
Abstract
Assessment of measurable residual disease (often referred to as “minimal residual disease”) has emerged as a highly sensitive indicator of disease burden during and at the end of treatment and has been correlated with time-to-event outcomes in chronic lymphocytic leukemia. Undetectable-measurable residual disease status at the end of treatment demonstrated independent prognostic significance in chronic lymphocytic leukemia, correlating with favorable progression-free and overall survival with chemoimmunotherapy. Given its utility in evaluating depth of response, determining measurable residual disease status is now a focus of outcomes in chronic lymphocytic leukemia clinical trials. Increased adoption of measurable residual disease assessment calls for standards for nomenclature and outcomes data reporting. In addition, many basic questions have not been systematically addressed. Here, we present the work of an international, multidisciplinary, 174-member panel convened to identify critical questions on key issues pertaining to measurable residual disease in chronic lymphocytic leukemia, review evaluable data, develop unified answers in conjunction with local expert input, and provide recommendations for future studies. Recommendations are presented regarding methodology for measurable residual disease determination, assay requirements and in which tissue to assess measurable residual disease, timing and frequency of assessment, use of measurable residual disease in clinical practice versus clinical trials, and the future usefulness of measurable residual disease assessment. Nomenclature is also proposed. Adoption of these recommendations will work toward standardizing data acquisition and interpretation in future studies with new treatments with the ultimate objective of improving outcomes and curing chronic lymphocytic leukemia.
Similar content being viewed by others

Introduction
In patients with chronic lymphocytic leukemia (CLL) treated with fixed-duration regimens, such as chemoimmunotherapy (CIT), end-of-treatment response by standard criteria correlates with time-to-event endpoints, including progression-free (PFS) and overall survival (OS) [1,2,3,4,5]. Complete remission (CR) by National Cancer Institute-sponsored Working Group/international Workshop on Chronic Lymphocytic Leukemia (NCI-WG/iwCLL) criteria is associated with superior outcomes. Achieving this depth of response has been the therapeutic goal. However, CR is not disease eradication. Advanced diagnostic methods have enabled detection of very low levels of disease in peripheral blood (PB) and bone marrow (BM). Low but measurable persistent CLL is present in many CIT-treated patients, including those achieving CR.
CLL cells are found in lymphoid tissues and circulate through blood and lymphatics. Measurable residual disease (MRD; often referred to as “minimal residual disease”) is distinct from standard response, providing additional independent prognostic information. MRD is a sensitive reflection of disease burden during and after fixed-duration treatment and has been correlated with PFS and OS (Table 1). Because Bruton tyrosine kinase inhibitor- and PI3K inhibitor-based treatments are continuous and responses are not deep, achieving undetectable-MRD (U-MRD) with such monotherapies is uncommon [6]. Depth of remission with BCL2 inhibitor-based monotherapy (venetoclax) or in combination with a CD20 monoclonal antibody (mAb) is greater, and such combination regimens are of fixed-duration. Furthermore, data with fixed-duration combined Bruton tyrosine kinase inhibitors and BCL2 inhibitors (with or without CD20 mAb) indicate high CR and U-MRD rates [7,8,9,10,11,12]. Further data are needed to clarify the association of MRD status with time-to-event endpoints in these regimens.
Given its utility in evaluating depth of response and identifying treatment superiority in randomized trials, MRD determination is increasingly adopted in CLL trials, frequently as a co-primary or secondary endpoint [13]. Although PFS and OS remain regulatory endpoints, MRD status is an important surrogate approved by the European Medicines Agency as an intermediate endpoint for CLL trials. iwCLL guidelines recommend that “in clinical trials aimed at maximizing the depth of remission, the presence of MRD after therapy should be assessed [14].” It is, however, not yet applicable to standard community care.
As highly effective treatments achieving deep remission emerge, it becomes necessary to standardize new methods for assessing disease and response. Many relevant questions must be addressed—e.g., whether it is optimal to assess MRD in PB or BM, at which timepoint(s) to evaluate MRD, by which analysis method, and at what sensitivity. These issues have been touched on by the iwCLL and US and European regulatory agencies [14,15,16].
Here, we present a consensus document from an international, multidisciplinary, 174-member panel convened to assemble critical questions on key issues pertaining to MRD in CLL, review available data, develop unified answers with local expert input, and provide recommendations for future efforts.
Methods
An international steering committee (ISC) was convened in Paris in March 2017. Key topics regarding the assessment and utility of MRD in CLL were identified, and a set of 84 pertinent questions was drafted. National Faculties (Supplementary Appendix) subsequently ranked these questions by importance (“How important does the National Faculty think this question is with regard to integrating MRD measurement into clinical practice?”) and timing (“How soon does the National Faculty predict that the answer to this question would have an impact on clinical practice?”). The sum of the scores for each question determined its ranking. An initial set of answers to the 13 highest-ranking questions based on ISC opinion and literature review was drafted and then refined by the National Faculties at a series of local/regional meetings (Supplementary Fig. S1). National Faculties voted on level of agreement with draft answers on a scale of 1–9, with agreement defined as ≥75% voting in the 7–9 range. If agreement was lacking after the first vote, the answer could be refined after discussion and National Faculties could vote a second time. If agreement was not achieved after a second vote, then no agreement was recorded. The consolidated feedback was reviewed by the ISC in Amsterdam in March 2018. Answers were finalized at a June 2018 meeting of the ISC and additional advisors (Supplementary Appendix) in Stockholm. The resulting report provided the basis for the consensus document presented herein. All authors reviewed/approved the submitted version of this manuscript.
The ISC was composed of 8 members (authors WGW, AR, FC, XB, JRB, PG, SS, PH), and the National Faculty was composed of 166 members for a total 174-member panel. ISC members were selected by AbbVie Global Medical Affairs after consideration of key markets, recognized international expertise, and any recommendations by another ISC member. National Faculties were nominated by AbbVie affiliates and reviewed by AbbVie Global Medical Affairs and the ISC. National Faculty selection criteria included the following: certified hematologist/oncologist; medical, clinical, and/or professional experience and scientists conducting relevant research; national, regional, or local distinction; credentials, including knowledge and experience in the applicable therapeutic or business area; and progressive approach to patient care.
Nomenclature
Recommended standardized nomenclature for MRD is provided (Table 2). Although “MRD” is often defined as “minimal residual disease,” the term “minimal” is subjective. “Measurable residual disease,” which is unambiguous when the detection limit is specified, is recommended. The MRD report should either state the percentage of disease involvement in the specified tissue or that disease was not detectable. In either situation, it is critical that the report also provides the detection limit for the sample, because this will depend on both the assay used as well as the sample quality, primarily based on number of cells or amount of DNA available for analysis. A categorical measure indicating the upper limit of MRD should also be noted. We recommend “MRD4” to identify cases with <10−4 MRD (<1 CLL cell per 10 000 leukocytes, or <0.01%), “MRD5” to identify cases with <10−5 MRD (<1 CLL cell per 100,000 leukocytes, or <0.001%), etc.
“Undetectable-MRD” (U-MRD) is preferable to “MRD-” or “MRD negative” as a general term to describe the inability to detect measurable disease at a specified reporting threshold, because disease may be detectable below this level. Because it is now common to use an assay with better sensitivity than the reporting threshold (e.g., clinical trials reporting the MRD4 rate often use an assay with a detection limit of 1 CLL cell in 100,000 or 1 million leukocytes), it is more informative to follow the approach recommended by the CML community [17] and use detectable/undetectable to provide additional information on assay sensitivity and whether disease is detectable below the reporting threshold. So, the category “MRD4” would indicate MRD < 0.01%/<1 CLL cell per 10,000 leukocytes but does not specify whether disease is detectable or not below this level. MRD4d (detectable) indicates that an assay capable of detecting disease at the 0.001% threshold and residual disease is above this level but <0.01% (between 10−4 and 10−5). MRD4u (undetectable) indicates that residual disease is <0.01%, but the assay was not capable of detecting 0.001% disease due to assay or sample limitations.
MRD level may differ in the PB compared with that in the BM and so the sampled tissue (e.g., PB, BM) must be specified. The method used to determine MRD (e.g., flow cytometry [flow], polymerase chain reaction [PCR], next-generation sequencing [NGS]) should be specified. When reporting U-MRD rates for clinical trials, rates should be calculated based on the full intention-to-treat population. Missing values should be counted as positive by default; other ways of reporting must be clearly explained.
Quantifying MRD
MRD methodology
With improved methodology, the sensitivity at which CLL cells can be measured continues to increase. Limitations exist regarding sample volume, assay time, and use of cellular versus molecular assays. The minimum sensitivity limit for response assessment is 10−4, demonstrated to be an independent prognostic factor in patients treated with first-line CIT [18,19,20,21,22,23,24]. Assays require prospective technical validation and must undergo cross-laboratory standardization with external quality assurance (EQA) procedure [25, 26]. Available assays include ≥4 color flow cytometry and immunoglobulin heavy-chain variable region (IGHV) real-time quantitative PCR (RQ-PCR) capable of quantifying MRD at the 10−5 level (Table 3). Still more sensitive technologies undergoing validation include high-throughput sequencing/NGS and droplet digital PCR [27, 28].
In clinical trials, European Research Initiative in CLL (ERIC)-compliant flow and EuroMRD-compliant RQ-PCR are most common, typically performed at specialized centers [12, 25, 29,30,31]. For flow cytometry, the four-color assay is the historical gold standard, but six- and eight-color flow are also available. Four-color flow (CD5/CD19 with CD20/CD38, CD81/CD22, and CD79b/CD43) and RQ-PCR have undergone clinical validation and cross-laboratory standardization [25, 26, 32]. Flow cytometry is optimal for rapid turnaround, whereas RQ-PCR requires a pretreatment sample to determine the target sequence and patient-specific primers, making it more suitable for batch processing (e.g., at trial completion). However, samples <48 h old are preferred for flow cytometry, though can be accepted to <72 h [33], whereas PCR and NGS can use stored DNA.
Consensus recommendations
Only validated assays are recommended. Validated methods include ERIC-compliant flow cytometry and EuroMRD-compliant RQ-PCR. The choice of assays depends upon the rationale for MRD determination. The minimum sensitivity required for regulatory approval is MRD4 (10−4), whereas evaluating curative approaches may require the most sensitive method available, based on local availability and/or economic restrictions. When reporting MRD data, the quantification and/or detection limit should be stated for each sample. Method validation and standardization information should be provided. Providing quantitative results for detectable MRD is also recommended.
MRD assay requirements
Standardization is the process of developing and implementing technical standards based on multiparty consensus. Documented standards for flow (ERIC) and RQ-PCR (EuroMRD) have been reported [25, 26]. Harmonization is the process of coordinating different systems, creating minimum requirements or standards to ensure global reproducibility [34]. Validation comprises analytical validation (for accuracy, precision, and reproducibility) and clinical validation, ensuring that the assay effectively associates with clinical outcome [34]. Verification evaluates whether a validated assay meets the required standard. Quality assurance (QA) or conformity schemes are required for localized testing [15].
Consensus recommendations
Harmonized MRD testing is essential for globally reproducible results. MRD testing may be performed in central laboratories, regional centers, or local hospital laboratories, provided the assay meets accepted standards. When using a proprietary method, adequate validation is required. Each testing center must be certified by internal quality assurance (IQA) and external quality assurance (EQA) procedures and demonstrate assay validation. QA, validation, proficiency testing, training to standardized techniques, and adequate infrastructure for appropriate sample handling are required.
Tissue for MRD assessment
In both PB and BM, MRD status is strongly prognostic for PFS and OS in patients with CLL treated with first-line CIT (Table 1). The multi-compartment nature of CLL (PB, BM, lymph nodes, liver, spleen), however, suggests the possibility of discordant MRD results when sampling different tissues; thus, the sampling site may affect its prognostic ability. Timing of sampling (along the course of disease) is also critical. Concordance between PB and BM MRD status is ~85% at the 10−4 threshold and can be affected by treatment type [18,19,20, 25, 35]. Based on clinical trials, BM assessment may be necessary for U-MRD with certain monoclonal antibody-containing regimens where a relevant discrepancy between PB and BM exists. Studies have shown low PB and BM concordance for patients receiving alemtuzumab (48.6% concordance) [25] or rituximab (79% concordance; 0.7 log lower disease in PB compared to BM) [18, 35]. BM is the most sensitive source for MRD assessment following CIT [25]. Residual disease in spleen, liver, and lymph nodes may, however, play a role in relapse. Current testing methods do not assess these sites [36].
Consensus recommendations
In clinical trials aimed at disease eradication, MRD status should be assessed in both PB and BM. Designing strategies and/or more sensitive techniques allowing PB to be used to monitor MRD is desirable. MRD assessment in PB is useful for screening and informing BM aspiration decisions. If MRD is detected in blood, no BM aspiration is needed. PB U-MRD, however, calls for BM aspiration for confirmatory purposes in regimens where a relevant discrepancy between PB and BM exists. Novel strategies to detect MRD across all disease compartments should be developed, and the utility of circulating cell-free DNA as an MRD measure should be explored. The interchangeability of various MRD methods, tissues, and compartments should be investigated.
Timing and frequency of MRD assessment
Most CLL trials reporting MRD data evaluated CIT regimens (Table 1). Targeted therapy MRD data are limited, as are data on optimal timing for MRD testing and MRD kinetics data. Identifying the optimal clinically relevant timepoint for MRD testing requires a greater understanding of CLL kinetics, including MRD clearance and re-emergence and timing of relapse following U-MRD achievement.
Consensus recommendations
For fixed-duration therapies, MRD testing should be aligned with response assessment, at least 2 months after completion of the last treatment [14]. For continuous treatment, MRD status should be tested when best clinical response has been achieved; fixed testing timepoints are recommended for clinical trial design. Importantly, patients with PR may achieve U-MRD; therefore, MRD assessment should not be limited to patients with CR. Prospective studies evaluating time-to-U-MRD and time-to-MRD relapse should be conducted to determine their value as secondary endpoints. Measuring clonal growth kinetics by serial MRD assessment could be important in considering U-MRD as a surrogate marker for PFS in fixed-duration therapies. Clinical trials should assess MRD kinetics and its correlation with time-to-event outcomes.
MRD in response assessment
U-MRD significance
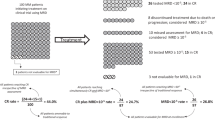
In first-line CIT treatment of CLL, MRD status was independently associated with extended treatment-free survival, PFS, and OS [19, 21,22,23,24, 37,38,39]. U-MRD is more accurately associated with survival than conventional responses after first-line CIT. In this setting, patients achieving U-MRD and PR may have a prognosis superior to that of MRD-positive patients achieving CR (Fig. 1) [21]. The impact of MRD status on treatment outcome after first-line CIT may, however, differ by disease biology (e.g., IGHV mutation status) [24].

PFS (A and C) and OS (B and D) at end of treatment by PB MRD and additional response status. Reprinted with permission from Kovacs G, Robrecht S, Fink AM, et al. Minimal residual assessment improves prediction of outcome in patients with chronic lymphocytic leukemia (CLL) who achieve partial response: comprehensive analysis of two-phase III studies of the German CLL Study Group. J Clin Oncol. 2016;31:3758–3765. BM, bone marrow; CR, complete response; MRD-, minimal residual disease negative; MRD+, minimal residual disease positive; OS, overall survival; PFS, progression-free survival; PR, partial response.
In relapsed/refractory CLL, U-MRD status correlates with longer PFS and OS [39,40,41,42]. These benefits, however, may be less than those seen in the first-line. Additional trial data are needed regarding MRD status correlation with survival benefit for novel agents and combinations.
Consensus recommendations
The relationship between clinical response (i.e., iwCLL response) and end-of-treatment MRD status requires clarification. Because this may differ between treatment strategies, it should be evaluated in a treatment-specific context. The relationship between end-of-treatment MRD status and PFS and OS may also depend on other factors. Further studies are needed to clarify effects of prior treatment, prior response, IGHV mutational status, del(17p)/mutated TP53 status, cytogenetic abnormalities, and other variables.
U-MRD as a potential surrogate endpoint
In first-line, defined-length CLL treatment, a prognostic association exists between U-MRD and improved outcomes [19, 21, 23, 24, 43, 44]. In relapsed/refractory CLL, preliminary evidence suggests a valid association between U-MRD and improved outcomes with BCL-2 inhibitor treatment (venetoclax ± rituximab), irrespective of del(17p) status [41, 42, 45, 46]. To date, no correlation between MRD status and time-to-event outcomes has been established for B cell receptor (BCR) inhibitor monotherapy or combinations with CD20 mAbs. BCR inhibitor-based treatment is uncommonly associated with U-MRD and is continuous and indefinite, which likely impacts this.
The value of U-MRD as a surrogate for time-to-event endpoints has not been proven in prospective trials. Consequently, MRD is currently not accepted as a surrogate endpoint by regulatory authorities. European Medicines Agency guidelines for anticancer medicinal products state that U-MRD in patients with CLL who are in clinical CR may be used as an intermediate endpoint for licensure in randomized well-controlled studies designed to show PFS superiority provided certain conditions are met [15]. Outside of clinical trials and the setting of allotransplantation, evidence is insufficient to support modification of patient management based on MRD assessment.
Consensus recommendations
Clinical trials should investigate U-MRD rates for different therapies, U-MRD duration, and its impact on outcome. The role of MRD as a surrogate for PFS, specific to treatment type, needs validation, requiring MRD assessment in as many trials as possible, independent of the likelihood of achieving U-MRD. Trials should evaluate whether U-MRD is more reliable in predicting outcomes than clinical response and whether MRD-driven management modifications lead to improved outcomes. U-MRD status may be a suitable endpoint in trials designed to evaluate depth of response (even in the absence of a comparator), and because of its correlation with PFS, it may also be used in trials of fixed-duration treatments to inform treatment-stopping decisions. Patients should be followed until progression and next CLL treatment.
Optimal method of response assessment in CLL
Although clinical response and MRD are correlated, each is independently prognostic for outcome [18, 19, 21]. With CIT, MRD is more strongly prognostic than clinical response for PFS [18, 19, 21]. Clinical response has varying significance, depending on MRD status [21].
Consensus recommendations
In clinical practice, the optimal response assessment method depends on individual patient status, type of treatment, and treatment goal. At a minimum, response assessment should include full blood count and clinical examination. BM examination and CT scan may also be included. In clinical trials, BM examination and CT scan should be assessed, and MRD assessment is recommended to inform prognosis and quality of response, and to potentially identify candidates for MRD-driven changes in treatment duration [23, 37, 47].
Treatments inducing U-MRD in CLL (Table 4)
Fludarabine, cyclophosphamide, and rituximab (FCR) treatment has a high likelihood of inducing U-MRD [19, 48,49,50]. First-line treatments inducing U-MRD include chlorambucil and obinutuzumab [44], chlorambucil and ofatumumab [51], bendamustine and rituximab [48], bendamustine and obinutuzumab [52, 53], and fludarabine, cyclophosphamide, and obinutuzumab [54]. BCR pathway inhibitor-based therapy (ibrutinib, idelalisib) rarely results in U-MRD status [55], but early data indicate that ibrutinib and venetoclax ± obinutuzumab can lead to U-MRD [9, 12, 56]. Venetoclax and CD20 mAbs can lead to U-MRD in first-line and relapsed/refractory CLL [57,58,59,60,61].
In relapsed/refractory CLL, CIT can sometimes induce U-MRD [62]. Ibrutinib combined with rituximab or obinutuzumab, or with bendamustine and rituximab, can induce U-MRD [7, 8, 63]. Venetoclax monotherapy can achieve U-MRD in patients with or without del(17p) [42, 64], but higher rates have been reported for venetoclax combined with either a CD20 mAb or ibrutinib [41, 61]. Data on U-MRD durability for most therapies are lacking.
Consensus recommendations
Clinical trials of defined-duration CLL treatments should assess MRD at least 2 months after the last treatment cycle is completed for correlation with PFS and OS.
Disease-related factors prognostic of U-MRD
Type of therapy and line of treatment have the strongest associations with achieving U-MRD [22]. CIT, mutated IGHV, wildtype TP53, and absence of del(17p) are associated with a higher likelihood of achieving U-MRD [19, 22,23,24, 49, 65]. Other factors may include cytogenetic abnormalities and age [22, 23, 65].
Consensus recommendations
Factors associated with achieving U-MRD should be identified for each agent/regimen. Collaborative efforts are important for elucidating MRD biology for emerging therapies and correlations with outcomes. Ideally, for each treatment strategy, U-MRD objectives and methods should be defined at treatment initiation.
MRD relapse
MRD relapse has not been defined. Reports have used newly detectable disease above the 10−4 threshold on two consecutive PB tests [18, 66]. Relapse dynamics may vary according to disease characteristics, prognostic factors, and/or treatment [67]. MRD relapse may be used as a marker of subclinical progression in clinical trials to inform on disease kinetics, treatment re-initiation, and relationship with clinical relapse. In clinical practice, MRD relapse status is not currently used to inform treatment decisions. The potential benefit of early treatment for MRD relapse (versus waiting for clinical relapse) has not been systematically investigated.
Consensus recommendations
Serial MRD testing is not indicated in routine practice; MRD relapse currently has no impact on treatment decisions for standard of care. We propose that it be defined as detectable MRD (>10−4) on at least two consecutive timepoints in PB. The optimal time between the two positive tests should be evaluated in trials. Further studies are needed to define what constitutes MRD relapse and should include correlation with subsequent clinical relapse and its timing. Trials evaluating the possible benefit of treating MRD relapse or asymptomatic, progressive disease are needed. These will likely require randomization to therapy versus observation until clinical relapse.
Clinical utility of MRD assessment
MRD in clinical practice versus clinical trials
Current guidelines do not recommend routine MRD testing in clinical practice [67, 68]. One trial demonstrated that patients achieving U-MRD early in first-line CIT treatment have a prognosis similar to those achieving U-MRD later in treatment [23]. Two trials demonstrated that patients achieving U-MRD had a longer duration of PFS compared to patients with detectable MRD, suggesting that MRD may be a prognostic factor with some therapies [60, 61]. Lacking further prospective studies, however, this is insufficient evidence to change clinical practice. Data relating MRD and outcomes for novel nonchemotherapeutic treatments are limited.
Consensus recommendations
In clinical trials, MRD may be explored for modifying treatment duration or for determining whether switching treatment strategies may be beneficial. To transition MRD assessment from trials into routine clinical practice, however, data demonstrating that such modifications lead to improved outcomes are needed.
U-MRD and quality of life (QoL)
Currently, no data associate U-MRD with QoL. Potentially, using U-MRD to inform when to stop long-term therapy could impact patient QoL. U-MRD is a prerequisite for curing CLL but does not in itself indicate cure.
Consensus recommendations
Data from CLL studies that included MRD and QoL assessments should be retrospectively analyzed for a possible link. Future trials assessing MRD status should evaluate the effect of achieving U-MRD on QoL parameters and include CLL-adapted QoL questionnaires. Durable disease control with minimal toxicity and improved QoL without U-MRD achievement may be a suitable treatment goal but requires clinical validation.
Future MRD utility
MRD status offers a highly sensitive endpoint that may be used to design novel treatment strategies.
Consensus recommendations
The usefulness of MRD status in clinical practice depends on available therapies and setting as well as the cost and infrastructure required for assessing MRD. Currently, there is no clear role for MRD assessment in routine practice. Once sufficient data are available, MRD status may potentially inform decisions on when to stop or adjust treatment. Trial data for MRD and “omics” may prove useful in elucidating the most effective treatments and differentiating between subgroups. Clinical trials with fixed-duration treatment should be designed to achieve the deepest remission in the highest proportion of patients, evaluating MRD status with a test in PB at a sensitivity of at least 10−4 to correlate with time-to-event endpoints.
Discussion
Advances in bioanalytics have provided sensitive techniques for quantifying depth of response in patients with CLL. A large body of evidence indicates that MRD status is independently prognostic for time-to-event outcomes (Table 1). Its significance in CLL is complex and multifactorial, but MRD testing has already become widespread in CLL clinical trials. Future studies will determine whether MRD status may inform treatment decisions and/or represents an accepted surrogate endpoint in clinical trials of new CLL therapies. The current utility and future potential of MRD in CLL underscore the need to use standardized, validated assays; adopt a routine, unambiguous nomenclature; and report assay results systematically. This international panel was convened to provide expert guidance on these issues.
We strongly recommended that trials of fixed-duration treatments designed to achieve deepest remission in the highest proportion of patients evaluate MRD status in PB at a sensitivity of at least 10−4 to correlate with time-to-event outcomes. At present, however, there is no clear role for MRD status determination in routine CLL clinical practice.
CLL treatment has markedly evolved over the last 10 years. Alkylating-agent and purine analog monotherapies have developed into combination chemotherapy. Subsequently, CD20 mAbs have been added. The most active CIT regimen, FCR, achieved CR in the majority of patients, with U-MRD in approximately one-half of first-line patients [48]. A challenge with FCR, however, is myelosuppression, limiting its use to younger, fit patients, and long-term toxicities can be concerning. Approximately 50–55% of treated first-line patients with mutated-IGHV are progression-free for >10 years post-therapy and may be cured [24]. These outcomes initiated the focus on MRD as an important endpoint.
Focus, however, shifted with development of the BCR signaling inhibitors ibrutinib and idelalisib [6, 69, 70]. These achieved extremely durable disease control, including in high-risk, relapsed/refractory patients, and even more durable responses in the first-line. Most patients achieved remission, but most responses were partial, and treatment needed to be administered indefinitely. Interest in U-MRD as an endpoint consequently waned.
More recently, venetoclax, a BCL2 inhibitor that potently induces apoptosis in CLL cells, was developed [57, 61, 64]. It is highly effective in eliminating disease and is FDA-approved for treatment of adults with CLL or small lymphocytic leukemia. Venetoclax may be used for continuous monotherapy or for fixed-duration treatment when combined with a CD20 mAb. Venetoclax-based treatment not only achieves a higher rate of U-MRD in first-line and relapsed CLL than CIT does, but it also is well-tolerated, including in older patients, making deep remission and U-MRD with fixed-duration treatment a realistic goal [41, 71]. Thus, the interest and clinical importance of MRD as a treatment objective are more important than ever for patients and clinicians.
References
Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–74.
Hillmen P, Skotnicki AB, Robak T, Jaksic B, Dmoszynska A, Wu J, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol. 2007;25:5616–23.
Kay NE, Geyer SM, Call TG, Shanafelt TD, Zent CS, Jelinek DF, et al. Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood. 2007;109:405–11.
Tam CS, O’Brien S, Wierda W, Kantarjian H, Wen S, Do KA, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112:975–80.
Wierda W, O’Brien S, Wen S, Faderl S, Garcia-Manero G, Thomas D, et al. Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab for relapsed and refractory chronic lymphocytic leukemia. J Clin Oncol. 2005;23:4070–8.
Ahn IE, Farooqui MZH, Tian X, Valdez J, Sun C, Soto S, et al. Depth and durability of response to ibrutinib in CLL: 5-year follow-up of a phase 2 study. Blood. 2018;131:2357–66.
Fraser G, Cramer P, Demirkan F, Silva RS, Grosicki S, Pristupa A, et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia. 2019;33:969–80.
Rawstron A, Munir T, Brock K, Webster N, Munoz Vicente S, Yates F, et al. Ibrutinib and obinutuzumab in CLL: improved MRD response rates with substantially enhanced MRD depletion for patients with >1 year prior ibrutinib exposure. Blood. 2018;132(Suppl 1):S181.
Wierda WG, Siddiqi T, Flinn I, Badoux XC, Kipps TJ, Allan JN, et al. Phase 2 CAPTIVATE results of ibrutinib (ibr) plus venetoclax (ven) in first-line chronic lymphocytic leukemia (CLL). J Clin Oncol. 2018;36(15_suppl):S7502.
Hillmen P, Rawstron A, Brock K, Munoz Vicente S, Yates F, Bishop R, et al. Ibrutinib plus venetoclax in relapsed/refractory CLL: results of the bloodwise TAP clarity study. Blood. 2018;132(Suppl 1):182.
Rogers KA, Huang Y, Ruppert AS, Awan FT, Heerema NA, Hoffman C, et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood. 2018;132:1568–72.
Jain N, Keating M, Thompson P, Ferrajoli A, Burger J, Borthakur G, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019;380:2095–103.
Bottcher S, Hallek M, Ritgen M, Kneba M. The role of minimal residual disease measurements in the therapy for CLL: is it ready for prime time? Hematol Oncol Clin North Am. 2013;27:267–88.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–60.
Agency EM. Appendix 4 to the guidelines on the evaluation of anticancer medicinal products in man. [acceessed 29 Oct 2018]; https://www.ema.europa.eu/documents/scientific-guideline/evaluation-anticancer-medicinal-products-man-appendix-4-condition-specific-guidance-rev2_en.pdf.
Administration UFaD. Hematologic malignancies: regulatory considerations for use of minimal residual disease in development of drug and biological products for treatment. Guidance for Industry. 2018 [accessed 20 Feb 2019]; https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM623333.pdf.
Cross NC, White HE, Colomer D, Ehrencrona H, Foroni L, Gottardi E, et al. Laboratory recommendations for scoring deep molecular responses following treatment for chronic myeloid leukemia. Leukemia. 2015;29:999–1003.
Abrisqueta P, Villamor N, Terol MJ, Gonzalez-Barca E, Gonzalez M, Ferra C, et al. Rituximab maintenance after first-line therapy with rituximab, fludarabine, cyclophosphamide, and mitoxantrone (R-FCM) for chronic lymphocytic leukemia. Blood. 2013;122:3951–9.
Bottcher S, Ritgen M, Fischer K, Stilgenbauer S, Busch RM, Fingerle-Rowson G, et al. Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol. 2012;30:980–8.
Fischer K, Cramer P, Busch R, Bottcher S, Bahlo J, Schubert J, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2012;30:3209–16.
Kovacs G, Robrecht S, Fink AM, Bahlo J, Cramer P, von Tresckow J, et al. Minimal residual disease assessment improves prediction of outcome in patients with chronic lymphocytic leukemia (Cll) who achieve partial response: comprehensive analysis of two phase III studies of the German CLL Study Group. J Clin Oncol. 2016;34:3758–65.
Santacruz R, Villamor N, Aymerich M, Martinez-Trillos A, Lopez C, Navarro A, et al. The prognostic impact of minimal residual disease in patients with chronic lymphocytic leukemia requiring first-line therapy. Haematologica. 2014;99:873–80.
Strati P, Keating MJ, O’Brien SM, Burger J, Ferrajoli A, Jain N, et al. Eradication of bone marrow minimal residual disease may prompt early treatment discontinuation in CLL. Blood. 2014;123:3727–32.
Thompson PA, Tam CS, O’Brien SM, Wierda WG, Stingo F, Plunkett W, et al. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood. 2016;127:303–9.
Rawstron AC, Villamor N, Ritgen M, Bottcher S, Ghia P, Zehnder JL, et al. International standardized approach for flow cytometric residual disease monitoring in chronic lymphocytic leukaemia. Leukemia. 2007;21:956–64.
van der Velden VH, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-Grumayer ER, et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21:604–11.
Drandi D, Kubiczkova-Besse L, Ferrero S, Dani N, Passera R, Mantoan B, et al. Minimal residual disease detection by droplet digital PCR in multiple myeloma, mantle cell lymphoma, and follicular lymphoma: a comparison with real-time PCR. J Mol Diagn. 2015;17:652–60.
Rawstron AC, Fazi C, Agathangelidis A, Villamor N, Letestu R, Nomdedeu J, et al. A complementary role of multiparameter flow cytometry and high-throughput sequencing for minimal residual disease detection in chronic lymphocytic leukemia: an European Research Initiative on CLL study. Leukemia. 2016;30:929–36.
Rawstron AC, Bottcher S, Letestu R, Villamor N, Fazi C, Kartsios H, et al. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia. 2013;27:142–9.
Michallet AS, Aktan M, Hiddemann W, Ilhan O, Johansson P, Laribi K, et al. Rituximab plus bendamustine or chlorambucil for chronic lymphocytic leukemia: primary analysis of the randomized, open-label MABLE study. Haematologica. 2018;103:698–706.
Roberts AW, Ma S, Kipps TJ, Coutre SE, Davids MS, Eichhorst B, et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood. 2019;134:111–22.
Bottcher S, Ritgen M, Pott C, Bruggemann M, Raff T, Stilgenbauer S, et al. Comparative analysis of minimal residual disease detection using four-color flow cytometry, consensus IgH-PCR, and quantitative IgH PCR in CLL after allogeneic and autologous stem cell transplantation. Leukemia. 2004;18:1637–45.
Shim YK, Rachel JM, Ghia P, Boren J, Abbasi F, Dagklis A, et al. Monoclonal B-cell lymphocytosis in healthy blood donors: an unexpectedly common finding. Blood. 2014;123:1319–26.
CLSI. Evaluation of detection capability for clinical laboratory measurement procedures; approved guideline—second edition. 2012 [accessed 20 Feb 2019]; https://clsi.org/media/1430/ep17a2_sample.pdf.
Rawstron AC, Howard D, McParland L, de Tute RM, Collett L, Phillips D, et al. Compartment effect on the prognostic significance of MRD detection in CLL: impact of treatment type and duration of follow-up. Blood. 2016;128:3226.
Thompson PA, Wierda WG. Eliminating minimal residual disease as a therapeutic end point: working toward cure for patients with CLL. Blood. 2016;127:279–86.
Appleby N, O’Brien D, Quinn FM, Smyth L, Kelly J, Parker I, et al. Risk adjusted therapy in chronic lymphocytic leukemia: a phase II cancer trials Ireland (CTRIAL-IE [ICORG 07-01]) study of fludarabine, cyclophosphamide, and rituximab therapy evaluating response adapted, abbreviated frontline therapy with FCR in non-del(17p) CLL. Leuk Lymphoma. 2018;59:1338–47.
Frankfurt O, Ma S, Gordon L, Winter JN, Horowitz JM, Rademaker A, et al. Phase II study of alemtuzumab-rituximab therapy in previously untreated patients with chronic lymphocytic leukemia: short- and long-term outcomes. Leuk Lymphoma. 2015;56:315–23.
Kwok M, Rawstron AC, Varghese A, Evans PA, O’Connor SJ, Doughty C, et al. Minimal residual disease is an independent predictor for 10-year survival in CLL. Blood. 2016;128:2770–3.
Moreton P, Kennedy B, Lucas G, Leach M, Rassam SM, Haynes A, et al. Eradication of minimal residual disease in B-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. J Clin Oncol. 2005;23:2971–9.
Seymour JF, Kipps TJ, Eichhorst B, Hillmen P, D’Rozario J, Assouline S, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107–20.
Stilgenbauer S, Eichhorst B, Schetelig J, Hillmen P, Seymour JF, Coutre S, et al. Venetoclax for patients with chronic lymphocytic leukemia With 17p deletion: results from the full population of a phase II pivotal trial. J Clin Oncol. 2018;36:1973–80.
Dimier N, Delmar P, Ward C, Morariu-Zamfir R, Fingerle-Rowson G, Bahlo J, et al. A model for predicting effect of treatment on progression-free survival using MRD as a surrogate end point in CLL. Blood. 2018;131:955–62.
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101–10.
Coutre S, Choi M, Furman RR, Eradat H, Heffner L, Jones JA, et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood. 2018;131:1704–11.
Jones JA, Mato AR, Wierda WG, Davids MS, Choi M, Cheson BD, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19:65–75.
Thompson PA, Peterson CB, Strati P, Jorgensen J, Keating MJ, O’Brien SM, et al. Serial minimal residual disease (MRD) monitoring during first-line FCR treatment for CLL may direct individualized therapeutic strategies. Leukemia. 2018;32:2388–98.
Eichhorst B, Fink AM, Bahlo J, Busch R, Kovacs G, Maurer C, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17:928–42.
Dartigeas C, Van Den Neste E, Berthou C, Maisonneuve H, Lepretre S, Dilhuydy MS, et al. Evaluating abbreviated induction with fludarabine, cyclophosphamide, and dose-dense rituximab in elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2016;57:328–34.
Munir T, Howard DR, McParland L, Pocock C, Rawstron AC, Hockaday A, et al. Results of the randomized phase IIB ADMIRE trial of FCR with or without mitoxantrone in previously untreated CLL. Leukemia. 2017;31:2085–93.
Hillmen P, Robak T, Janssens A, Babu KG, Kloczko J, Grosicki S, et al. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet. 2015;385:1873–83.
Sharman JP, Yimer HA, Boxer M, DiBella N, Babu S, Li J, et al. Results of a phase II multicenter study of obinutuzumab plus bendamustine in pts with previously untreated chronic lymphocytic leukemia (CLL). J Clin Oncol. 2017;35(15_suppl):S7523.
Stilgenbauer S, Leblond V, Foa R, Bottcher S, Ilhan O, Knauf W, et al. Obinutuzumab plus bendamustine in previously untreated patients with CLL: a subgroup analysis of the GREEN study. Leukemia. 2018;32:1778–86.
Leblond V, Cantin G, Cortelezzi A, Knauf W, Tiab M, Turgut M, et al. Safety and efficacy of obinutuzumab plus fludarabine and cyclophosphamide in previously untreated patients with chronic lymphocytic leukemia: subgroup analysis of the GREEN study. Blood. 2017;130(Suppl 1):S3015.
Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379:2517–28.
Rogers KA, Huang Y, Stark A, Awan FT, Maddocks KJ, Woyach JA, et al. Initial results of the phase 2 treatment naive cohort in a phase 1b/2 study of obinutuzumab, ibrutinib, and venetoclax in chronic lymphocytic leukemia. Blood. 2017;130(Suppl 1):S431.
Fischer K, Al-Sawaf O, Fink AM, Dixon M, Bahlo J, Warburton S, et al. Venetoclax and obinutuzumab in chronic lymphocytic leukemia. Blood. 2017;129:2702–5.
Stilgenbauer S, Morschhauser F, Wendtner C-M, Cartron G, Hallek M, Eichhorst BF, et al. Phase Ib study (GO28440) of venetoclax with bendamustine/rituximab or bendamustine/obinutuzumab in patients with relapsed/refractory or previously untreated chronic lymphocytic leukemia. Blood. 2016;128:4393.
Cramer P, von Tresckow J, Bahlo J, Robrecht S, Langerbeins P, Al-Sawaf O, et al. Bendamustine followed by obinutuzumab and venetoclax in chronic lymphocytic leukaemia (CLL2-BAG): primary endpoint analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19:1215–28.
Fischer K, Ritgen M, Al-Sawaf O, Robrecht S, Tandon M, Fink A-M, et al. Quantitative analysis of minimal residual disease (MRD) shows high rates of undetectable mrd after fixed-duration chemotherapy-free treatment and serves as surrogate marker for progression-free survival: a prospective analysis of the randomized CLL14 trial. Blood. 2019;134(Supplement_1):S36.
Kater AP, Seymour JF, Hillmen P, Eichhorst B, Langerak AW, Owen C, et al. Fixed duration of venetoclax-rituximab in relapsed/refractory chronic lymphocytic leukemia eradicates minimal residual disease and prolongs survival: post-treatment follow-up of the MURANO phase III study. J Clin Oncol. 2019;37:269–77.
Al-Sawaf O, Fischer K, Herling CD, Ritgen M, Bottcher S, Bahlo J, et al. Alemtuzumab consolidation in chronic lymphocytic leukaemia: a phase I/II multicentre trial. Eur J Haematol. 2017;98:254–62.
Burger JA, Sivina M, Jain N, Kim E, Kadia T, Estrov Z, et al. Randomized trial of ibrutinib versus ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood. 2019;133:1011–9.
Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–22.
Raponi S, Del Giudice I, Ilari C, Cafforio L, Della Starza I, De Propris MS, et al. The role of genetic-based prognostic factors in predicting minimal residual disease negativity in chronic lymphocytic leukemia patients treated with fludarabine, cyclophosphamide, and ofatumumab. In Proceedings of the 22nd European Hematology Association Congress; 2017; Madrid, Spain; 2017.
Bouvet E, Borel C, Oberic L, Compaci G, Cazin B, Michallet AS, et al. Impact of dose intensity on outcome of fludarabine, cyclophosphamide, and rituximab regimen given in the first-line therapy for chronic lymphocytic leukemia. Haematologica. 2013;98:65–70.
Eichhorst B, Robak T, Montserrat E, Ghia P, Hillmen P, Hallek M, et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26:78–84. Suppl 5.
Ladetto M, Buske C, Hutchings M, Dreyling M, Gaidano G, Le Gouill S, et al. ESMO consensus conference on malignant lymphoma: general perspectives and recommendations for prognostic tools in mature B-cell lymphomas and chronic lymphocytic leukaemia. Ann Oncol. 2016;27:2149–60.
Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123:3390–7.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42.
Fischer K, Al-Sawaf O, Bahlo J, Fink AM, Tandon M, Dixon M, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380:2225–36.
Egle A, Steurer M, Melchardt T, Weiss L, Gassner FJ, Zaborsky N, et al. Fludarabine and rituximab with escalating doses of lenalidomide followed by lenalidomide/rituximab maintenance in previously untreated chronic lymphocytic leukaemia (CLL): the REVLIRIT CLL-5 AGMT phase I/II study. Ann Hematol. 2018;97:1825–39.
Short NJ, Keating MJ, Wierda WG, Faderl S, Ferrajoli A, Estrov Z, et al. Fludarabine, cyclophosphamide, and multiple-dose rituximab as frontline therapy for chronic lymphocytic leukemia. Cancer. 2015;121:3869–76.
Greil R, Obrtlikova P, Smolej L, Kozak T, Steurer M, Andel J, et al. Rituximab maintenance versus observation alone in patients with chronic lymphocytic leukaemia who respond to first-line or second-line rituximab-containing chemoimmunotherapy: final results of the AGMT CLL-8a Mabtenance randomised trial. The Lancet Haematology. 2016;3:e317–29.
Rawstron AC, de Tute R, Jack AS, Hillmen P. Flow cytometric protein expression profiling as a systematic approach for developing disease-specific assays: identification of a chronic lymphocytic leukaemia-specific assay for use in rituximab-containing regimens. Leukemia. 2006;20:2102–10.
Letestu R, Cartron G, Lepretre S, Le Garff-Tavernier M, Solly F, Campos L, et al. Minimal Residual Disease (MRD) By 8-Color Flow Cytometry (Flow-MRD) and IGH Clonospecific Quantitative PCR (ASO RQPCR) Reached Similar Performances for Evaluation of CLL Treatment in a Phase II Clinical Trial: Cross Validation of the Methods. Blood. 2014;124:3307.
Sartor MM, Gottlieb DJ. A single tube 10-color flow cytometry assay optimizes detection of minimal residual disease in chronic lymphocytic leukemia. Cytometry B Clin Cytom. 2013;84:96–103.
clonoSEQ®. [technical summary]. Seattle, WA: Adaptive Biotechnologies; 2020. https://clonoseq.com/technical-summary.
Thompson PA, Srivastava J, Peterson C, Strati P, Jorgensen JL, Hether T, et al. Minimal residual disease undetectable by next-generation sequencing predicts improved outcome in CLL after chemoimmunotherapy. Blood. 2019;134:1951–9.
Dartigeas C, Van Den Neste E, Leger J, Maisonneuve H, Berthou C, Dilhuydy MS, et al. Rituximab maintenance versus observation following abbreviated induction with chemoimmunotherapy in elderly patients with previously untreated chronic lymphocytic leukaemia (CLL 2007 SA): an open-label, randomised phase 3 study. The Lancet Haematology. 2018;5:e82–94.
Acknowledgements
Medical writing support was provided by Robert Rydzewski, CMPP, and Devon Roll, Ph.D. of BioConnections, LLC, funded by AbbVie.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
WGW received consulting fees from Genzyme and has conducted contracted research for GSK/Novartis, AbbVie, Genentech, Karyopharm, Pharmacyclics, Gilead Sciences, Juno Therapeutics, Kite Pharma, Sunesis, Miragen, Oncternal Therapeutics, Cyclacel, Loxo Oncology, Janssen, and Zencor; AR received grants from AbbVie, Gilead, Janssen, Pharmacyclics, Roche, and Celgene, consulting fees/honoraria from AbbVie, BD Biosciences, Beckman Coulter, Gilead, Janssen, Pharmacyclics, Roche, and Celgene, nonfinancial support from BD Biosciences, Beckman Coulter, and Janssen, and royalties from BD Biosciences; FC received consulting fees from Roche, Janssen, Gilead, AbbVie, AstraZeneca, and Sunesis, grant funding from Sunesis, honoraria for lectures from AbbVie and Janssen, and travel accommodations from Roche, Janssen, Gilead, and AbbVie, XB received funding from AbbVie; DR received honoraria from AbbVie, AstraZeneca, Gilead, Janssen, Roche, and research grants from AbbVie, Gilead, Janssen, Cellestia; JB received consulting fees from AbbVie, Acerta, Beigene, Genentech/Roche, Gilead, Juno/Celgene, Kite, Loxo, Novartis, Pfizer, Pharmacyclics, Sunesis, TG Therapeutics, Verastem, AstraZeneca, Octapharma, Catapult Therapeutics, and Dynamo Therapeutics, honoraria from Janssen and Teva, research funding from Gilead, Loxo Oncology, Verastem, and Sun, and served on the Data Safety Monitoring Board for Morphosys and Invectys; AE received consulting fees and nonfinancial support from AbbVie, Janssen, and Roche; VA received advisory board honoraria, research funding, and travel grants from AbbVie and advisory board honoraria from Janssen; YH has received consulting fees from Janssen and AbbVie; SPM has participated in advisory boards for AbbVie and Janssen, and has been an invited lecturer for Janssen; CN has received grants from AbbVie, Janssen, Roche, and AstraZeneca/Acerta, consulting fees from AbbVie, Janssen, AstraZeneca/Acerta, Sunesis, and CSL Behring; TS received consulting fees and travel grants from AbbVie; RS received consulting fees/honoraria from AbbVie, BMS, Kyowa-Hakko Kirin, Chugai Pharmaceuticals, Shionogi, Takeda, Meiji Seika Pharma, MSD, Ohtsuka, Sawai, Celgene, Sumitomo Dainippon, Eisai, Alexion, Sanofi, Gilead, Mundi, Jazz, Ono, and Janssen; HTTT received consulting fees from AbbVie; SJW received grants from Roche and AbbVie; CO received consulting fees/honoraria from AbbVie, AstraZeneca, Janssen, Merck, Roche, Teva, and Gilead, SS received fees for consulting, drug/equipment, funding, and writing assistance from AbbVie, AstraZeneca, Celgene, Gilead, GlaxoSmithKline, Hoffman La-Roche, Janssen, Novartis, Pharmacyclics, and Sunesis; PG has participated in advisory boards for AbbVie, Acerta/AstraZeneca, Arqule, Dynamo, Gilead, Janssen, Juno/Celgene, and Sunesis, has been an invited lecturer for AbbVie, Janssen, and Gilead, and has received research grants from AbbVie, Gilead, Janssen, Novartis, and Sunesis; PH received grants from AbbVie, Janssen, Pharmacyclics, Roche, and Gilead, and speaking fees from AbbVie and Janssen; EC and CPD have nothing to disclose. AbbVie supported participant attendance at the consensus meetings and provided financial support for third-party medical writing but otherwise had no role in the development of consensus guidelines. AbbVie also participated in the review of the manuscript. No honoraria or payments were made for authorship.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wierda, W.G., Rawstron, A., Cymbalista, F. et al. Measurable residual disease in chronic lymphocytic leukemia: expert review and consensus recommendations. Leukemia 35, 3059–3072 (2021). https://doi.org/10.1038/s41375-021-01241-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-021-01241-1
This article is cited by
-
Ibrutinib plus fludarabine, cyclophosphamide and rituximab (iFCR) as initial treatment in chronic lymphocytic leukemia/ small lymphocytic leukemia with or without TP53 aberrations: a prospective real-world study in Chinese cohort
Blood Cancer Journal (2023)
-
Measurable residual disease in chronic lymphocytic leukemia. Where do we stand?
Leukemia (2023)
-
Relapsed/Refractory Chronic Lymphocytic Leukemia (CLL)
Current Hematologic Malignancy Reports (2023)
-
Measurable residual disease in Japanese patients with relapsed or refractory chronic lymphocytic leukemia treated with venetoclax
International Journal of Hematology (2023)
-
Association of hematologic response and assay sensitivity on the prognostic impact of measurable residual disease in acute myeloid leukemia: a systematic review and meta-analysis
Leukemia (2022)
