
Abstract
While the past decade has seen meaningful improvements in clinical outcomes for multiple myeloma patients, a subset of patients does not benefit from current therapeutics for unclear reasons. Many gene expression-based models of risk have been developed, but each model uses a different combination of genes and often involves assaying many genes making them difficult to implement. We organized the Multiple Myeloma DREAM Challenge, a crowdsourced effort to develop models of rapid progression in newly diagnosed myeloma patients and to benchmark these against previously published models. This effort lead to more robust predictors and found that incorporating specific demographic and clinical features improved gene expression-based models of high risk. Furthermore, post-challenge analysis identified a novel expression-based risk marker, PHF19, which has recently been found to have an important biological role in multiple myeloma. Lastly, we show that a simple four feature predictor composed of age, ISS, and expression of PHF19 and MMSET performs similarly to more complex models with many more gene expression features included.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is a hematological malignancy of terminally differentiated plasma cells (PCs) that reside within the bone marrow [1]. It arises as a result of complex chromosomal translocations or aneuploidy with ~25,000–30,000 patients diagnosed annually in the United States [2, 3]. The disease’s clinical course depends on a complex interplay of molecular characteristics of the PCs and patient socio-demographic factors. While progress has been made with novel treatments extending the time to disease progression (and overall survival (OS)) for the majority of patients, a subset of 15–20% of newly diagnosed MM patients are characterized by an aggressive disease course with rapid disease progression and poor OS regardless of initial treatment [4,5,6]. Accurately predicting which newly diagnosed patients are at high risk is critical to designing studies that will lead to a better understanding of myeloma progression and facilitate the discovery of novel therapies that meet the needs of these patients.
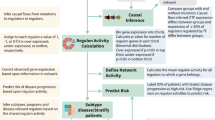
To date most MM risk models use patient demographic data, clinical laboratory results and cytogenetic assays to predict clinical outcome. High risk defining cytogenetic alterations typically include deletion of 17p (del(17p)) and gain of 1q as well as t(14;16), t(14;20), and most commonly t(4;14), which leads to juxtaposition of MMSET with the immunoglobulin heavy chain locus enhancer, resulting in overexpression of the MMSET oncogene [4]. While cytogenetic assays, in particular fluorescence in situ hybridization (FISH), are widely available, their risk prediction is sub-optimal and recently developed classifiers have used gene expression data to more accurately predict risk [7,8,9]. To investigate possible improvements to models of newly diagnosed myeloma progression, we organized the crowd sourced Multiple Myeloma DREAM Challenge, focusing on predicting high risk, defined as disease progression or death prior to 18 months from diagnosis. This benchmarking effort combined eight datasets, four which provided participants with clinical, cytogenetic, demographic and gene expression data to facilitate model development (N = 1624), and four hidden, independent data sets (N = 823) for unbiased validation. Over 800 people participated in this Challenge, resulting in the submission of 171 predictive algorithms for objective evaluation. Several models submitted to the Challenge demonstrated improved accuracy over existing state-of-the-art, published models.
Analysis of top-performing methods identified high expression of PHF19, a histone methyltransferase, as the gene with the strongest association with myeloma progression, with greater predictive power than the expression level of the known high-risk gene MMSET. We developed a four-parameter model using age, ISS, and PHF19 and MMSET expression that performs as well as more complex models having many more gene features. Significantly, we showed that knock down of PHF19 shifts myeloma cell lines into a less proliferative state. To our knowledge, this is the first DREAM Challenge to both nominate and experimentally validate a candidate biomarker and, as such, demonstrates the biological and clinical impact of crowdsourced efforts.
Methods
Datasets: The Challenge includes five microarray and three RNA-seq expression datasets, annotated with clinical characteristics such as gender, age, International Staging System stage (ISS), and cytogenetics (Table 1) [9,10,11,12,13,14]. In all datasets, expression assays were performed on CD138+PCs isolated from bone marrow aspirates or blood of newly diagnosed patients. Data were split into training and validation datasets (Table 1).
Three institutes provided RNA-sequencing data. The Multiple Myeloma Research Foundation (MMRF) provided an additional training dataset from its publicly available CoMMpass study (release IA9). Collaborating with the Myeloma Genome Project/Dana Farber Cancer Institute (MGP-DFCI) access was provided to data from their clinical trial where patients were randomized into a standard treatment arm and an aggressive treatment arm that included autologous stem cell transplant and high dose therapy [15]. An additional dataset from the Oncology Research Information Exchange Network (ORIEN) was made available through a collaboration with Moffitt Cancer Center and M2Gen (See Supplementary Information for more details on datasets).
Assessing model performance
To identify top-performing teams we employed two metrics to assess the accuracy of submitted models within a given validation cohort: the integrated area under the curve (iAUC) and balanced accuracy curve (BAC). While the AUC is a widely accepted metric of prediction accuracy, it is sensitive to the specific time threshold used to differentiate high and low patient risk. The myeloma research community has not yet reached a consensus on the time point that best separates patients into risk groups, though there is a general agreement that it lies between 1 and 2 years for progression-free survival (PFS). We, therefore, chose the iAUC “centered” on 18 months as a more robust primary metric. The iAUC range began 6 months prior to 18 months and continued to 6 months past with a sliding PFS threshold moving from12 to 24 months at weekly increments. iAUCs computed in each validation cohort were combined into a weighted average (wiAUC) with each cohort iAUC weighted by the square of the number of high-risk patients in order to ensure larger studies with more high-risk patients did not overwhelm smaller studies while still weighting them more heavily.
Using the wiAUC we computed the Bayes factor, K, to identify statistically tied top-performing predictors (see Supplemental Methods). Predictors with Kp < 3 are considered tied with the top-scoring model and the weighted BAC (wBAC) was used as a tie-breaking metric in order to determine the final top-performing model, with weighting by the square of the number of high-risk patients in each dataset (see Supplemental text).
In vitro studies for functional assessment of PHF19
Studies to determine the functional importance of PHF19 were performed using standard assays and are described in the Supplemental text. In brief, tetracycline-inducible short hairpin RNA (TRIPZ shRNA) was used to knockdown (KD) PHF19 expression in two MM cell lines (JJN3 and ARP1). A non-silencing scrambled TRIPZ shRNA was used as a control. PHF19 KD after doxycycline induction was measured by quantitative real-time polymerase chain reaction (qRT-PCR) and western blotting. Cell viability (Cell Titer Blue, Promega), cell cycle (Vybrant DyeCycle Stain, Thermo Scientific) and apoptosis (Annexin V, Biolegend) were assessed and differences between the PHF19 KD cells and control group were analyzed.
Results
Top Challenge models outperform baseline and published myeloma predictors
To develop and assess prognostic models of high risk in MM, we assembled eight data sets totaling 2447 patients annotated with OS and PFS (Table 1). We asked participants to predict whether a patient was high risk as defined as disease progression or death prior between 12–24 months since diagnosis (see Methods). Participants developed prognostic models using clinical features (e.g., age, sex, ISS, and cytogenetic features) and gene expression utilizing four training datasets. Challenge participants submitted models to be evaluated against four blinded validation cohorts (Fig. 1, Table 1, see Supplemental text). Model predictions were benchmarked against each other and comparator models (Table 2, Supplemental Table 1) using weighted-integrated AUC (wiAUC), with statistical ties resolved using weighted balanced accuracy (wBAC) (see Methods and Supplemental text).

Containerized models are submitted to cloud, ran on training datasets and risk predictions are scored.
Of 42 finalized models submitted to the Challenge, 11 exceeded the performance of the age plus ISS baseline model (Fig. 2, wiAUC = 0.6207). The top-performing predictor, developed by researchers at the Genome Institute of Singapore (GIS), outperformed all Challenge participant models (wiAUC = 0.6721) as well as the published comparator models UAMS-70 (wiAUC = 0.6414) and EMC-92 (wiAUC = 0.6042, Fig. 2).

a Box plots show distributions of bootstrapped model performances for each team. Comparator models are shown with text marked in blue for baseline models, green for published models and red for published models extended to include clinical features. The dashed red line indicates the median of the best performing comparator model. Barplots to the right show the tie-breaking metric, wBAC, for each model. Amongst statistically tied models, GIS has the highest wBAC and was declared the top-performer. Asterisk indicates internal collaborator’s comparator model. b Kaplan–Meier curve of UAMS-70 comparator model with and without age and ISS added.
Combining clinical features and gene expression features improves myeloma risk prediction accuracy
After the Challenge submission period ended, Challenge organizers and top-performing teams assessed which features had the largest impact on model performance. ISS was the most important model feature in GIS’s top-performing model as measured by the mean decrease in Gini coefficient (see Methods). A DNA repair signature previously associated with poor prognosis [16] was the second most important feature, while age ranked third (Supplemental Fig. 1).
To assess whether age and/or ISS explained the difference in model performance between the top-performing model and published comparator models, we extended the UAMS-70 [7] and EMC-92 [17] models to include age and/or ISS and assessed their performance (Fig. 2; see Supplemental Methods). The addition of these clinical features improved performance of both published models: EMC-92 wiAUC = from 0.6042 vs. EMC-92 + age + ISS wiAUC = 0.658 and UAMS-70 wiAUC = 0.6414 vs. UAMS-70 + ISS wiAUC = 0.6667 as has been suggested previously [13]. Adding ISS to the UAMS-70 model improved its accuracy such that it was tied with the top-performing model (i.e., its Bayes factor K < 3; Fig. 2a, b).
Top-performing Challenge methods identify PHF19 as a novel myeloma high-risk biomarker
The top-performing model implemented a wisdom of the crowd approach, utilizing clinical features and published myeloma signatures that summarize the expression of gene sets. The second-place “Stanford University Go” (SUGO) model instead included individual genes as features, utilizing a univariate-based feature selection approach to identify genes to include in their model. In each of the four training datasets the SUGO team computed each gene’s effect size, z, via the concordance index between OS and the gene’s expression. These effect sizes were combined across training sets using Stouffer’s method [18] without weighting to yield a single meta-z per gene. The meta concordance index was calculated using two expression normalization procedures, with one nominating PHF19 as the most important gene and the second identifying CDKN3 (See Supplemental Methods).
We replicated SUGO’s feature selection approach in both the training and validation expression datasets. We used the primary end point, PFS, in place of OS when computing each gene’s concordance index given that more patients will progress than die, which results in increased statistical power. We also weighted studies according to their number of high-risk patients when applying Stouffer’s method. This univariate analysis found that previously described myeloma risk genes MMSET and CKS1B as well as many proliferation genes had large values in the tail of the meta-z distribution (Fig. 3a and Supplemental Table 2), validating this approach. The meta-z values of these genes and all other genes were surpassed by PHF19, the top-ranked gene regardless of normalization procedure (not shown) in both the training and validation datasets (Fig. 3a).

a Two-dimensional histogram of PFS concordance index-based univariate effect sizes (z) in training and validation cohorts where colors represent the number of genes in a given hexagonal bin. PHF19 and well-known myeloma genes noted. b PHF19 and MMSET expression in relation to t(4;14). c A simple four feature model performs as well as UAMS-70 combined with age and ISS.
Incorporating PHF19 and MMSET expression with age and ISS identifies a simple model of high-risk MM
Given that both PHF19 and MMSET are histone modifiers playing a role in H3K36 methylation we checked whether their expression is correlated. As has previously been shown, MMSET expression is clearly affected by the t(4;14) translocation with the immunoglobulin enhancer driving high MMSET expression, while expression of PHF19 is not associated with the t(4;14) translocation (Fig. 3b). However, subsetting by t(4;14) reveals a modest linear relationship between MMSET and PHF19 expression in samples lacking the translocation (r = 0.423) while there is no such relationship in the t(4;14) positive samples (r = −0.067). We also found that higher PHF19 expression was associated with multiple high-risk genetic factors such as IGH translocation groups, non-hyperdiploidy, TP53 mutations and the overall number of drivers per sample (p < 0.001, Supplemental Fig. 2).
Given the impact of PHF19 on model performance and MMSET’s status as a reported myeloma risk marker, we checked to see if a Cox proportional-hazards model composed of age, ISS, PHF19 and MMSET could perform as well as a similar model using the features of the top-performing extended comparator model (UAMS-70 plus age plus ISS). We constructed Cox proportional-hazards models of the two feature sets and found that the four-parameter model (wiAUC = 0.693, Supplemental Table 3) out-performed the UAMS-70-based model in the validation cohort (wiAUC = 0.667; Fig. 3c) placing it on par with the winning algorithm.
Knockdown of PHF19 leads to decreased proliferation through cell cycle arrest in multiple myeloma cell lines
To determine whether PHF19 is functionally important for the malignant growth of MM cells, we used lenti viral-expressed shRNA directed against PHF19. We transduced JJN3 and ARP1 MM cell lines with a shRNA targeting PHF19 or a scrambled control shRNA and selected out transduced cells. shRNA induced cells showed KD of >90% PHF19 RNA and protein relative to the control after 72 h and 168 h for the JJN3 and ARP1 cell lines, respectively (Fig. 4a, b). KD of PHF19 led to significant inhibition of proliferation in the JJN3 and ARP1 MM cell lines compared with scrambled shRNA control (Fig. 4c, d) confirming the recent finding of PHF19’s effect on proliferation in MM cell lines [19]. To identify the mechanism of growth inhibition, we performed cell cycle analysis and observed a significant arrest of MM cells in the G0/G1 stage with PHF19 KD compared with the scrambled control shRNA (Supplemental Fig. 3a). This was seen consistently in both cell lines examined (Supplemental Fig. 3b). We further investigated the effect of PHF19 KD on apoptosis and necrosis, but did not find significant differences at the examined time points (Supplemental Fig. 3c–e). These results demonstrate that PHF19 is functionally relevant in MM and that reduction of PHF19 leads to a decrease in cell proliferation via cell cycle arrest.

a PHF19 knockdown, relative to scrambled shRNA control, was confirmed using qRT-PCR and b western blotting. c, d Cell proliferation was significantly decreased in MM cells with PHF19 knockdown compared with scrambled control for JJN3 and ARP1 cell lines.
Discussion
In the course of the crowd sourced Multiple Myeloma DREAM Challenge we benchmarked 171 prediction models and found that the accuracy of gene expression-based models benefited from the addition of clinical data, specifically: age and ISS improved AUC-based metrics by ~6%. As such, expression-based patient stratification efforts should incorporate age and ISS.
In addition, we show for the first time that expression of PHF19 is a stronger predictor of MM progression than the expression level of the high-risk marker MMSET, which is particularly overexpressed in patients with the high-risk translocation t(4;14). This strong association was likely missed in earlier studies given that PHF19 expression is not associated with any specific cytogenetic feature while several therapeutic advances over the last 20 years have made it difficult to model outcome across multiple studies from different periods. Furthermore, PHF19 has not been found to be significantly mutated in sequencing-based studies [20, 21], suggesting that its overexpression is not directly related to genomic alterations within the PHF19 gene. We also show that a simple four feature predictor composed of age, ISS, and expression of PHF19 and MMSET performs similarly to more complex models that are based on RNA-seq and gene expression analysis, which have been difficult to implement into the clinical setting (Supplemental Fig. 4). In contrast, the quantitative expression of PHF19 and MMSET could be easily measured by readily available methods such as real-time PCR, suggesting that the simplicity of the current model could be more easily adopted in a general clinical setting.
Apart from its prognostic value, we show that PHF19 has functional importance in MM. KD of PHF19 led to a significant reduction of growth and cell cycle arrest ex vivo, suggesting that PHF19 may play a role in transitioning cells into a highly proliferative state in MM. PHF19 has been shown to be a major modulator of histone methylation thereby regulating transcriptional chromatin activity [22] with a known role in B-cell differentiation into plasma cells within the germinal center [23, 24]. PHF19 directly recruits the polycomb repressive complex 2 (PRC2) via binding to H3K36me3 and leads to activation of enhancer of zeste homolog 1 and 2 (EZH1/EZH2) as enzymatic subunits of PRC2, thereby resulting in tri-methylation of H3K27 [25, 26]. This process has been shown to enforce gene repression and is known to promote tumor growth in a variety of cancers [27]. While MMSET has also been shown to regulate histone methylation, its role as an epigenetic modulator is less well understood. Some reports have suggested that MMSET leads to transcriptional repression through generation of H4K20me [28], H3K27me3 [29] or H3K36me3 [29], while other studies show that MMSET enhances transcription through generation of H4K20me2 [30] and H3K36me2 [29]. In contrast to MMSET, PHF19 expression is present in all MM subgroups and is preferentially overexpressed in high-risk MM. These results are indicative of a strong correlation between increased histone methylation, in particular H3k27 trimethylation, and disease aggressiveness. Further work will be necessary to elucidate the mechanisms of PHF19 in MM biology and any interplay with MMSET.
References
Palumbo A, Anderson K. Multiple myeloma. N. Engl J Med. 2011;364:1046–60.
Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Melton LJ 3rd. Incidence of multiple myeloma in Olmsted County, Minnesota: trend over 6 decades. Cancer. 2004;101:2667–74.
Kyle RA, Rajkumar SV. Multiple myeloma. N. Engl J Med. 2004;351:1860–73.
Sonneveld P, Avet-Loiseau H, Lonial S, Usmani S, Siegel D, Anderson KC, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127:2955–62.
Jethava Y, Mitchell A, Zangari M, Waheed S, Schinke C, Thanendrarajan S, et al. Dose-dense and less dose-intense Total Therapy 5 for gene expression profiling-defined high-risk multiple myeloma. Blood Cancer J. 2016;6:e453.
Pawlyn C, Morgan GJ. Evolutionary biology of high-risk multiple myeloma. Nat Rev Cancer. 2017;17:543–56.
Shaughnessy JD Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–84.
Hervé A-L, Florence M, Philippe M, Michel A, Thierry F, Kenneth A, et al. Molecular heterogeneity of multiple myeloma: pathogenesis, prognosis, and therapeutic implications. J Clin Oncol. 2011;29:1893–7.
Broyl A, Hose D, Lokhorst H, de Knegt Y, Peeters J, Jauch A, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood. 2010;116:2543–53.
Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol. 2010;28:827–38.
Kryukov F, Nemec P, Radova L, Kryukova E, Okubote S, Minarik J, et al. Centrosome associated genes pattern for risk sub-stratification in multiple myeloma. J Transl Med. 2016;14:150.
Dickens NJ, Walker BA, Leone PE, Johnson DC, Brito JL, Zeisig A, et al. Homozygous deletion mapping in myeloma samples identifies genes and an expression signature relevant to pathogenesis and outcome. Clin Cancer Res. 2010;16:1856–64.
Meissner T, Seckinger A, Rème T, Hielscher T, Möhler T, Neben K, et al. Gene expression profiling in multiple myeloma-reporting of entities, risk, and targets in clinical routine. Clin Cancer Res. 2011;17:7240–7.
Popovici V, Chen W, Gallas BG, Hatzis C, Shi W, Samuelson FW, et al. Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. 2010;12:R5.
Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N. Engl J Med. 2017;376:1311–20.
Kassambara A, Gourzones-Dmitriev C, Sahota S, Rème T, Moreaux J, Goldschmidt H, et al. A DNA repair pathway score predicts survival in human multiple myeloma: the potential for therapeutic strategy. Oncotarget. 2014;5:2487–98.
Kuiper R, Broyl A, de Knegt Y, van Vliet MH, van Beers EH, van der Holt B, et al. A gene expression signature for high-risk multiple myeloma. Leukemia. 2012;26:2406–13.
Liptak T. On the combination of independent tests. Magy Tud Akad Mat Kut Int Kozl. 1958;3:171–97.
Ren Z, Ahn JH, Liu H, Tsai Y-H, Bhanu NV, Koss B, et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation. Blood. 2019; https://doi.org/10.1182/blood.2019000578
Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587–97.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101.
Cai L, Rothbart SB, Lu R, Xu B, Chen W-Y, Tripathy A, et al. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol Cell. 2013;49:571–82.
Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol. 2015;16:663–73.
Ning F, Wang C, Niu S, Xu H, Xia K, Wang N. Transcription factor Phf19 positively regulates germinal center reactions that underlies its role in rheumatoid arthritis. Am J Transl Res. 2018;10:200–11.
Brien GL, Gambero G, O’Connell DJ, Jerman E, Turner SA, Egan CM, et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat Struct Mol Biol. 2012;19:1273–81.
Ballaré C, Lange M, Lapinaite A, Martin GM, Morey L, Pascual G, et al. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat Struct Mol Biol. 2012;19:1257–65.
Yamagishi M, Uchimaru K. Targeting EZH2 in cancer therapy. Curr Opin Oncol. 2017;29:375–81.
Marango J, Shimoyama M, Nishio H, Meyer JA, Min D-J, Sirulnik A, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111:3145–54.
Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–91.
Kang H-B, Choi Y, Lee JM, Choi K-C, Kim H-C, Yoo J-Y, et al. The histone methyltransferase, NSD2, enhances androgen receptor-mediated transcription. FEBS Lett. 2009;583:1880–6.
Laganà A, Perumal D, Melnekoff D, Readhead B, Kidd BA, Leshchenko V, et al. Integrative network analysis identifies novel drivers of pathogenesis and progression in newly diagnosed multiple myeloma. Leukemia. 2018;32:120–30.
Went M, Sud A, Speedy H, Sunter NJ, Försti A, Law PJ, et al. Genetic correlation between multiple myeloma and chronic lymphocytic leukaemia provides evidence for shared aetiology. Blood Cancer J. 2018;9:1.
Chattopadhyay S, Thomsen H, Yadav P, da Silva Filho MI, Weinhold N, Nöthen MM, et al. Genome-wide interaction and pathway-based identification of key regulators in multiple myeloma. Commun Biol. 2019;2:89.
Boyd KD, Ross FM, Walker BA, Wardell CP, Tapper WJ, Chiecchio L, et al. Mapping of chromosome 1p deletions in myeloma identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as being genes in regions associated with adverse survival. Clin Cancer Res. 2011;17:7776–84.
Wu P, Walker BA, Brewer D, Gregory WM, Ashcroft J, Ross FM, et al. A gene expression-based predictor for myeloma patients at high risk of developing bone disease on bisphosphonate treatment. Clin Cancer Res. 2011;17:6347–55.
Moreaux J, Rème T, Leonard W, Veyrune J-L, Requirand G, Goldschmidt H, et al. Development of gene expression-based score to predict sensitivity of multiple myeloma cells to DNA methylation inhibitors. Mol Cancer Ther. 2012;11:2685–92.
Maes K, De Smedt E, Kassambara A, Hose D, Seckinger A, Van Valckenborgh E, et al. In vivo treatment with epigenetic modulating agents induces transcriptional alterations associated with prognosis and immunomodulation in multiple myeloma. Oncotarget. 2015;6:3319–34.
Acknowledgements
Work completed by CS was funded by NCATS grant, KL2TR000063. A complete list of the members of the Multiple Myeloma DREAM Consortium appears in the supplement text.
Author information
Authors and Affiliations
Consortia
Contributions
MJM, CS, CLPE, FT, FG and AD contributed equall to this work. CS, VL and KB performed experiments; MJM, CS, CLPE, FT, FG, BSW, AP and ECN analyzed results and made the figures; CLPE, BSW, AP, YG, HC, YC, BL, FG, BH and MO built top-performing and comparator models for predicting high risk; TY, ECN, MJM, MT, JGu and BSW designed and built compute infrastructure and model performance metrics for benchmarking methods; HYD, WSD, HG, HA-L, MS, PS, KHS, NM, DA, GM, DH and BAW provided key test datasets without which this investigation would not be possible; DR, EF, MJM, and KM processed and curated datasets; MJM, AD, DB, FS, CS, FT, BSW, AT, BAW, JGu designed the research; MJM, AD, CS, FT, BSW, AT, BAW, JGu wrote the manuscript. DB, JG, BAW, AT, and JGu contributed equally to this work.
Corresponding author
Ethics declarations
Conflict of interest
Please note the following byline authors: FT, KM, KB, FS, EF, AR, SAD, AD, DB, MT, AT have employment and/or equity ownership in Celgene. AD also had equity in Twinstrand Biosciences. WSD has employment with M2Gen. HG has research support (Amgen, BMS, Celgene, Chugai, Janssen, Molecular Partners, MSD, Sanofi, Mundipharma, Takeda and Novartis), advisory boards (Adaptive Biotechnology, Amgen, BMS, Celgene, Janssen, Sanofi and Takeda), honoraria (ArtTempi, BMS, Celgene, Chugai, Janssen, Novartis and Sanofi). NM has consulted for Abbvie, Adaptive, Amgen, Celgene, Janssen, Takeda and Oncopep and also has equity in Oncopep. KHS is on advisory boards for: Celgene, BMS, Amgen, Takeda, Janssen and Sanofi Genzyme, has received research funding from AbbVie, and has consulted for Adaptive Biotechnologies.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the Multiple Myeloma DREAM Consortium and their affiliations are listed in the Supplemental text.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mason, M.J., Schinke, C., Eng, C.L.P. et al. Multiple Myeloma DREAM Challenge reveals epigenetic regulator PHF19 as marker of aggressive disease. Leukemia 34, 1866–1874 (2020). https://doi.org/10.1038/s41375-020-0742-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-0742-z
This article is cited by
-
Enhancing prognostic power in multiple myeloma using a plasma cell signature derived from single-cell RNA sequencing
Blood Cancer Journal (2024)
-
Identifying novel mechanisms of biallelic TP53 loss refines poor outcome for patients with multiple myeloma
Blood Cancer Journal (2023)
-
Plasma cells expression from smouldering myeloma to myeloma reveals the importance of the PRC2 complex, cell cycle progression, and the divergent evolutionary pathways within the different molecular subgroups
Leukemia (2022)
-
A simple additive staging system for newly diagnosed multiple myeloma
Blood Cancer Journal (2022)
-
Insights into high-risk multiple myeloma from an analysis of the role of PHF19 in cancer
Journal of Experimental & Clinical Cancer Research (2021)
