
Abstract
Multiple myeloma (MM) is a plasma cell malignancy that is often driven by chromosomal translocations. In particular, patients with t(4;14)-positive disease have worse prognosis compared to other MM subtypes. Herein, we demonstrated that t(4;14)-positive cells are highly dependent on the mevalonate (MVA) pathway for survival. Moreover, we showed that this metabolic vulnerability is immediately actionable, as inhibiting the MVA pathway with a statin preferentially induced apoptosis in t(4;14)-positive cells. In response to statin treatment, t(4;14)-positive cells activated the integrated stress response (ISR), which was augmented by co-treatment with bortezomib, a proteasome inhibitor. We identified that t(4;14)-positive cells depend on the MVA pathway for the synthesis of geranylgeranyl pyrophosphate (GGPP), as exogenous GGPP fully rescued statin-induced ISR activation and apoptosis. Inhibiting protein geranylgeranylation similarly induced the ISR in t(4;14)-positive cells, suggesting that this subtype of MM depends on GGPP, at least in part, for protein geranylgeranylation. Notably, fluvastatin treatment synergized with bortezomib to induce apoptosis in t(4;14)-positive cells and potentiated the anti-tumor activity of bortezomib in vivo. Our data implicate the t(4;14) translocation as a biomarker of statin sensitivity and warrant further clinical evaluation of a statin in combination with bortezomib for the treatment of t(4;14)-positive disease.
Similar content being viewed by others


Introduction
Translocations between the immunoglobulin heavy chain (IGH) gene on chromosome 14 and putative oncogenes on other chromosomes are hallmarks of multiple myeloma (MM) [1, 2]. In particular, translocations between IGH and chromosome 4 affect ~15% of MM patients, and are associated with poor progression-free and overall survival [3,4,5]. Despite recent improvements in the management of t(4;14)-positive disease, it remains difficult to treat and is largely incurable [6]. This is partly due to the fact that the precise driver of t(4;14)-positive MM remains debated.
Evidence supports that statins, common cholesterol-lowering drugs, have anti-cancer activity. Statins are inhibitors of HMG-CoA reductase (HMGCR), the rate-limiting enzyme of the mevalonate (MVA) pathway, which is responsible for the synthesis of cholesterol and non-sterol isoprenoids that are crucial for cell growth and survival (Fig. 1a) [7]. We, and others, have demonstrated that statins induce apoptosis in a subset of MM cell lines by directly inhibiting HMGCR, indicating that some MM cells are dependent on the MVA pathway for survival [8,9,10,11]. These preclinical data are supported by recent retrospective analyses, where statin use was associated with reduced MM-specific mortality [12, 13]. Moreover, phase I/II trials in patients with relapsed/refractory MM have reported promising responses when a statin was combined with standard-of-care therapies in some, but not all, patients [14, 15]. Collectively, these data suggest that these safe and inexpensive drugs may be effective anti-MM agents, but highlight the need for a predictive biomarker of statin sensitivity for patient stratification.

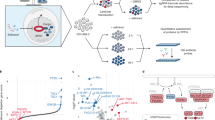
a Schematic representation of the MVA pathway and its sterol-regulated feedback loop. Statins inhibit the rate-limiting enzyme of the MVA pathway, HMGCR. Statin-mediated cholesterol depletion activates the SREBP2 transcription factor, which induces genes involved in MVA metabolism, including HMGCR. b List of lovastatin-sensitive and insensitive MM cell lines previously characterized by Wong et al. [10]. c Differential gene expression analysis between statin-sensitive and insensitive MM cell lines listed in (b). The blue and red dots represent differentially expressed genes with a log2(fold change) >2 or <−2. The genes in red further pass the adjusted p value cut-off when corrected for multiple testing (Bonferroni correction). Black dots represent genes with no significant difference in expression between statin-sensitive and insensitive MM cell lines. d Sensitivity to fluvastatin determined by MTT assays following 48 h of treatment. Fluvastatin IC50 value and 95% confidence interval (CI) for each cell line are shown. The t(4;14) translocation status is indicated and t(4;14)-positive cell lines are shaded in gray. e Sensitivity to fluvastatin, lovastatin, and simvastatin in t(4;14)-positive or negative MM cell lines mined from the CTRPv2 database. Percent area above the drug dose-response curve (% AAC) values are plotted as a box plot with whiskers representing minimum and maximum values. A higher % AAC indicates greater drug sensitivity (unpaired, two-tailed Wilcoxon rank-sum test comparing t(4;14)-positive and negative MM cell lines). f Primary plasma cells from t(4;14)-positive or negative patients were cultured in the presence of 5 μM fluvastatin or ethanol as a solvent control. After 72 h, cells were labeled with PE-conjugated anti-CD138 and FITC-conjugated Annexin V, and then cells were analyzed by flow cytometry. The percent change in primary CD138+ cell viability is plotted. The data are represented as the mean ± SD, p value = 0.14 (unpaired, two-tailed Wilcoxon rank-sum test).
Using transcriptomic and drug sensitivity data, we show here that MM cells driven by a t(4;14) translocation are particularly sensitive to statin-induced apoptosis. We further demonstrate that fluvastatin can synergize with bortezomib to induce apoptosis in t(4;14)-positive MM cells. Taken together, our data implicate the t(4;14) translocation as a biomarker of statin sensitivity and warrant further evaluation of the statin-bortezomib combination for the treatment of t(4;14)-positive disease.
Materials and methods
Cell lines, primary cells, and reagents
Cell lines were cultured as previously described [10]. All other cell lines were cultured in Iscove’s Modified Dulbecco’s Medium (Gibco) with 10% fetal bovine serum, 100 units/mL penicillin, and 100 mg/mL streptomycin. Cell lines were routinely confirmed to be mycoplasma-free (MycoAlert, Lonza). Primary human MM cells were obtained from bone marrow aspirates or peripheral blood draws from consenting MM patients with approval from the University Health Network’s Research Ethics Board. Isolated cells were processed and analyzed as previously described [11]. See “Supplemental Information” for reagent details.
Cell viability and Annexin V assays
MTT assays were performed as previously described [11]. Cell lines were seeded at 10,000–20,000 cells/well in 96-well plates and treated with 0–50 μM fluvastatin for 48 h. For Annexin V assays, 750,000 cells were seeded in six-well plates and treated as indicated. Cells were processed using the FITC Annexin V Apoptosis Detection Kit (BD Biosciences) as per the manufacturer’s protocol.
Animal experiments
In all, 7–9-week-old female NOD/SCID mice were injected with 5 million cells subcutaneously in the flank in a 1:1 mixture with Matrigel (Corning). Mice were treated as indicated with fluvastatin (orally, in phosphate-buffered saline) and/or bortezomib (intraperitoneally, in saline). Animal experiments were approved by the University Health Network’s Research Ethics Board and performed in accordance with Canadian Council on Animal Care regulations.
Supplemental methods
Method descriptions for quantitative RT-PCR (qRT-PCR), immunoblotting, live-cell imaging, RNA interference, RNA sequencing (RNA-seq), and differential expression analysis are available in “Supplemental Information.”
Results
The t(4;14) chromosomal translocation is associated with statin sensitivity in MM
We previously screened a panel of 17 MM cell lines for their response to lovastatin treatment [10]. We observed a dichotomized response, where approximately half of the cell lines underwent apoptosis in response to lovastatin exposure and the other half remained viable [10]. Using these drug sensitivity data in combination with basal RNA-seq data, we performed differential gene expression analysis comparing seven statin-sensitive and seven insensitive cell lines (Fig. 1b). One of the differentially expressed genes between the two groups was fibroblast growth factor receptor 3 (FGFR3), where FGFR3 was more highly expressed in statin-sensitive cell lines (Fig. 1c and Table S1). FGFR3 expression is deregulated in ~15% of MM patients as the result of a translocation between chromosome 4 and the IGH locus at chromosome 14q32, which places FGFR3 under the control of the 3′ IGH enhancer [16, 17]. Given our observation that statin-sensitive MM cells express high levels of FGFR3, we decided to evaluate whether t(4;14) translocation status was associated with statin sensitivity.
We built an independent validation panel of MM cell lines and evaluated their sensitivity to fluvastatin. We chose to evaluate fluvastatin instead of lovastatin, as lovastatin has been reported to have HMGCR-independent activities through its interaction with P-glycoprotein [18, 19] and the proteasome [20]. In contrast, fluvastatin is more specific to HMGCR and is less likely to participate in unwanted drug–drug interactions compared to many of the other statins [18, 21]. We treated cells with a range of fluvastatin concentrations and compared the IC50 values derived from the dose-response curves after 48 h of treatment. Consistent with our hypothesis, cells that were t(4;14)-positive had physiologically achievable [22, 23], low micromolar IC50 values compared to t(4;14)-negative cell lines, which were predominantly insensitive to fluvastatin at the concentrations evaluated (Fig. 1d).
We next mined publicly available drug sensitivity data from the Cancer Therapeutics Response Portal version 2 (CTRPv2) database using PharmacoDB [24, 25]. In CTRPv2, data for three statins (fluvastatin, lovastatin, and simvastatin) were available. We extracted statin sensitivity data as area above the drug dose-response curve (AAC) for all MM cell lines that were evaluated (20, 21, and 18 for fluvastatin, lovastatin, and simvastatin, respectively) (Fig. 1e). When stratified based on t(4;14) translocation status, MM cell lines that were t(4;14)-positive had higher AAC values for all three statins compared to t(4;14)-negative cell lines, indicating greater drug sensitivity (Fig. 1e). Ten MM cell lines in the CTRPv2 database were unique from those that we independently validated for statin sensitivity [10] (Fig. 1d), and the remaining cell lines were concordant in their response to statin treatment between data sets.
To establish whether fluvastatin could also kill primary cells derived from patients with t(4;14)-positive MM, we exposed primary cells to fluvastatin ex vivo. After 72 h of treatment, viable MM cells (CD138+/Annexin V−) were quantified by flow cytometry. Consistent with our cell line data, a trend toward increased sensitivity was observed in primary cells derived from t(4;14)-positive patients (Figs. 1f and S1).
Finally, we grew t(4;14)-positive NCI-H929 (hereafter referred to as H929) cells as xenografts in mice. Once tumors reached ~200 mm3, the mice were randomized to receive treatment with vehicle control, 20 or 50 mg/kg/day fluvastatin. Consistent with our in vitro data, we observed a dose-dependent decrease in tumor volumes over time in response to treatment (Fig. S2). Taken together, these data clearly demonstrate an association between t(4;14) status and statin sensitivity in MM.
Fluvastatin sensitivity in t(4;14)-positive MM cells is independent of FGFR3 and MMSET
Since we observed higher FGFR3 expression in statin-sensitive MM cell lines and an association between t(4;14) translocation status and statin sensitivity, we next evaluated whether the deregulated expression of FGFR3 in t(4;14)-positive cells was necessary for the greater statin sensitivity observed in these cells. We generated KMS11 sublines (statin sensitive and t(4;14)-positive) that express doxycycline-inducible short hairpin RNAs (shRNAs) against FGFR3 or a non-targeting shRNA control. Treatment of these sublines with doxycycline for 48 h was sufficient to reduce FGFR3 expression, but did not alter fluvastatin sensitivity (Fig. S3). In addition to FGFR3, the histone methyltransferase MMSET (NSD2) is also deregulated as a result of the t(4;14) translocation; however, depletion of MMSET in KMS11 cells, with previously characterized shRNAs [26] (Fig. S4), similarly had no effect on fluvastatin-induced apoptosis (Fig. S3).
A small proportion of t(4;14)-positive MM cells also have FGFR3-activating mutations [17]. Only three cell lines in our panel (KMS11, OPM2, and KMS18) had a mutation in FGFR3, and no correlation was observed between FGFR3 mutation status and statin sensitivity (data not shown). While mutations in MMSET are not as common in MM, aberrant activation of MMSET occurs as a result of a point mutation in acute lymphoblastic leukemia (ALL) cells [27]; however, we observed no differences in statin sensitivity between wild-type and E1099K MMSET-expressing ALL cell lines (Fig. S5). Collectively, these data support that statin sensitivity is independent of FGFR3 and MMSET.
The t(4;14) translocation is a novel and independent biomarker of statin sensitivity
We previously reported that sensitivity to lovastatin in MM was associated with an impaired sterol-regulated feedback response in statin-sensitive cells [11]. When cells are exposed to a statin, the depletion of intracellular cholesterol triggers the activation of the sterol regulatory element-binding protein (SREBP) family of transcription factors (Fig. 1a). The SREBPs subsequently induce the expression of genes involved in MVA and sterol metabolism, including HMGCR and HMGCS1, to restore homeostasis. In a subset of cell lines, this feedback loop fails to engage in response to statin treatment, and therefore the working model is that these cells are unable to compensate for the loss of crucial MVA-derived metabolites, and thereby undergo apoptosis.
To test whether t(4;14)-positive cells were sensitive to statins because of impaired feedback regulation of the MVA pathway, we evaluated the induction of HMGCR and HMGCS1 in a panel of t(4;14)-positive and negative MM cell lines following fluvastatin treatment. No association between t(4;14) translocation status and impaired feedback regulation of the MVA pathway was observed (Fig. S6), providing evidence that the t(4;14) translocation and impaired feedback regulation of the MVA pathway are independent predictors of statin sensitivity in MM.
Fluvastatin induces the integrated stress response (ISR) in t(4;14)-positive cells
To better understand this unique dependency of t(4;14)-positive MM cells on the MVA pathway, we performed RNA-seq to identify genes that were perturbed in response to fluvastatin treatment in t(4;14)-positive MM cell lines. Gene ontology analysis of fluvastatin-perturbed genes revealed a number of upregulated and downregulated biological processes (Fig. S7). Fluvastatin-downregulated genes were largely involved in processes such as RNA splicing and ribosome biogenesis, whereas a number of fluvastatin-upregulated genes were involved in response to stress. In particular, genes involved in the ISR, including ATF4, ATF3, and DDIT3 (also known as CHOP), were induced following fluvastatin treatment. The ISR is activated in response to various stressors, including endoplasmic reticulum (ER) stress and nutrient deprivation [28]. These stresses result in the activation of kinases that converge on the phosphorylation of eIF2α at serine 51 [28]. Phosphorylation of eIF2α leads to the global attenuation of mRNA translation, while selectively allowing for the translation of a subset of proteins to aid in cell recovery and survival, including the transcription factor ATF4 [28]. ATF4 then induces the expression of a number of downstream target genes, including ATF3, PPP1R15A (also known as GADD34), and CHOP, the latter of which induces apoptosis if the initiating stress remains unresolved [28].
Currently, one of the most important classes of anti-MM therapeutics is the proteasome inhibitors, including bortezomib [29], which remain important agents for the improved management of t(4;14)-positive disease [30]. Bortezomib is known to activate the eIF2α-ATF4 signaling axis via the induction of ER stress [31], and studies have shown that activation of this signaling axis is important for the proapoptotic activity of bortezomib [32,33,34]. Hence, we decided to further validate whether inhibition of the MVA pathway by statins could result in activation of the ISR in t(4;14)-positive cells.
We performed gene set enrichment analysis to compare our list of fluvastatin-perturbed genes to a published list of ATF4 target genes [35]. Of the 472 previously reported ATF4 targets, 307 were differentially expressed between solvent control and fluvastatin-treated t(4;14)-positive cells, with a significant enrichment of ATF4 target genes among the genes upregulated in response to fluvastatin treatment (Fig. 2a). Moreover, fluvastatin treatment resulted in the phosphorylation of eIF2α and induction of both ATF4 and ATF3 expression by immunoblotting, which was comparable to the increase observed following treatment with tunicamycin, a known inducer of ER stress (Fig. 2b). We then evaluated mRNA expression of two ATF4 target genes, CHOP and GADD34. We observed a time-dependent increase in both CHOP and GADD34 expression in t(4;14)-positive cell lines following fluvastatin exposure; however, in t(4;14)-negative cells, these genes were either not induced or, in some lines, weakly induced compared to t(4;14)-positive lines (Fig. 2c).

H929 and KMS11 cells were treated with 2 μM fluvastatin or ethanol as a solvent control for 24 h, after which RNA was isolated and RNA-seq was performed. a Gene set enrichment analysis of ATF4 target genes in a gene list of fluvastatin-perturbed genes, ranked by fold change between fluvastatin-treated and ethanol-treated cells. b H929 and KMS11 cells were treated with solvent controls, 4 μM fluvastatin or 0.5 μg/mL tunicamycin for 24 h, after which protein was isolated and immunoblotting was performed to evaluate phosphorylated eIF2α, ATF4, and ATF3 expression. c t(4;14)-positive cells (H929, KMS11, OPM2, and LP1; in blue) and t(4;14)-negative cells (JJN3, SKMM1, U266, and EJM; in orange) were treated with ethanol control (0 h fluvastatin) or 4 μM fluvastatin for 16 or 24 h, after which RNA was isolated for qRT-PCR. The ATF4 target genes CHOP and GADD34 were evaluated and expression was normalized to RPL13A. The data are represented as the mean + SD, n = 3, *p < 0.05 (one-way ANOVA with Bonferroni’s multiple comparisons test, where each group was compared to the ethanol control).
To evaluate whether fluvastatin activated the ISR via induction of ER stress in t(4;14)-positive cells, we evaluated the expression of GRP78 and XBP1 splicing, which are induced together with eIF2α-ATF4 signaling as part of the unfolded protein response [36]. The concentration of fluvastatin that induced ATF4 target gene expression in t(4;14)-positive cells had no effect on GRP78 expression or XBP1 splicing compared to tunicamycin, suggesting that fluvastatin induces the ISR via a mechanism independent of ER stress (Fig. S8).
Geranylgeranyl pyrophosphate (GGPP) rescues statin-induced apoptosis and ISR activation in t(4;14)-positive cells
Statin-induced apoptosis is known to be an on-target response, as exogenous MVA can fully rescue cell viability [10, 22, 37, 38]. Studies that have attempted to rescue statin-induced apoptosis with other MVA-derived metabolites have found that exogenous GGPP, and sometimes farnesyl pyrophosphate (FPP), can rescue statin-induced cell death [10, 38, 39]. Consistent with these data, the addition of GGPP, but not FPP, was sufficient to fully rescue fluvastatin-induced apoptosis in t(4;14)-positive H929 and KMS11 cells (Fig. 3a), indicating that these cells depend on the MVA pathway for the synthesis of GGPP.

a t(4;14)-positive H929 or KMS11 cells were treated with 4 μM fluvastatin ± 2 μM GGPP or 10 μM FPP for 48 h, and apoptosis was determined by Annexin V staining. The data are represented as the mean + SD, n = 3, *p < 0.05 (one-way ANOVA with Bonferroni’s multiple comparisons test, where each group was compared to the solvent controls group). b H929 or KMS11 cells were treated with 4 μM fluvastatin ± 2 μM GGPP or 10 μM FPP for 24 h, after which b protein was isolated and immunoblotting was performed to evaluate ATF4 expression or c RNA was isolated for qRT-PCR. The ATF4 target genes CHOP and GADD34 were evaluated and expression was normalized to RPL13A. The data are represented as the mean + SD, n = 3, *p < 0.05 (one-way ANOVA with Bonferroni’s multiple comparisons test, where each group was compared to the solvent controls group). d H929, KMS11, U266, and EJM cells were treated with solvent controls, 4 μM fluvastatin, 10 μM GGTI-298, or 20 μM FTI-277 for 24 h, after which RNA was isolated for qRT-PCR. The ATF4 target genes CHOP and GADD34 were evaluated and expression was normalized to RPL13A. The data are represented as the mean + SD, n = 3, *p < 0.05 (one-way ANOVA with Bonferroni’s multiple comparisons test, where each group was compared to the solvent controls group).
Since fluvastatin treatment induced a robust ISR in t(4;14)-positive cells, we next evaluated whether this response was similarly on-target and circumvented by exogenous GGPP. Indeed, exogenous GGPP, but not FPP, fully rescued fluvastatin-induced ATF4 expression (Fig. 3b) and upregulation of CHOP and GADD34 (Fig. 3c), suggesting that GGPP depletion triggers the ISR in t(4;14)-positive cells.
FPP and GGPP are used as substrates for the posttranslational prenylation (farnesylation and geranylgeranylation, respectively) of various proteins, which enables their proper membrane localization and function [40]. GGPP can further serve as a precursor for the synthesis of ubiquinone and dolichols [7]. Our lab previously demonstrated that statin-induced MM cell death can be phenocopied by a geranylgeranylation inhibitor (GGTI), which specifically blocks the transfer of GGPP onto target proteins [10]. Hence, one working model is that statin-sensitive MM cells depend on GGPP for protein prenylation. To test whether inhibition of protein geranylgeranylation could similarly phenocopy statin-induced activation of the ISR in t(4;14)-positive cells, we treated cells with a GGTI or farnesyltransferase inhibitor (FTI), which blocks the transfer of FPP onto target proteins, as a negative control. Treatment of t(4;14)-positive H929 and KMS11 cells with GGTI-298, but not FTI-277, was sufficient to induce CHOP and GADD34 expression (Fig. 3d). In contrast, neither GGTI-298 nor FTI-277 were able to induce the ISR in t(4;14)-negative EJM cells, and GGTI-298 only weakly induced CHOP and GADD34 expression in t(4;14)-negative U266 cells (Fig. 3d). Collectively, these data support that t(4;14)-positive cells are dependent on the MVA pathway for the production of GGPP, and suggest that GGPP may be required, at least in part, for protein prenylation.
Fluvastatin and bortezomib induce a robust ISR in t(4;14)-positive cells and synergize to induce apoptosis
Given that bortezomib also induces the ISR, we sought to determine whether combining these two clinically approved agents would potentiate apoptosis. We treated t(4;14)-positive H929 and t(4;14)-negative EJM cells with a range of fluvastatin and/or bortezomib concentrations for 48 h, after which cells were stained with Hoechst-33342 (DNA dye), TMRE (marker of active mitochondria), and Annexin V (marker of apoptotic cells). The stained cells were then imaged by confocal microscopy (Fig. S9), and the images were subjected to linear classification analysis to determine the percentage of dead cells for each treatment condition (Fig. 4a, b). The dose-response matrices were then used to compute synergy scores using SynergyFinder [41]. We observed synergy between physiologically achievable [22, 23] concentrations of fluvastatin and low nanomolar concentrations of bortezomib in H929 cells, where fluvastatin reduced the concentration of bortezomib required to induce MM cell death (Fig. 4c, e). This same effect was not observed in t(4;14)-negative EJM cells, where synergy with bortezomib was only observed at higher concentrations of fluvastatin (Fig. 4d, e).

a H929 and b EJM cells were treated with fluvastatin (ranging from 0 to 4 μM for H929 and 0 to 8 μM for EJM) or bortezomib (ranging from 0 to 5 nM for both cell lines) as single agents or in combination for 48 h. The dose-response matrices, as determined by live-cell imaging and quantification of % dead cells, are representative of three independent experiments and depict the mean % dead cells ± SD. The mean % dead cell values for each dose combination were used to compute Bliss synergy scores for each cell line. Synergy plots for c H929 and d EJM cells are shown, where red represents synergy and green represents antagonism. e The Bliss synergy scores from the three independent experiments are plotted for H929 and EJM cells. The data are represented as the mean ± SD. The mean Bliss synergy score and 95% confidence interval (CI) of the mean for each cell line are reported in the table.
To further validate these results, we assayed MM cell lines treated with fluvastatin and/or bortezomib for Annexin V staining by flow cytometry and ISR activation by qRT-PCR (Fig. 5). Consistent with our synergy data, the combination of fluvastatin and bortezomib, at doses that had a minimal effect when used as single agents, resulted in significantly enhanced apoptosis in both H929 and LP1 cells (Fig. 5a, b). This was associated with a greater increase in CHOP and GADD34 expression when the two drugs were used in combination, compared to their effects on the ISR as single agents (Fig. 5d, e). Intriguingly, H929 cells have an impaired sterol-regulated feedback response and are highly sensitive to statins, whereas LP1 cells have a very robust feedback response that reduces their sensitivity to statins [11, 42] (Fig. S6). This reveals that the statin-bortezomib combination can induce apoptosis in t(4;14)-positive cells independent of feedback regulation of the MVA pathway. Moreover, bortezomib had no effect on fluvastatin-induced expression of HMGCR or HMGCS1 in LP1 cells (Fig. S10), indicating that bortezomib cooperates with fluvastatin to induce apoptosis via a mechanism that is independent of SREBP and the sterol-regulated feedback response of the MVA pathway.

a H929, b LP1, or c EJM cells were treated with solvent controls, fluvastatin or bortezomib (BTZ) at the indicated concentrations for 48 h, and apoptosis was determined by Annexin V staining. The data are represented as the mean + SD, n = 3–4, *p < 0.05 (one-way ANOVA with Tukey’s multiple comparisons test, or Kruskal–Wallis test with Dunn’s multiple comparisons test (a), where each group was compared to the solvent controls group), #p < 0.05 (one-way ANOVA with Tukey’s multiple comparisons test, comparing the two indicated groups). d H929, e LP1, or f EJM cells treated with solvent controls, fluvastatin, or BTZ at the indicated concentrations for 24 h, and RNA was isolated for qRT-PCR. The ATF4 target genes CHOP and GADD34 were evaluated and expression was normalized to RPL13A. The data are represented as the mean + SD, n = 3, *p < 0.05 (one-way ANOVA with Tukey’s multiple comparisons test, where each group was compared to the solvent controls group), #p < 0.05 (one-way ANOVA with Tukey’s multiple comparisons test, comparing the two indicated groups).
In contrast, we did not observe the same level of potentiation when EJM cells were treated with fluvastatin and bortezomib; however, a slight, but statistically significant, increase in apoptosis was observed when a higher dose (10 μM) of fluvastatin was combined with 3 nM bortezomib (Fig. 5c). Consistent with the apoptosis data, no additional increases in CHOP or GADD34 expression were observed when EJM cells were treated with bortezomib in combination with fluvastatin (Fig. 5f). Notably, bortezomib alone was sufficient to induce CHOP and GADD34 expression in EJM cells, highlighting that bortezomib and fluvastatin converge on the ISR via distinct mechanisms (Fig. 5f). Collectively, these data demonstrate that the addition of fluvastatin to bortezomib augments activation of the ISR in t(4;14)-positive MM cells, and that physiologically achievable concentrations of fluvastatin can synergize with bortezomib to induce t(4;14)-positive MM cell death.
Fluvastatin potentiates bortezomib activity in a t(4;14)-positive tumor model
Given that fluvastatin and bortezomib synergized to induce cell death in t(4;14)-positive cells in vitro, we decided to evaluate this drug combination in vivo. We grew t(4;14)-positive H929 cells as xenografts in mice. Once tumor volumes reached ~500 mm3, the mice were randomized to receive treatment with a clinically relevant dose of bortezomib (1 mg/kg) as a single agent or in combination with 50 mg/kg fluvastatin (Fig. 6a). A 50 mg/kg dose of fluvastatin in a mouse is approximately equivalent to 4 mg/kg in a human [43], which has been shown to be well-tolerated [44]. Consistent with our in vitro data, fluvastatin treatment significantly sensitized H929 tumors to bortezomib, with no overt added toxicity (Fig. 6b, c).

a NOD/SCID mice were injected with 5 million H929 cells subcutaneously (s.c.) in the flank. Once tumor volumes reached ~500 mm3, the mice were randomized to receive either bortezomib in combination with phosphate-buffered saline (PBS; vehicle control) or 50 mg/kg fluvastatin. Bortezomib was delivered twice per week at 1 mg/kg via intraperitoneal (i.p.) injection, up to a total of three doses. Fluvastatin was resuspended in PBS and delivered daily via oral gavage (per os; p.o.). Tumor measurement assessments were not blinded. b Percent change in tumor growth over time. The data are represented as the mean + SD, n = 5 mice per treatment group. *p < 0.05 (unpaired, two-tailed Wilcoxon rank-sum test). c Percent change in mouse body weight over the course of treatment. d Schematic diagram detailing the potential for statins to be used in combination with bortezomib for the personalized treatment of t(4;14)-positive MM.
Discussion
Statins have been shown to induce apoptosis in subsets of cancer cells across a wide variety of tumor types [10, 19, 22, 38, 45]; however, biomarkers that predict which cancer cells will respond to statin treatment are lacking. Our lab previously characterized a panel of 17 MM cell lines for their sensitivity to lovastatin, but at the time was unable to correlate statin sensitivity with any single MM-associated genetic abnormality [10]. In that study, five cell lines were t(4;14)-positive, four of which were characterized as lovastatin sensitive. In our current study, we took an unbiased, genome-wide approach to interrogate differentially expressed transcripts between statin-sensitive and insensitive MM cell lines. Through this approach, we independently identified that high FGFR3 expression was associated with increased statin sensitivity in MM, which prompted us to evaluate the t(4;14) translocation as a possible biomarker. Indeed, we observed a clear association between the t(4;14) translocation and statin sensitivity in MM, which we validated by analyzing an independent panel of cell lines and publicly available drug sensitivity data. In total, 36 MM cell lines were evaluated, 11 of which were t(4;14)-positive.
Although the precise driver of t(4;14)-positive MM remains unclear, we evaluated whether oncogenes known to be deregulated in t(4;14)-positive disease functionally contribute to statin sensitivity. Interestingly, we found that statin sensitivity was independent of FGFR3 and MMSET (Fig. S3). While other genes flank the genomic breakpoints at 4p16, their expression is less consistently deregulated in t(4;14)-positive cells [46]. It is possible that a complex interaction between multiple deregulated genes, on chromosome 4 or elsewhere, confers statin sensitivity in t(4;14)-positive MM. Alternatively, this translocation could be a surrogate marker for another co-occurring genetic abnormality. For example, the t(4;14) translocation has been associated with deletions of chromosome 13q and mutations in PRKD2 and DIS3 [47, 48]. Further research is needed to identify the driver(s) of statin sensitivity in t(4;14)-positive MM cells.
We previously showed that statin sensitivity is associated with an impaired ability to induce the expression of MVA pathway genes following statin treatment [11, 22]. This was not the case here, as we demonstrated that statin sensitivity in t(4;14)-positive cells was not simply due to a lack of feedback regulation of the MVA pathway. While some t(4;14)-positive lines failed to induce the expression of HMGCR and HMGCS1 in response to fluvastatin exposure, others significantly upregulated the expression of these genes (Fig. S6). We previously demonstrated that inhibiting this feedback response with the drug dipyridamole sensitizes MM cells, including t(4;14)-positive cells, to statin-induced apoptosis [42]. In the present study, we identified that fluvastatin and bortezomib also synergize to induce apoptosis in t(4;14)-positive cells (Figs. 4 and 5). In contrast to dipyridamole, the statin-bortezomib interaction was independent of feedback regulation of the MVA pathway, as apoptosis was potentiated in both feedback-impaired (e.g., H929) and feedback-intact (e.g., LP1) t(4;14)-positive cell lines, and bortezomib did not function to inhibit the sterol-regulated feedback loop of the MVA pathway (Fig. S10).
We showed that t(4;14)-positive MM cells are dependent on the MVA pathway for the synthesis of GGPP, and that the depletion of GGPP triggers the ISR in these cells. Moreover, co-treatment with bortezomib, a drug already used to treat patients with t(4;14)-positive MM, augments this response and synergizes with statin treatment to induce t(4;14)-positive cell death. While statin-mediated activation of the ISR has been reported in other cancer cell types [49, 50], our study uncovered a clinically relevant biomarker capable of identifying MM cells that will induce this proapoptotic mechanism in response to statin treatment.
Although GGPP is important for various biological processes [7], we demonstrated that treatment of t(4;14)-positive MM cells with a GGTI phenocopies statin treatment and induces the ISR, thus implicating protein prenylation in t(4;14)-positive cell survival. Hundreds of proteins are predicted to be prenylated in mammalian cells [40, 51], and therefore it is not surprising that attempts to rescue statin-induced apoptosis with select individual prenylated proteins have largely failed [10]. More recent evidence supports a “class effect,” where statins deplete GGPP and hinder the prenylation of multiple proteins important for cell survival [52]. Further investigation is required to elucidate the importance of protein prenylation in t(4;14)-positive MM, and delineate the relationship between GGPP metabolism and the ISR.
Our findings have important clinical implications. Our in silico and in vitro data strongly support an association between the t(4;14) translocation and statin sensitivity in MM; however, further clinical validation is necessary prior to advancing statins to clinical trials in t(4;14)-positive patients. Unfortunately, no transgenic or patient-derived models of t(4;14)-driven MM have been developed. As an alternative approach, we evaluated the response of primary patient-derived MM cells to fluvastatin treatment ex vivo. While we demonstrated a promising trend toward increased statin sensitivity in t(4;14)-positive samples (Figs. 1f and S1), this association was not statistically significant in our small patient cohort. Regrettably, at the time of revision, the on-going COVID-19 pandemic limited our access to additional primary samples. A more rigorous analysis of statin sensitivity in primary MM cells should be the focus of future studies. Given that statins are already widely prescribed for cholesterol management, a retrospective comparison of t(4;14)-positive patient outcomes between statin users and nonusers may also validate our preclinical results.
To date, few studies have evaluated statins in combination with bortezomib in MM [14, 53]. Notably, our study is the first to describe a molecular subtype of MM in which these two drugs interact synergistically, at physiologically relevant concentrations. These data reveal that t(4;14)-positive patients in particular may benefit from the addition of a statin to their standard treatment regimen, which includes bortezomib (Fig. 6d). Hence, clinical validation of this immediately available drug combination for the treatment of t(4;14)-positive MM is warranted.
Data availability
The basal RNA-seq data are a Keats lab resource (https://www.keatslab.org/data-repository). The fluvastatin treatment RNA-seq data were deposited in the Gene Expression Omnibus (GEO) database (GSE152327). The code used to analyze these data can be found at: https://github.com/bhklab/StatinsMM.
References
Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene. 2001;20:5611–22.
Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005;23:6333–8.
Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, et al. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003;101:1520–9.
Jaksic W, Trudel S, Chang H, Trieu Y, Qi X, Mikhael J, et al. Clinical outcomes in t(4;14) multiple myeloma: a chemotherapy-sensitive disease characterized by rapid relapse and alkylating agent resistance. J Clin Oncol. 2005;23:7069–73.
Rajkumar SV, Gupta V, Fonseca R, Dispenzieri A, Gonsalves WI, Larson D, et al. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia. 2013;27:1738–44.
Chan H, Phillips M, Maganti M, Farooki S, Piza Rodriguez G, Masih-Khan E, et al. Single-center experience in treating patients with t(4;14) multiple myeloma with and without planned frontline autologous stem cell transplantation. Clin Lymphoma Myeloma Leuk. 2018;18:225–34.
Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016;16:718–31.
van de Donk NWCJ, Kamphuis MMJ, Lokhorst HM, Bloem AC. The cholesterol lowering drug lovastatin induces cell death in myeloma plasma cells. Leukemia. 2002;16:1362–71.
van de Donk NWCJ, Kamphuis MMJ, van Kessel B, Lokhorst HM, Bloem AC. Inhibition of protein geranylgeranylation induces apoptosis in myeloma plasma cells by reducing Mcl-1 protein levels. Blood. 2003;102:3354–62.
Wong WW-L, Clendening JW, Martirosyan A, Boutros PC, Bros C, Khosravi F, et al. Determinants of sensitivity to lovastatin-induced apoptosis in multiple myeloma. Mol Cancer Ther. 2007;6:1886–97.
Clendening JW, Pandyra A, Li Z, Boutros PC, Martirosyan A, Lehner R, et al. Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma. Blood. 2010;115:4787–97.
Sanfilippo KM, Keller J, Gage BF, Luo S, Wang TF, Moskowitz G, et al. Statins are associated with reduced mortality in multiple myeloma. J Clin Oncol. 2016;34:4008–14.
Brånvall E, Ekberg S, Eloranta S, Wästerlid T, Birmann BM, Smedby KE. Statin use is associated with improved survival in multiple myeloma: a Swedish population-based study of 4315 patients. Am J Hematol. 2020;95:652–61.
Schmidmaier R, Baumann P, Bumeder I, Meinhardt G, Straka C, Emmerich B. First clinical experience with simvastatin to overcome drug resistance in refractory multiple myeloma. Eur J Haematol. 2007;79:240–3.
Hus M, Grzasko N, Szostek M, Pluta A, Helbig G, Woszczyk D, et al. Thalidomide, dexamethasone and lovastatin with autologous stem cell transplantation as a salvage immunomodulatory therapy in patients with relapsed and refractory multiple myeloma. Ann Hematol. 2011;90:1161–6.
Chesi M, Bergsagel PL, Kuehl WM. The enigma of ectopic expression of FGFR3 in multiple myeloma: a critical initiating event or just a target for mutational activation during tumor progression. Curr Opin Hematol. 2002;9:288–93.
Kalff A, Spencer A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: prognostic implications and current clinical strategies. Blood Cancer J. 2012;2:e89.
Goard CA, Mather RG, Vinepal B, Clendening JW, Martirosyan A, Boutros PC, et al. Differential interactions between statins and P-glycoprotein: Implications for exploiting statins as anticancer agents. Int J Cancer. 2010;127:2939–48.
Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010;10:103.
Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci USA. 1999;96:7797–802.
Kapur NK, Musunuru K. Clinical efficacy and safety of statins in managing cardiovascular risk. Vasc Health Risk Manag. 2008;4:341–53.
Longo J, Mullen PJ, Yu R, van Leeuwen JE, Masoomian M, Woon DTS, et al. An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer. Mol Metab. 2019;25:119–30.
Longo J, Hamilton RJ, Masoomian M, Khurram N, Branchard E, Mullen PJ, et al. A pilot window-of-opportunity study of preoperative fluvastatin in localized prostate cancer. Prostate Cancer Prostatic Dis. 2020. https://doi.org/10.1038/s41391-020-0221-7.
Smirnov P, Kofia V, Maru A, Freeman M, Ho C, El-Hachem N, et al. PharmacoDB: an integrative database for mining in vitro anticancer drug screening studies. Nucleic Acids Res. 2018;46:D994–1002.
Smirnov P, Safikhani Z, El-Hachem N, Wang D, She A, Olsen C, et al. PharmacoGx: an R package for analysis of large pharmacogenomic datasets. Bioinformatics. 2016;32:1244–6.
Martinez-Garcia E, Popovic R, Min D-J, Sweet SMM, Thomas PM, Zamdborg L, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211–20.
Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia. 2014;28:198–201.
Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–95.
Moreau P, Attal M, Facon T. Frontline therapy of multiple myeloma. Blood. 2015;125:3076–84.
Sonneveld P, Lonial S, Usmani S, Siegel D, Anderson KC, Donk NWCJ, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2017;127:2955–63.
Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–16.
Schewe DM, Aguirre-Ghiso JA. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res. 2009;69:1545–52.
Suraweera A, Münch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell. 2012;48:242–53.
Narita T, Ri M, Masaki A, Mori F, Ito A, Kusumoto S, et al. Lower expression of activating transcription factors 3 and 4 correlates with shorter progression-free survival in multiple myeloma patients receiving bortezomib plus dexamethasone therapy. Blood Cancer J. 2015;5:e373.
Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–90.
Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6.
Wong WWL, Tan MM, Xia Z, Dimitroulakos J, Minden MD, Penn LZ. Cerivastatin triggers tumor-specific apoptosis with higher efficacy than lovastatin. Clin Cancer Res. 2001;7:2067–75.
Xia Z, Tan MM, Wong WW, Dimitroulakos J, Minden MD, Penn LZ. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia. 2001;15:1398–407.
Yu R, Longo J, van Leeuwen JE, Mullen PJ, Ba-Alawi W, Haibe-Kains B, et al. Statin-induced cancer cell death can be mechanistically uncoupled from prenylation of RAS family proteins. Cancer Res. 2018;78:1347–57.
Wang M, Casey PJ. Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol. 2016;17:110–22.
Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017;33:2413–5.
Pandyra A, Mullen PJ, Kalkat M, Yu R, Pong JT, Li Z, et al. Immediate utility of two approved agents to target both the metabolic mevalonate pathway and its restorative feedback loop. Cancer Res. 2014;74:4772–82.
Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–61.
Sabia H, Prasad P, Smith HT, Stoltz RR, Rothenberg P. Safety, tolerability, and pharmacokinetics of an extended-release formulation of fluvastatin administered once daily to patients with primary hypercholesterolemia. J Cardiovasc Pharm. 2001;37:502–11.
Goard CA, Chan-Seng-Yue M, Mullen PJ, Quiroga AD, Wasylishen AR, Clendening JW, et al. Identifying molecular features that distinguish fluvastatin-sensitive breast tumor cells. Breast Cancer Res Treat. 2014;143:301–12.
Keats JJ, Maxwell CA, Taylor BJ, Hendzel MJ, Chesi M, Bergsagel PL, et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood. 2005;105:4060–9.
Fonseca R, Oken MM, Greipp PR, Group on behalf of the ECOGM. The t(4;14)(p16.3;q32) is strongly associated with chromosome 13 abnormalities in both multiple myeloma and monoclonal gammopathy of undetermined significance. Blood. 2001;98:1271–2.
Walker BA, Mavrommatis K, Wardell CP, Cody Ashby T, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587–97.
Niknejad N, Morley M, Dimitroulakos J. Activation of the integrated stress response regulates lovastatin-induced apoptosis. J Biol Chem. 2007;282:29748–56.
Niknejad N, Gorn-Hondermann I, Ma L, Zahr S, Johnson-Obeseki S, Corsten M, et al. Lovastatin-induced apoptosis is mediated by activating transcription factor 3 and enhanced in combination with salubrinal. Int J Cancer. 2014;134:268–79.
Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, Eisenhaber F. Towards complete sets of farnesylated and geranylgeranylated proteins. PLoS Comput Biol. 2007;3:634–48.
Jiao Z, Cai H, Long Y, Sirka OK, Padmanaban V, Ewald AJ, et al. Statin-induced GGPP depletion blocks macropinocytosis and starves cells with oncogenic defects. Proc Natl Acad Sci USA. 2020;117:4158–68.
Terzi H, Altun A, Şencan M. In vitro comparison of the cytotoxic effects of statins on U266 myeloma cell line. Indian J Med Res. 2019;150:630–4.
Acknowledgements
This work was inspired by Dr. Richard Hill. We thank the members of the Penn lab, Dr. Rodger Tiedemann, Dr. Catherine O’Brien, and Dr. Michael Tomasson for helpful discussions. We also thank Jarkko Ylanko for technical support. KMS12-PE, KMS28-BM, KMS28-PE, and FR4 cell lines were generously provided by Dr. Rodger Tiedemann (University Health Network, Toronto, Canada). This study was supported by research funding from a Canadian Cancer Society Innovation Grant (706394; LZP and ST), the Canadian Institutes of Health Research (CIHR) (FRN: 142263; LZP), the Terry Fox Research Institute—New Frontiers Program Project Grant (1064; LZP and BHK), the US National Institutes of Health (R01 CA195732; JDL), and a Leukemia & Lymphoma Society Specialized Center of Research Grant (JDL). JL and PS were supported by CIHR Doctoral Research Awards. LZP holds a Tier 1 Canada Research Chair in Molecular Oncology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Longo, J., Smirnov, P., Li, Z. et al. The mevalonate pathway is an actionable vulnerability of t(4;14)-positive multiple myeloma. Leukemia 35, 796–808 (2021). https://doi.org/10.1038/s41375-020-0962-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-0962-2
This article is cited by
-
Acetyl-CoA metabolism in cancer
Nature Reviews Cancer (2023)
-
Geranylgeranyl diphosphate synthase inhibitor and proteasome inhibitor combination therapy in multiple myeloma
Experimental Hematology & Oncology (2022)
-
Computational pharmacogenomic screen identifies drugs that potentiate the anti-breast cancer activity of statins
Nature Communications (2022)
-
Statins and prostate cancer—hype or hope? The biological perspective
Prostate Cancer and Prostatic Diseases (2022)
