of these DMRs with chromatin states in CD34+ cells [36] demonstrated that 44% occurred in enhancers, which was a twofold enrichment over regions commonly hypermethylated (Fig. 5A). This enrichment was not observed in analyses on DMRs identified in other AML subtypes (Fig. S5A–C). We further defined this association using ChIP-seq peaks for active, weak, and poised enhancers using ChIP-seq data for H3K27ac, H3K4me1, and H3K27me3 modifications from 16 primary AML samples, including two with IDH mutations. This demonstrated that 47% of the IDHmut DMRs overlapped an active enhancer, compared to 3% and 1% that overlapped poised and weak regions, respectively (Fig. 5B–D). In comparison, commonly hypermethylated regions showed less overlap with active enhancers (13% of DMRs) and greater intersection with repressive H3K27me3 marks (Fig. 5D). Analysis of IDHmut-specific DMRs for transcription factor (TF) binding motifs identified binding sites for hematopoietic-associated TFs, including SPI1, RUNX1, and MYC (Fig. 5E), further supporting the occurrence of IDHmut-specific hypermethylation in regions with potential regulatory activity. However, quantitative analysis of H3K27ac signal over these regions in samples with and without IDH mutations did not identify appreciable differences (P = 0.24, Fig. 5F), suggesting that hypermethylation does not modify H3K27ac levels within these regions.

A Distribution of ChromHMM chromatin states from CD34+ cells represented in IDHmut-specific DMRs. Enrichment of chromatin states within IDHmut-specific DMRs is shown with respect to the frequency of states overlapping regions of common CpG island hypermethylation. B Enhancer-based annotation of common hypermethylated regions, IDH1mut, and IDH2mut DMRs, where DMRs overlapping an H3K27ac peak alone or in combination with H3K4me1 were called active enhancers, those overlapping a H3K27ac peak in combination with H3K27me3 were called poised enhancers, and those overlapping a H3K4me1 alone were called weak enhancers. C Examples of intragenic and genic enhancer regions exhibiting IDH1mut, IDH2mut, or IDH1/2mut hypermethylation compared with CD34+ cells and other AML subtypes. D Heatmap of enhancer histone modifications and heterochromatin modifications over IDHmut-specific DMRs (left) and generic hypermethylation (right) in CD34+ cells (N = 4 H3K27ac, N = 7 H3K3me1, and N = 7 H3K27me3), IDHmut AML (n = 3), and IDHwt AML samples (N = 9 H3K27ac, N = 10 H3K3me1, and N = 24 H3K27me3). E HOMER motif enrichment analysis of IDH1/2mut-specific DMRs with respect to a background set of generically hypermethylated regions. F Differential active enhancer signal (H3K27ac) for all AML-associated putative enhancers (black points) compared with putative enhancers intersecting an IDH1/2mut-specific DMR (red points).
IDH mut-specific DMRs occur in enhancers that form direct interactions with highly expressed genes in AML cells
We next asked whether enhancers with IDHmut DMRs could be involved in controlling gene expression relevant for AML pathogenesis. To directly link these enhancers to their target genes, we analyzed 3D genome interactions generated using in situ HiC from both normal CD34+ cells [32] and three primary AML samples [31] (all were wild type for DNMT3A, IDH1, IDH2, and TET2). This analysis demonstrated that 26% (1158/4541) of all IDHmut-specific DMRs and 30% (602/2000) of the DMRs in putative enhancers overlapped the “loop anchor” of a genome interaction (Fig. 6A, Fig. S6A). IDHmut DMRs in these loop anchors were highly enriched in “superenhancers”, with between 37 and 39% of superenhancers defined in three primary IDHmut AML samples containing at least one IDHmut-specific DMR (Fig. 6B, C, Fig. S6B, C). We next analyzed gene expression in 750 genes with promoters that formed 3D interactions with IDHmut-specific DMRs using RNA-seq data from 179 AML samples from the TCGA AML study. This showed that the genes linked to IDHmut-specific DMRs were highly expressed, with 68% of these genes ranked in the top 25th percentile of gene expression (Fig. 6D, Fig. S6D). Further analysis of 3D genome interactions containing IDHmut-specific DMRs identified known and novel enhancers of genes important in hematopoiesis and AML, including an enhancer of MYC [40,41,42] (Fig. 6E), and previously unreported putative enhancers that form interactions with ETV6 (Fig. 6F), DOT1L, and SRSF3 (Fig. S6E, F). Although we did not observe significant changes in expression of these genes between IDHmut and IDHwt AMLs, their high expression in AML samples and CD34+ cells was consistent with the enrichment of IDHmut-specific DMRs in enhancers of active genes (Fig. 6D–F).

A Schematic of a DMR and an enhancer-associated DMR (eDMR) and their interaction with target genes based on intersection HiC-defined genome loops. B Rank ordered enhancer regions based on H3K27ac signal in a representative IDHmut AML sample, annotated by the presence of overlapping IDHmut-specific DMRs (absence of DMRs indicated by green points, greater than one DMR indicated by orange points) and computationally-defined “superenhancer” (above the red line). Enhancers of specific hematopoietic genes are labeled. C Distribution of the number of IDHmut-specific DMRs overlapping a set of AML consensus superenhancers from H3K27ac data from four primary samples (N = 779). D Distribution of normalized gene expression values for all expressed genes (orange histogram) and a set of 750 eDMR target genes (blue histogram) in IDHmut AML samples. E Example IDHmut-eDMR locus displaying interactions with the MYC promoter. A zoomed-in view of the locus demonstrates focal enhancer hypermethylation in IDH1mut (purple) and IDH2mut (green) samples compared with CD34+ cells (blue). Normalized MYC expression is shown for 17 CD34+ cord blood cell samples, 6 and 14 IDH1mut and IDH2mut samples, and 91 IDHwt samples. F Example IDHmut-DMR locus in a candidate enhancer that displays robust interactions with the ETV6 promoter. A zoomed-in locus view demonstrates focal enhancer hypermethylation in IDH1mut (purple) and IDH2mut (green) samples compared with CD34+ cells (blue). Normalized ETV6 expression is shown for CD34+ cells, IDH1mut, and IDH2mut samples, and IDHwt samples (see E for sample sizes).
Discussion
Recurrent gain-of-function IDH mutations increase DNA methylation, but the genomic locations and functional consequences of these changes have not previously been clearly defined. Our analysis of WGBS data from primary AML samples shows that methylation changes caused by these mutations are not widespread but instead manifest as thousands of focal regions that are uniquely hypermethylated compared to normal CD34+ cells and AML cells without IDH mutations. These regions had lower CpG density and fewer CGIs than loci that are commonly hypermethylated in AML, suggesting that IDHmut-associated hypermethylation is caused by a distinct mechanism. The IDH2mut AMLs in our dataset had less pronounced hypermethylation than those with IDH1 mutations, but both were hypermethylated at a highly overlapping set of loci. AMLs with biallelic inactivating TET2 mutations had a far less dramatic methylation phenotype, although many of the hypermethylated DMRs identified in these samples overlapped an IDHmut-specific DMR. Further, oxidative bisulfite sequencing demonstrated increased levels of 5hmC in these regions in AML samples that were wild type for TET2, IDH1, and IDH2; 5hmC levels were significantly lower in IDHmut or TET2mut samples in these regions, providing evidence that these mutations cause increased DNA methylation by impairing TET-mediated DNA demethylation. Regions with IDHmut-specific hypermethylation were enriched for active enhancers, many of which formed direct interactions with highly expressed AML genes, including MYC and ETV6. Although increased methylation at these loci was not associated with repressed chromatin or lower gene expression in IDHmut AML samples, this finding demonstrates that IDHmut-associated hypermethylation affects the regulatory sequences of genes that may contribute to AML pathogenesis.
This study adds new context to the dynamics of de novo DNA methylation and active demethylation pathways in normal hematopoietic cells and in AML. The fact that IDHmut-associated hypermethylation occurs at regions with low levels of DNA methylation in normal CD34+ cells suggests that de novo DNA methylation and TET-mediated demethylation are both active in these regions, despite their low steady-state methylation levels. This is supported by the observation that AML samples with co-occurring IDH and DNMT3AR882 mutations show significantly attenuated hypermethylation, and that IDHmut-specific DMRs have high levels of 5hmC, which is produced from 5mC as a substrate. Remodeling of DNA methylation by these processes in specific regions has been reported previously in studies of embryonic stem cells, which have shown that methylation and active demethylation are in equilibrium at many loci [1, 2], and may be maintained by the occupancy of methylation and demethylation complexes [43]. Our analysis suggests this equilibrium also exists in normal hematopoietic stem/progenitor cells and is disrupted in the presence of mutant IDH alleles, leaving de novo DNA methylation unopposed. The focal nature of IDHmut-associated hypermethylation implies that activity (or occupancy) of DNMT3A and TET enzymes is not diffuse and may instead be targeted to specific genomic regions. The genomic or epigenetic features directing this activity are unclear [44], but the enrichment of IDHmut DMRs in active enhancers suggests that components of active chromatin may recruit methylation and demethylation machinery. The convergence of these processes at enhancers could provide clues as to why mutations with opposite effects on DNA methylation both contribute to AML development, perhaps via dysregulation of common target genes.
Our analysis of 3D genome interactions involving IDHmut-specific DMRs found that these sequences directly interact with genes that are highly expressed in hematopoiesis and AML (e.g., MYC and ETV6). Contrary to the canonical relationship between DNA methylation and activity, hypermethylation in the IDHmut AML samples does not appear to repress either the enhancer elements or the expression of their target genes. Other regulatory factors may therefore be dominant to DNA methylation at these loci, and result in persistently high gene expression. It is also possible that regions of active chromatin, such as enhancers (and superenhancers), have high rates of methylation turnover, and are therefore more susceptible to perturbations in methylation and demethylation [1, 2]. Focal hypermethylation may occur in DNA elements bound by factors that contribute to “fine-tuning” these enhancers in specific cellular or developmental contexts, but that do not drive their activity in AML cells. Additional studies will be necessary to understand whether enhancer hypermethylation is a consequence of decreased occupancy of these modulating factors [45], or whether it directly prevents proper regulation in ways that contribute to AML development.
Data availability
All raw data from primary AML samples presented in this study are available in dbGaP (accession number phs000159). Processed data from this study are available for public download at the following site: https://wustl.box.com/v/wilsonIDHmethylation.
References
Charlton J, Jung EJ, Mattei AL, Bailly N, Liao J, Martin EJ, et al. TETs compete with DNMT3 activity in pluripotent cells at thousands of methylated somatic enhancers. Nat Genet. 2020;52:819–27.
Ginno PA, Gaidatzis D, Feldmann A, Hoerner L, Imanci D, Burger L, et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity. Nat Commun. 2020;11:2680.
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23.
Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014;25:442–54.
Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RS, et al. CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell 2017;168:801–816.e13.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. Plos Genet. 2012;8:e1002781.
Losman J-A, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013;339:1621–5.
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30.
Glass JL, Hassane D, Wouters BJ, Kunimoto H, Avellino R, Garrett-Bakelman FE, et al. Epigenetic identity in AML depends on disruption of nonpromoter regulatory elements and is affected by antagonistic effects of mutations in epigenetic modifiers. Cancer Discov. 2017;7:868–83.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24.
Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012;488:656.
Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14:329–41.
Yoshimi A, Lin K-T, Wiseman DH, Rahman MA, Pastore A, Wang B, et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019;574:273–7.
Ley TJ, Miller C, Ding L. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl J Med 2013;368:2059–74.
GitHub—huishenlab/biscuit: BISulfite-seq CUI Toolkit [Internet]. [cited 2021 Apr 7]. Available from: https://github.com/huishenlab/biscuit.
Feng H, Conneely KN, Wu H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014;42:e69–e69.
Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87.
Schmidl C, Rendeiro AF, Sheffield NC, Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat Methods. 2015;12:963–5.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; https://arxiv.org/abs/1303.3997.
Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–5.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Hahne F, Ivanek R. Methods in molecular biology. Methods Mol Biol Clifton N. J. 2016;1418:335–51.
Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34.
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013;153:307–19.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000research. 2016;4:1521.
Pabst C, Bergeron A, Lavallée V-P, Yeh J, Gendron P, Norddahl GL, et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood 2016;127:2018–27.
MacRae T, Sargeant T, Lemieux S, Hébert J, Deneault É, Sauvageau G. RNA-Seq reveals spliceosome and proteasome genes as most consistent transcripts in human cancer cells. Plos One. 2013;8:e72884.
Ghasemi R, Struthers H, Wilson ER, Spencer DH. Contribution of CTCF binding to transcriptional activity at the HOXA locus in NPM1-mutant AML cells. Leukemia. 2020;1–13.
Zhang X, Jeong M, Huang X, Wang XQ, Wang X, Zhou W, et al. Large DNA methylation nadirs anchor chromatin loops maintaining hematopoietic stem cell identity. Mol Cell. 2020;78:506–521.e6.
Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–8.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2.
Klco JM, Miller CA, Griffith M, Petti A, Spencer DH, Ketkar-Kulkarni S, et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. Jama 2015;314:811–22.
Ernst J, Kellis M. Chromatin-state discovery and genome annotation with ChromHMM. Nat Protoc. 2017;12:2478–92.
Wu H, Xu T, Feng H, Chen L, Li B, Yao B, et al. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015;43:gkv715.
Kernaleguen M, Daviaud C, Shen Y, Bonnet E, Renault V, Deleuze J-F, et al. Epigenome editing, methods and protocols. Methods Mol. Biol. 2018;1767:311–49.
Smith AM, LaValle TA, Shinawi M, Ramakrishnan SM, Abel HJ, Hill CA, et al. Functional and epigenetic phenotypes of humans and mice with DNMT3A overgrowth syndrome. Nat Commun. 2021;12:4549.
Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe J-S, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Gene Dev. 2013;27:2648–62.
Pulikkan JA, Hegde M, Ahmad HM, Belaghzal H, Illendula A, Yu J, et al. CBFβ-SMMHC inhibition triggers apoptosis by disrupting MYC chromatin dynamics in acute myeloid leukemia. Cell 2018;174:172–186.e21.
Bahr C, Paleske L, von, Uslu VV, Remeseiro S, Takayama N, Ng SW, et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018;553:515–20.
Dixon G, Pan H, Yang D, Rosen BP, Jashari T, Verma N, et al. QSER1 protects DNA methylation valleys from de novo methylation. Science 2021;372:eabd0875.
Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 2019;573:281–6.
Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature 2012;489:75.
Acknowledgements
This work was supported by the National Cancer Institute (K08CA190815) and The Cancer Research Foundation Young Investigator Award to D.H.S. Support for procurement, annotation, and sequencing of human samples was provided by the Genomics of AML Program Project (P01CA101937, to Dr. Ley) and the Specialized Program of Research Excellence in AML (P50CA171963, to Dr. Link) from the NCI.
Author information
Authors and Affiliations
Contributions
Conception and design: E.R.W., D.H.S. Acquisition of data: S.E.H., R.S.F., J.S.W., J.E.P., P.W., M.J.W., J.F.D., D.C.L., C.A.M., T.J.L. Data processing, statistical and computational analysis: E.R.W., D.H.S. Manuscript preparation: E.R.W., D.H.S. Scientific input and manuscript comments: J.S.W., T.J.L. Administrative, technical, or material support: S.E.H., R.S.F., J.E.P., P.W., M.J.W., J.F.D., D.C.L., C.A.M., T.J.L., D.H.S. Study supervision: D.H.S.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wilson, E.R., Helton, N.M., Heath, S.E. et al. Focal disruption of DNA methylation dynamics at enhancers in IDH-mutant AML cells. Leukemia 36, 935–945 (2022). https://doi.org/10.1038/s41375-021-01476-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-021-01476-y
This article is cited by
-
Bridging tissue repair and epithelial carcinogenesis: epigenetic memory and field cancerization
Cell Death & Differentiation (2024)
-
Crosstalk between DNA methylation and hypoxia in acute myeloid leukaemia
Clinical Epigenetics (2023)
-
TET2 lesions enhance the aggressiveness of CEBPA-mutant acute myeloid leukemia by rebalancing GATA2 expression
Nature Communications (2023)
-
Isocitrate dehydrogenase 1 mutation drives leukemogenesis by PDGFRA activation due to insulator disruption in acute myeloid leukemia (AML)
Leukemia (2023)
-
Perturbed epigenetic transcriptional regulation in AML with IDH mutations causes increased susceptibility to NK cells
Leukemia (2023)
