
Abstract
Recurrent loss-of-function mutations of BCL6 co-repressor (BCOR) gene are found in about 4% of AML patients with normal karyotype and are associated with DNMT3a mutations and poor prognosis. Therefore, new anti-leukemia treatments and mouse models are needed for this combinatorial AML genotype. For this purpose, we first generated a Bcor−/− knockout mouse model characterized by impaired erythroid development (macrocytosis and anemia) and enhanced thrombopoiesis, which are both features of myelodysplasia/myeloproliferative neoplasms. We then created and characterized double Bcor−/−/Dnmt3a−/− knockout mice. Interestingly, these animals developed a fully penetrant acute erythroid leukemia (AEL) characterized by leukocytosis secondary to the expansion of blasts expressing c-Kit+ and the erythroid marker Ter119, macrocytic anemia and progressive reduction of the thrombocytosis associated with loss of Bcor alone. Transcriptomic analysis of double knockout bone marrow progenitors revealed that aberrant erythroid skewing was induced by epigenetic changes affecting specific transcriptional factors (GATA1-2) and cell-cycle regulators (Mdm2, Tp53). These findings prompted us to investigate the efficacy of demethylating agents in AEL, with significant impact on progressive leukemic burden and mice overall survival. Information gained from our model expands the knowledge on the biology of AEL and may help designing new rational treatments for patients suffering from this high-risk leukemia.
Similar content being viewed by others


Introduction
The BCL6 co-repressor (BCOR) gene is located on chromosome Xp11.4 and encodes a transcription regulatory factor that was initially identified as an interactor partner of the germinal center-associated BCL6 protein [1, 2]. The BCOR protein is located in the nucleus [3] where exerts its function as a member of the non-canonical multimeric polycomb group repressive complex 1 (PRC1) which is recruited to the target sites independently of H3K27me3 [4]. This complex is involved in the control of various biological processes, including pluripotency, reprogramming, and hematopoiesis [4, 5]. Relatively high frequency of BCOR mutations has been reported in aplastic anemia suggesting that these genetic events may confer a selective advantage in the context of aplastic anemia autoimmune environment, although they do not appear to be associated with an increased risk of secondary AML/MDS [6]. Moreover, various aberrations of the BCOR gene, such as internal tandem duplications of the PCGF Ub-like fold discriminator domain, gene fusions, and loss-of-function mutations play a role in promoting hematological and extra-hematological malignancies [7, 8].
In 2011, we discovered recurrent loss-of-function mutations of BCOR in AML (about 4% of cases with normal karyotype) and found that they were usually mutually exclusive of FLT3-ITD and NPM1 mutations, co-occurred with DNA methyl transferase (DNMT3A) mutations and were associated with a poor outcome [9]. Our findings have been subsequently confirmed by other investigators both in AML and MDS [10,11,12,13,14]. Interestingly, one study on Japanese patients reported a preferential co-occurrence of BCOR mutations with K-RAS, N-RAS, and RUNX1 mutations [11]. BCOR 1 ligand (BCORL1) gene has been also found to be mutated in 3.7–6% of AML patients [11, 15].
Bcor somatic heterozygous mutations in AML are similar to the germline mutations that in females cause rare genetic syndrome characterized by cranio-facial, ocular and cardiac abnormalities [16]. The disruptive nature of these mutations that usually results in a premature stop codon and non-sense mediated decay or protein truncation are consistent with a tumor-suppressive role of the Bcor gene in myeloid malignancies. Accordingly, mice lacking Bcor exons 9 and 10, which encode for a carboxyl-terminal truncated Bcor unable to interact with the PCR1 core effector components, are characterized by expansion of myeloid progenitors [17], enhanced cell proliferation, and myeloid differentiation associated with upregulation of HoxA cluster [18].
However, Bcor deficiency is not itself sufficient to promote leukemia [17, 18], strongly suggesting that other mutations are required to induce myeloid malignancies. The type of myeloid neoplasm developing in Bcor-deficient mouse may vary depending on the co-occurring genetic lesions. For example, compound mice carrying concurrent full deletion of Tet2 develop lethal MDS [19] whilst Bcor loss cooperates with KrasG12D to drive AML [17].
We previously found that about 40% of AML patients with BCOR mutations also carry mutations of DNMT3a [9] catalyzing the addition of methyl groups to CpG dinucleotides. Because BCOR and DNMT3A are both epigenetic modifiers [4, 20, 21], mutations of these genes could promote AML through a synergistic mechanism [9]. Murine hematopoietic cells lacking Dnmt3a have thousands of focal, “canonically” located, hypomethylated regions that are amenable to be “repaired” with partial correction of dysregulated gene expression and myeloid skewing [22]. Dnmt3a loss with its hypomethylated phenotype is known to promote expansion and immortalization of hematopoietic stem cells, block in hematopoietic differentiation [23] and development of myeloid and lymphoid malignancies after a long period of latency [23,24,25]. Mx1-Cre-mediated Dnmt3a ablation led to the development of a lethal, fully penetrant myelodysplasia/myeloproliferative (MDS/MPN) neoplasm characterized by peripheral cytopenias and marked extramedullary hematopoiesis with liver involvement [26]. Thus, Dnmt3a deficiency establish an epigenetic state somehow predisposing to the emergence of cooperating mutations leading to overt leukemia in mice [25]. Indeed, co-expression of mutated Dnmt3a with other disease alleles (including Tet2, Flt3, Npm1) promote overt leukemic transformation in mice [2, 27]. However, information on the cooperation between Dnmt3a and Bcor is still missing.
Therefore, we generated a conditional mouse model of Bcor inactivation to explore its function in normal and leukemic hematopoiesis either alone or in combination with Dnmt3a loss. Bcor-deficient mice demonstrated impaired erythroid development and enhanced thrombopoiesis. Notably, the Bcor−/−Dnmt3a−/− double knockout mice developed a fully penetrant acute erythroid leukemia (AEL) sensitive to demethylating agents. Information gained from this model expand our knowledge on the biology of AEL and may help to design new rational treatments for patients suffering from this high-risk leukemia.
Methods
Mouse strains
Mice were bred and housed by the “Service center of Preclinical Research” of Perugia’s animal house facility, and mouse manipulations were performed according to the protocol reviewed and approved by the Italian Health Ministry (generation on Supplementary Methods).
Peripheral blood counts
Mice were anesthetized with isoflurane followed by retro-orbital bleeding. Peripheral blood (PB) was taken into glass capillary tubes. Complete blood count was performed using an XE-2100 hematology automated analyzer (Dasit).
Flow Cytometry and Cell Sorting
Bone marrow, spleen, and PB cells were stained with antibodies from eBioscience (see list on Supplementary Methods). Cell acquisition and analysis were performed on BD FACS CANTO and BD FORTESSA. Sorting experiments (LSK and MEP subpopulations) were performed using the FACS AriaIII cell sorter. Gates were drawn to exclude nonviable cells and debris. Part of the flow cytometry data was analyzed with FlowJo software (Tree Star, Ashland, OR).
Histology and cytospins
BM, spleen, liver, kidney, lung, and spinal cord were harvested from moribund mice. Spinal cord was decalcified, then spleen, liver, kidney, lung, and spinal cord were paraffin embedded, and sections were stained with hematoxylin and eosin and cytospins were stained with Giemsa. Cytospins of BM and spleen were performed both before and after red blood cell (RBC) lysis.
Gene expression profiling and accession numbers
Total RNA was extracted from LSK and MEP BM cells using an RNA extraction kit (RNeasy plusMicro kit, Qiagen) and samples analyzed for Gene Expression Profiling. Starting from the results of the differential analysis, the comparisons of all three mutated genotypes with the wild type were taken into consideration and the genes were filtered considering only those with a p value < 0.05 and an absolute fold change value >1.5.
The three resulting lists were compared to evaluate both common and exclusive genes for each genotype. To show these similarities and differences we have resorted to Venn diagrams (GSE158018).
In vivo treatments
Mice were treated through intra-peritoneal injections with a maximum tolerated dose of the demethylating agent 5-aza-2’-deoxycytidine DEC (2.5 mg/kg, every 3 days 5 doses) and Cytarabine ARA-C (50 mg/kg for 6 days).
Immunoblotting analysis
Gata1 protein was detected by western blot analysis on lysates from 1 to 2 × 106 cells with an anti-rat primary antibody against GATA-1 (N6) clone sc-265 (Santa Cruz biotechnology). After washing, blots were incubated with HPR-conjugated secondary antibody anti-IgG (Sigma). Mouse a-tubulin antibody was used as control and obtained from Sigma.
Results
Loss of Bcor induces red blood cells changes and expansion of the megakaryocyte compartment
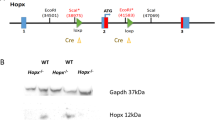
We generated a conditional knockout mouse model in which Bcor deletion mimics truncating Bcor mutations observed in AML. Our Bcor conditional knockout mouse model was developed deleting exons from 8 to 10, resulting in frameshift and premature stop codon in exon 11. Originated mRNA preserves the polyA tail but is extremely unstable due to the presence of five splicing junctions between the STOP codon and the polyA tail (Supplementary Fig. 1A, B). To test the impact of Bcor deletion in adult hematopoiesis, mutant mice were crossed with Mx1-Cre mice carrying an interferon-inducible Cre recombinase transgene under an hematopoietic stem cell promoter. PCR genotyping of the offspring allowed the identification of both the wild type and mutant alleles from tails DNA (Supplementary Fig. 1Ci) and the recombination of loxP-containing target alleles in Bcor-flox/flox-Cre + BM after pIpC induction of the Cre (Supplementary Fig. 1Cii). To confirm that the induced Mx1-Cre + Bcor mutant mice did not express Bcor mRNA, RNA from BM Lin-Sca+c-Kit + (LSK) was reverse transcribed and amplified by PCR. Bcor mRNA was not detectable by this method in Bcor-flox/flox-Cre + (Bcor−/−) homozygous and Bcor-flox/Y-Cre + (bcor−/−) hemyzigous mice (Supplementary Fig. 2Ai). Western blot analysis of BM lin- cells from Bcor-flox/flox-Cre + (Bcor−/−) homozygous and hemyzigous demonstrated loss of Bcor protein (Supplementary Fig. 2Aii).
To determine the effect of Bcor loss in mice hematopoietic system, we performed serial complete blood counts that showed leukopenia (mainly due to B-cell lymphopenia (Supplementary Fig. 3Ai–v), red blood cells’ (RBC) reduction (Fig. 1Ai) with increased mean corpuscle volume (MCV) (Fig. 1Aii), and platelet counts’ progressive increase (Fig. 1Aiii; Supplementary Fig. 3B, C). Resulting thrombocytosis derived from the accumulation of both megakaryocytic-erythroid (MEP, Lin−/Sca1−/Kit+CD34−FCyRII/IIIlo/−) and megakaryocytic progenitors (MkPs, Lin–c-Kit+Sca-1–CD150 + CD41+) (Fig. 1Bi–iii;) relied on a decrease of apoptosis (Fig. 1Biv) within BM cavity. To assess the impact of Bcor loss on survival, we monitored our mice cohort for an extended period (18 months). Although Kaplan-Meier analyses showed low survival of Bcor null mice (Fig. 1Ci), post-mortem pathological examinations did not reveal any leukemia infiltration in hematopoietic organs (BM, liver, spleen) (Fig. 1Cii). Taken together, these data confirmed that Bcor loss negatively influences mice survival by specifically subverting normal hematopoietic compartments and PB output. The absence of a frank acute leukemia phenotype clearly suggested the need of additional cooperative leukemogenic events.

A RBCs count (i) mean corpuscolar volume (MCV) (ii) and platelets count (iii) in PB of Bcor−/−, Bcor+/− and WT from 12 months old mice. B (i) Megakaryocyte-erythroid progenitors (MEP Lin − /Sca1-/Kit+CD34-FCgRII/IIIlo/-) in BM of Bcor−/− and WT 6 months old mice. (ii) Representative flow-cytometric analysis of immature megakaryocytic compartments, megakaryocyte progenitor (MkP) and total number of MkP (iii) in BM of Bcor−/−, Bcor+/− and WT mice. (iv) total number of CD41 + cells in BM of Bcor−/−, Bcor+/− and WT 6 months old mice. (iv) Apoptosis in CD41 cells in WT and Bcor−/− 3 months old mice. 1 × 106 cells from BM were plated for 6 h in RPMI + BRDU, than counted and stained. C (i) Kaplan–Mayer plot of mouse survival according to the indicated genotypes (n = 44 WT,19 Bcor+/−, 37 Bcor−/−). (ii) Pie charts showing the different causes of mortality: 56% of deaths occurred in the Bcor mutant cohort in the presence of well-defined hematological abnormalities. In the remaining mice, pathological examinations were consistent with the occurrence of hepatic carcinoma (8%), intestinal/lung tumors (4%), possibly due to Cre-recombinase leakiness. Notably, 32% of deaths were of unknown origin. *p < 0.05, **p < 0.01; ***p < 0.001 unpaired t-test with Welch’s correction.
Bcor and Dnmt3a loss induces a highly penetrant acute erythroid leukemia (AEL)
We crossed Bcor (Bcor−/−) and Dnmt3a (Dnmt3−/−) conditional knockout mice with Mx1-Cre transgenic mutant to generate Bcor−/−Dnmt3a−/−double knockout mice, single ko Bcor−/−and Dnmt3−/− mice expressing Mx1-Cre and mice with untargeted genes as wild type controls.
Bcor−/−Dnmt3a−/− mice developed a fully penetrant and lethal leukemic phenotype with a median survival of 135 days (range from 59 to 234 days), significantly shorter than the other groups (Fig. 2A). Interestingly, we found a Bcor deletion both in the single knockout and in the leukemic double Bcor−/−Dnmt3a−/− knockout mice (Supplementary Fig. 4A), while regarding to Dnmt3a, the deletion by Mx1Cre was partial in Dnmt3a−/− knockout, while its complete loss occurred only in double mutant leukemic mice. Interestingly, in preleukemic mice we detected a strong but not the complete loss of Dnmt3a expression. This suggests that the Bcor absence may influence the loss of the Dnmt3a gene (Supplementary Fig. 4B, C).

A Kaplan–Mayer plot of mice survival according to the indicated genotypes (n = 8 to 24 per genotype) Bcor−/−Dnmt3a−/− mice display a median survival of 135 days (p < 0.0001, Logrank Test). B WBCs number (i) and MCV (ii) values in Bcor−/− Dnmt3a−/− leukemic mice compared to other control. (iii) Platelets number in Bcor−/−, Dnmt3a−/−, Bcor−/−Dnmt3a−/− and WT in PB of 3/4 months old mice. C i BM cytospin (top panel) hematossilin and eosin of the spleen (middle panel) and liver (bottom panel) in Bcor−/−, Dnmt3a−/− and WT control mice, showing the blasts infiltration. ×600 of magnification. ii Spleen weight to total body weight ratio in the indicated genotypes. Spleen ratio in Bcor−/−Dnmt3a−/− (n = 9) mice was two fold greater than in Dnmt3a−/−(n = 4), Bcor−/− (n = 6) and WT control (n = 8) (0.03378 ± 0.0077 and 0.0025 ± 0.000022 vs 0.0042 ± 0.00036 and 0.0035 ± 0.0026 p < 0.001 by one-way ANOVA analysis). *p < 0.05, **p < 0.01; ***p < 0.001 unpaired t-test with Welch’s correction.
The leukemia diagnosis was primarily based on the presence of leukocytosis and marked macrocytic anemia (Fig. 2Bi, ii). Moreover, the compound mutants showed a consistent drop in platelets number (about 50%) comparing to the preleukemic phase. Conversely, in the other genotypes, platelet numbers remained stable during the entire follow-up period (Fig. 2Biii).
Consistently with the diagnosis of acute leukemia, BM, PB, and spleen cytospins showed an expansion of nucleated cells with blastic appearance (Fig. 2Ci, top panel, Supplementary Fig. 5ai, ii). The latter was also supported by the presence of paraspinal masses (Supplementary Fig. 5Bi, ii). Disease aggressiveness was confirmed by multiple organ infiltration (including lung, liver, and spleen) and the monotonous blastic populations that partially subverted the local tissue architecture (Fig. 2Ci, ii, middle panel/bottom panel and Supplementary Fig. 5Ci, ii). Flow cytometric analysis demonstrated that leukemic cells co-expressed Ter119 and c-Kit (Fig. 3A), clearly indicating they belonged to the erythroid cell lineage and supporting the diagnosis of acute erythroid leukemia (AEL).

A TER119 + cKIT+ number of cells in BM (left) and spleen (right) in Bcor−/−Dnmt3a−/− leukemic mice and preleukemic compared to other control of 3/4 months old mice (n = 9, 6, 7, 6, 7). B Kaplan–Mayer plot of mice transplanted with 3 × 106 Bcor−/−Dnmt3a−/− leukemic cells n = 12. C Total number of cKIT+, Ter119+cKIT+, GR1 + MAC1+, MAC1 + GR−, CD3 and B220+, pre PipC and 3 months post PipC induction in PB of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/− and WT control (pre n = 8,4,4,7; post n = 14,8,7,18). *p < 0.05, **p < 0.01; ***p < 0.001 unpaired t-test with Welch’s correction.
This AEL phenotype was transplantable up to 9 secondary recipients, which developed a lethal AEL with a median survival of 59 days (range 18–78 days) (Fig. 3B). These secondary recipients displayed similar phenotypic characteristics of the primary tumor. Furthermore, the self-renewal capability of the Bcor−/−/Dnmt3a−/− BM cells was evaluated in replating experiments using colony-forming unit assay (CFU) demonstrating that double knockout cells can regenerate for long time period as compared to other groups (Supplementary Fig. 6).
Bcor and Dnmt3a loss induces a displacement towards the erythroid profile starting from early leukemia stages
To better understand the cellular effects of Bcor and Dnmt3a loss in vivo, we analysed PB and BM samples of unmutated, single and double knockout mice at both early and overt leukemia stages.
At leukemia onset, it was observed an expansion of white blood cells (WBC) in Bcor−/−Dnmt3a−/− due to increased numbers of Gr1 + Mac1+ granulocytes, Gr1+Mac- monocytes and CD3 + lymphocytes together with a progressive expansion of a population of immature cells co-expressing c-Kit and the erythroid marker Ter119 (Fig. 3C). Double knockout mice exhibited a constant increase of WBC count and a drop in the hemoglobin levels (not shown) associated with an increased MCV (Fig. 4A). However, while Bcor−/− mice showed a progressive increase of platelets counts, the Bcor−/−Dnmt3a−/− mutant only showed an initial expansion followed by a significant decline associated with leukemic phase (Fig. 4B).

A Changes in WBC counts (left) and MCV (right) of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/−and WT control during six months. B Changes in platelets number (left) and Platelets Ratio between 3 and 6 months (right) of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/−, and WT control during 6 months. *p < 0.05, **p < 0.01; ***p < 0.001 unpaired t-test with Welch’s correction.
Cellular alterations of BM and spleen were analysed 2–3 months after activation of conditional mutations in all genotypes to investigate early leukemic stages. For comparison, we included in the analysis Bcor−/−Dnmt3a−/− mice developing leukemia 4–6 months after pIpC induction of the Cre-recombinase. Lin−/Sca1+/Kit+ (LSK) stem cell compartment examination revealed a significant accumulation of long term hematopoietic stem cells (LT-HSC Lin−/Sca1+/Kit+CD34lo/−FLT3−) in leukemic Bcor−/−Dnmt3a−/− BM compared to other groups, a feature not found at early leukemic stages (Fig. 5Ai, ii Supplementary Fig. 7).

A Representative flow-cytometric analysis (i) of stem cell compartment, long term hematopoietic stem cells (LT-HSC Lin−/Sca1+/Kit+CD34lo/−FLT3-), short term hematopoietic stem cells (ST-HSC Lin−/Sca1+/Kit+CD34+FLT3-) and multipotent progenitors (MPP Lin−/Sca1+/Kit+CD34+FLT3+) in BM of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/−and WT mice. Total number (Right panel) of LT-HSC (ii), ST-HSC (iii) and MPP (iv) in BM of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/−and WT mice. B Representative flow-cytometric analysis (i) of progenitors cell compartment, granulocyte-macrophage progenitors (GMP Lin−/Sca1−/Kit+CD34+FCgRII/III+), common-myeloid (CMP Lin−/Sca1−/Kit+CD34+FCgRII/III−) and megakaryocyte-erythroid progenitors (MEP Lin−/Sca1−/Kit+CD34−FCgRII/IIIlo/−) in BM of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/−and WT mice. Total number of GMP (ii), CMP (iii) and MEP (iv) in BM of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/− and WT mice. (n = 16, 12, 10, 9) (v) Total number of (Pre-MegE Lin−/FCgRII/III− CD150+CD105−) and Pre-CFUe (Lin−/FCgRII/III- CD150+CD105+) in BM of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/− and WT mice. (n = 16, 12, 10, 9) C Summary of BM hemopoietic development in mice; red arrows indicate the deregulated populations. *p < 0.05, **p < 0.01; ***p < 0.001 unpaired t-test with Welch’s correction. .
Short term hematopoietic stem cells (ST-HSC Lin−/Sca1+/Kit+CD34+FLT3−) were markedly lower only in Bcor−/− genotype. Multipotent progenitors (MPP Lin−/Sca1+/Kit+CD34+FLT3+) were lower in double mutant mice both at early and overt leukemic stages. However, statistical significance was only reached when MPPs numbers were compared to wild type animals (Fig. 5Aiii, iv). The analysis of changes occurring during lineage commitment and maturation revealed a striking 5-fold increase of megakaryocyte-erythroid progenitors (MEP Lin−/Sca1−/Kit+CD34−FCgRII/IIIlo/−) in preleukemic and leukemic Bcor−/−Dnmt3a−/− mice (Fig. 5Bi, iv). Accordingly, only leukemic mice displayed a marked expansion of other more committed erythroid-restricted progenitors including the bipotent pre-megakaryocyte erythrocyte (Pre-MegE Lin−/FCgRII/III− CD150+CD105−) and Pre-CFUe (Lin−/FCgRII/III− CD150+CD105+) (Fig. 5Bv).
Given the erythroid skewing of HSC differentiation of Bcor−/−Dnmt3a−/− mice, we used CD71 and Ter119 staining to further characterize later downstream stages of RBCs development within the BM. Our data demonstrated an accumulation of proerythroblasts both at preleukemic and leukemic stages, while the increase of early basophilic, late basophilic, chromatophilic, and orthochromatophilic erythroblasts was detected only in double knockout leukemic mice (Supplementary Fig. 8A).
Altogether, these data suggest that the development of overt leukemia is predated by changes in the composition of hematopoietic stem/progenitor cells compartment, more detectable among myeloid and erythroid progenitors (Fig. 5C).
No significant differences emerged in total number of different myeloid committed progenitors including granulocyte-macrophage (GMP Lin−/Sca1−/Kit+CD34+FCgRII/III+), common-myeloid (CMP Lin−/Sca1−/Kit+CD34+FCgRII/III−) (Fig. 5Bii, iii) and pre-granulocyte-monocyte progenitors (Pre-GM Lin−/FCgRII/III− CD150−CD105−), as well as more mature BM granulocytes and monocytes, among all littermate groups (data not shown).
Bcor and Dnmt3a loss determines altered GATA factor switching and changes of p53 family members in LSK and MEP compartments
Next, we performed RNA-seq experiments to get insight on the molecular changes of Bcor and Dnmt3a cooperation in AEL development. We performed a meta-analysis of the differentially regulated genes from the following pairwise comparisons: leukemic Bcor−/−Dnmt3a−/− vs wild type, Bcor−/− vs wild type, Dnmt3a−/− vs wild type. Moreover, we searched for significant two-way and three-way overlaps among the differentially expressed gene lists. Analysis was done on both LSK and MEP populations, given the significant expansion of these cells in the BM of leukemic mice.
Bcor−/−Dnmt3a−/− LSK and MEP showed a large number of differentially expressed genes (560 and 269, respectively). Within LSK population, 106/560 were upregulated and 454/560 were downregulated (Fig. 6Ai, Table S1) while, within MEP compartment 133/269 were upregulated and 136/269 downregulated (Fig. 6Ai, Supplementary Fig. 9A, Table S2). There were 35 genes for LSK and 17 for MEP transcripts commonly altered between Bcor−/−Dnmt3a−/− and Bcor−/−, on the other hand, there were no transcripts commonly altered in all pairwise comparisons with Dnmt3a−/− (Fig. 6Ai and Supplementary Fig. 9A).

A (i) Overlap of differently gene expression (RNAseq) in the LSK cells of Bcor−/−Dnmt3a−/−, Bcor−/−, Dnmt3a−/−compared to WT control (n = 3 mice for each genotype). (ii) mRNA expression in the LSK cells of the indicated genotypes for the most up- and downregulated genes. B Heatmaps of apoptosis pathway (left), cell cycle (middle), cancer (right) in LSK cell for the indicated genotype. C Enrichr bar plot (https://amp.pharm.mssm.edu/Enrichr/). Bars represent the proportion of genes upregulated in both human AEL samples (n = 137) and Bcor−/−Dnmt3a−/− LSK (n = 3) whose promoter is bound by the indicated transcription factor.
Interestingly, GATA1 scored as one of the most upregulated genes in LSK compartment in line with an aberrant erythroid skewing originating in the hematopoietic compartment of leukemic mice. In these cells, GATA2 levels were highly downregulated (Fig. 6Aii) proving further evidence of an altered switching of GATA factors as a driving event in Bcor−/−Dnmt3a−/− mice with AEL (Table S1). The dysregulation on GATA family members was not detected in MEP compartment of leukemic mice, confirming that this defect was directly linked to the leukemic stem cell population (Table S2, Supplementary Fig. 9B).
Additional pathway analysis of LSK and MEP cells from Bcor−/−Dnmt3a−/− mice showed the enrichment of deregulated genes involved in cellular apoptosis, such as BID and BAK pro-apoptotic genes and the anti-apoptotic Bcl2 family genes. The PARP1 pro-survival gene was strongly upregulated in LSK only (Fig. 6Aii and Supplementary Fig. 9B, C). In addition, we found a notable decline of p53 levels, that has been driven by the marked elevation of MDM2, that is its master antagonist in the cell cycle machinery [28] (Fig. 6Aii, b and Supplementary Fig. 9B, C).
In order to search for shared molecular alterations, RNA-seq data on mouse LSK cells were used as a backbone for comparison with human AEL samples [29]. By interpolating these data with the list of deregulated genes in our leukemic mice, we found 104 genes, commonly upregulated, and 136 genes, commonly downregulated, between mouse LSK cells and human AEL (Supplementary Fig. 10A, B, Supplementary Fig. 11, Table 1). Within genes upregulated in both human and mouse cells, there was a striking enrichment of genes regulated by GATA1 (Fig. 6C), suggesting a pivotal role of this transcription factor in driving AEL. These findings provided evidence that our AEL mouse may serve as an operational platform for human Bcor−/−Dnmt3a−/− leukemia modeling.
Decitabine exerts inhibitory effects on the Bcor/Dnmt3a null leukemic mice
In order to assess the impact of anti-leukemic therapies on Bcor−/−Dnmt3a−/−AEL mice, we tested the therapeutic efficacy of the standard chemotherapeutic agent cytarabine compared to the demethylating agent decitabine in lethally irradiated CD45.1 recipient mice transplanted with leukemic cells. The rationale for using decitabine was based on the fact that the Bcor transcriptional repression activity is mediated by histone demethylase and the function of Dnmt3a is to catalyse the addition of methyl groups to CpG dinucleotides. Thus, in our double knockout mice, we expected a hypomethylation status with leukemogenic potentials.
Mouse treatments started at leukemia onset, defined by the presence of WBC count above 20,000 cells/μl and/or high MCV values and/or low platelets number. Cytarabine was administered for 7 days continuously, while decitabine for a total of 5 administrations twice a week, followed by the assessment of disease burden (Fig. 7Ai). WBC count was significantly reduced at the end of decitabine treatment compared to vehicle, while chemotherapy determined only a modest impact on leukocytosis (Fig. 7Aii). Two weeks after the end of treatments, WBC count was significantly lower in decitabine group, compared to cytarabine and vehicle ones. PB flow cytometry showed a significant reduction of immature c-KIT and Ter119 + c-KIT + cells after decitabine compared to other treatments (Fig. 7Bi, ii), as also confirmed by post-mortem examination on pathological splenic specimens (Fig. 7Biii), thus indicating that Bcor−/−Dnmt3a−/− leukemic cells were more sensitive to hypomethylating agent than to chemotherapy. Moreover, there was a tendency for decitabine treated mice toward the achievement of a longer survival, compared to cytarabine and vehicle groups (Fig. 7C).

A WBC count changes (i) in leukemic Bcor−/−Dnmt3a−/− mice pre and 3 days after the end of the treatment with decytabine (DEC), cytarabine (ARAC) and vehicle (VEI). (ii) WBC follow up (right) during and post the treatment with DEC, ARAC and VEI in leukemic mice. B (i) Total number of GR1 + MAC1 + , MAC1 + GR1-, cKIT+, CD41+, B220+ and CD3+ cells in PB 3 days after DEC, ARAC and vehicle treatment in leukemic Bcor−/−Dnmt3a−/−, mice (n = 11,11,12). (ii) Total number of TER119+ and TER119 + /cKIT+ cells in PB 3 days after DEC, ARAC and vehicle treatment in leukemic Bcor−/−Dnmt3a−/− mice (n = 11,11,12). (iii) Spleen weight to total body weight ratio in Bcor−/−Dnmt3a−/− leukemic mice after each indicated treatment (N = 4,4,3). C Kaplan–Mayer plot of leukemic Bcor−/−Dnmt3a−/− mice survival after DEC, ARAC and VEI treatment to the indicated genotypes (n = 10 to 9 per genotype) (p < 0.0001, Logrank Test). *p < 0.05, **p < 0.01; ***p < 0.001 Wilcoxon matched pairs test.
Discussion
Somatic recurrent loss-of-function mutations of Bcor have been detected in AML [7] but their precise role in normal and malignant hematopoiesis is still under investigation. Here, we demonstrate for the first time that Bcor deficiency perturbs erythro-megakaryopoiesis and cooperates with Dnmt3a loss (a phenotype partially recapitulating that of heterozygous Dnmt3aR662H mutations [30]) in promoting AEL in mice.
Perturbation of the erythroid compartment in our Bcor-deficient mice was characterized by a decrease in the total number of red blood cells and macrocytosis, confirming previous observations from Tara et al. [19] who demonstrated mild macrocytic anemia in mice after the deletion of Bcor exons 9 and 10. Besides the negative effects on erythroid cells, we also found a strong increase in platelets counts due to a significant expansion of the MKP population in BM.
Contribution of Bcor loss in promoting megakaryocytic proliferation in mice is a novel finding that is also of potential clinical relevance. A missense Bcor mutation has been reported in a patient with triple-negative essential thrombocytemia and normal karyotype, suggesting a possible role of this variant in the pathogenesis of the disease [31]. Moreover, Bcor mutations may occur in myelodysplastic/myeloproliferative neoplasms (MDS/MPNs) that share dysplastic and proliferative features [32]. MDS/MPN disorders include chronic myelomonocytic leukemia (CMML) and MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) in which Bcor is recurrently mutated in 7.4% [33] and 24% of cases [34], respectively. Although the most frequent mutation in MDS/MPN-RS-T is that affecting the SF3B1 gene [35, 36], the presence of variants in epigenetic genes, such as Bcor, may contribute to the development of this myeloid neoplasm. Co-occurrence of erythroid blood cell alterations and thrombocytosis in our conditional knockout Bcor mice support this hypothesis.
Similar to previous models [17, 19], Bcor deficiency alone was not sufficient to drive a myeloid malignancy in our mice. However, we could demonstrate that the combined Bcor and Dnmt3a loss promoted a fully penetrant AEL phenotype that killed mice in 5–6 months. Leukemic mice showed leukocytosis due to the expansion of c-Kit+ blasts expressing the erythroid marker Ter119, macrocytic anemia, and progressive reduction of the thrombocytosis driven by the single Bcor deletion. The analysis of BM subpopulations showed an erythroid skewing of HSC differentiation demonstrating that AEL was driven by loss of Bcor and Dnmt3a.
The 2016 WHO classification of hematopoietic tumors has adopted rather restrictive criteria for the diagnosis of pure erythroid leukemia (or AEL), moving cases with an increased percentage of myeloid blasts (previously named erythroleukemia) in the group of MDS [37,38,39]. Thus AEL, described for the first time by Di Guglielmo in 1928 under the term of “acute erythremic myelosis” [40], is now included as an entity within the category of AML not otherwise specified (NOS) of WHO-2016 [37]. However, targeted next-generation sequencing has clearly demonstrated the molecular heterogeneity of this apparently homogeneous morphological form of AML [41, 42] that shows a mutational spectrum intermediate between MDS and AML [29]. Notably, two cases of AEL were characterized by the co-occurrence of Bcor and Dnmt3a mutations only [29], thus supporting the findings described in our mice model.
In order to define the gene expression signature of AEL in our mice, we performed RNAseq analysis on the LSK population of the four different genotypes. We identified an altered switching of the Gata factors that are known to play a key role in controlling mechanisms underlying erythroid differentiation [43]. Specifically, Gata1 was one of the most upregulated genes in leukemic mice compared to the other genotypes whereas Gata2 was downregulated. The latter finding is unexpected since in physiological conditions [44], the transcription factor Gata2 is highly expressed in hematopoietic stem cells, whereas its expression declines after erythroid commitment of progenitors [43]. In contrast, the start of Gata1 expression coincides with the erythroid commitment and increases along with the erythroid differentiation. Collectively, these data provide evidence for an imbalance towards an erythroid phenotype starting from the LSK stage and point to Gata factors deregulation as an early event altering the HSC fate and sensitizing cells to further malignant transformation in the context of concurrent Bcor and Dnmt3a deficiency, as it has been also very recently demonstrated in both human and mouse AEL samples [45].
Our AEL mouse model also exhibited dysregulation of various oncogenes and tumor suppressor genes involved in cancer pathways, including decline of Tp53 levels and marked elevation of Mdm2. Accordingly, an in vivo mouse model demonstrated a key driver leukemogenic function of Tp53 supported by the potent interaction with Ntrk1 [29]. Moreover, the presence of more than a single TP53 abnormality seems to play a key role in the molecular pathogenesis of AEL in patients [46].
AEL is usually poorly responsive to intensive chemotherapy and shows a dismal outcome [47] that may be due, at least in part, to the accumulative impaired TP53 function and consequent genomic instability. Similarly, our AEL mice were resistant to the nucleoside analog cytarabine but showed significantly improved survival when treated with the epigenetic drug decitabine. The benefit of this drug suggests an important role of epigenetics in promoting AEL in our Bcor−/−Dnmt3a−/− compound mice. Accordingly, the presence of TP53 mutations appears to be associated with a high degree of decitabine sensitivity in AML patients in one study [48], although it has not been confirmed in another study [46].
In conclusion, we provide the first demonstration that concurrent Bcor and Dnmt3a loss promotes AEL in mice and sheds light, at least in part, on the cellular and molecular features underlying this leukemia. Our model also represents a potential platform for the identification and validation of drugs for improving therapy of AEL patients.
References
Huynh KD, Fischle W, Verdin E, Bardwell VJ. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000;14:1810–23.
Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunological Rev. 2012;247:172–83.
Surapornsawasd T, Ogawa T, Moriyama K. Identification of nuclear localization signals within the human BCOR protein. Febs Lett. 2015;589:3313–20.
Isshiki Y, Iwama A. Emerging role of noncanonical polycomb repressive complexes in normal and malignant hematopoiesis. Exp Hematol. 2018;68:10–14.
Vidal M, Starowicz K. Polycomb complexes PRC1. and their function in hematopoiesis. Exp Hematol. 2017;48:12–31.
Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yohida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35–47.
Tiacci E, Grossmann V, Martelli MP, Kohlmann A, Haferlach T, Falini B. The corepressors BCOR and BCORL1: two novel players in acute myeloid leukemia. Haematologica. 2012;97:3–5.
Astolfi A, Fiore M, Melchionda F, Indio V, Bertuccio SN, Pession A. BCOR involvement in cancer. Epigenomics. 2019;11:835–55.
Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118:6153–63.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik V, Paschka P, Roberts N, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Terada K, Yamaguchi H, Ueki T, Usuki K, Kobayashi Y, Tajika K, et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosomes Cancer. 2018;57:401–8.
Shiba N, Yoshida K, Shiraishi Y, Okuno Y, Yamato G, Hara Y, et al. Whole-exome sequencing reveals the spectrum of gene mutations and the clonal evolution patterns in paediatric acute myeloid leukaemia. Br J Haematol. 2016;175:476–89.
de Rooij JDE, van den Heuvel-Eibrink MM, Hermkens MCH, Verboon LJ, Arentsen-Peters S, Fornerod M, et al. BCOR and BCORL1 mutations in pediatric acute myeloid leukemia. Haematologica. 2015;100:e194–5.
Herold T, Metzeler KH, Vosberg S, Hartmann L, Rollig C, Stolzel F, et al. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood. 2014;124:1304–11.
Li M, Collins R, Jiao YC, Ouillette P, Bixby D, Erba H, et al. Somatic mutations in the transcriptional corepressor gene BCORL1 in adult acute myelogenous leukemia. Blood. 2011;118:5914–7.
Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, et al. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet. 2004;36:411–6.
Kelly MJ, So J, Rogers AJ, Gregory G, Li J, Zethoven M, et al. Bcor loss perturbs myeloid differentiation and promotes leukaemogenesis. Nat Commun. 2019;10:1347.
Cao Q, Gearhart MD, Gery S, Shojaee S, Yang H, Sun H, et al. BCOR regulates myeloid cell proliferation and differentiation. Leukemia. 2016;30:1155–65.
Tara S, Isshiki Y, Nakajima-Takagi Y, Oshima M, Aoyama K, Tanaka T, et al. Bcor insufficiency promotes initiation and progression of myelodysplastic syndrome. Blood. 2018;132:2470–83.
Ley TJ, Ding L, Walter MJ, McLellan M, Lamprecht T, Larson D, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33.
Brunetti L, Gundry MC, Goodell MA. DNMT3A in Leukemia. Cold Spring Harb Perspect Med. 2017;7:a030320.
Ketkar S, Verdoni AM, Smith AM, Bangert CV, Leight ER, Chen DY, et al. Remethylation of Dnmt3a (-/-) hematopoietic cells is associated with partial correction of gene dysregulation and reduced myeloid skewing. Proc Natl Acad Sci USA. 2020;117:3123–34.
Challen GA, Sun DQ, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–43.
Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015;125:629–38.
Sportoletti P, Varasano E, Rossi R, Mupo A, Tiacci E, Vassiliou G, et al. Mouse model of NPM1-mutated acute myeloid leukemia: biological and clinincal implications. Leukemia. 2015;29:269–78.
Guryanova OA, Lieu YK, Garrett-Bakelman FE, Spitzer B, Glass JL, Shank K, et al. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia. 2016;30:1133–42.
Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med. 2016;22:1488–95.
Wang SM, Zhao YJ, Aguilar A, Bernard D, Yang CY. Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb Perspect Med. 2017;7:a026245.
Iacobucci I, Wen J, Meggendorfer M, Choi JK, Shi L, Pounds SB, et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat Genet. 2019;51:694–704.
Cole CB, Russler-Germain DA, Ketkar S, Verdoni AM, Smith AM, Bangert CV, et al. Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J Clin Invest. 2017;127:3657–74.
Zaidi U, Shahid S, Fatima N, Ahmed S, Sufaida G, Nadeem M, et al. Genomic profile of a patient with triple negative essential thrombocythemia, unresponsive to therapy: a case report and literature review. J Adv Res. 2017;8:375–8.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitiz M, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia (vol 127, pg 2391, 2016). Blood. 2016;128:462–3.
Damm F, Chesnais V, Nagata Y, Yoshida K, Scouriz L, Okuno Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122:3169–77.
McClure RF, Ewalt MD, Crow J, Temple-Smolkin R, Pullambhatla M, Sargent R, et al. Clinical significance of DNA variants in chronic myeloid neoplasms a report of the association for molecular pathology. J Mol Diagnostics. 2018;20:717–37.
Patnaik MM, Tefferi A. Refractory anemia with ring sideroblasts (RARS) and RARS with thrombocytosis: “2019 update on diagnosis, risk-stratification, and management”. Am J Hematol. 2019;94:475–88.
Li P, Shahmarvand N, Lynch D, Gotlib JR, Merker JD, Zehnder J, et al. Revisiting diagnostic criteria for myelodysplastic/myeloproliferative neoplasms with ring sideroblasts and thrombocytosis: borderline cases without anemia exist. Int J Lab Hematol. 2019;41:345–52.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitiz M, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Arber DA. Revisiting erythroleukemia. Curr Opin Hematol. 2017;24:146–51.
Pellagatti A, Armstrong RN, Steeples V, Sharma E, Repapi E, Singh S, et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood. 2018;132:1225–40.
Balduini CL. 100-Year old haematologica images: Di Guglielmo disease or pure erythroid leukemia. Haematologica. 2020;105:525.
Grossmann V, Bacher U, Haferlach C, Schnittger S, Potsinger F, Weissmann S, et al. Acute erythroid leukemia (AEL) can be separated into distinct prognostic subsets based on cytogenetic and molecular genetic characteristics. Leukemia. 2013;27:1940–3.
Cervera N, Carbuccia N, Garnier S, Guille A, Adelaide J, Murati A, et al. Molecular characterization of acute erythroid leukemia (M6-AML) using targeted next-generation sequencing. Leukemia. 2016;30:966–70.
Katsumura KR, Bresnick EH. The GATA factor revolution in hematology. Blood. 2017;129:2092–102.
Sportoletti P, Celani L, Varasano E, Rossi R, Sorcini D, Rompietti C, et al. GATA1 epigenetic deregulation contributes to the development of AML with NPM1 and FLT3-ITD cooperating mutations. Leukemia. 2019;33:1827–32.
Fagnan A, Otzen Bagger F, Piquè-Borras M, Ignacimouttou C, Caulier A, Lopez CK, et al. Human erythroleukemia genetics and transcriptomes identify master transcription factors as functional disease drivers. Blood. 2020. https://doi.org/10.1182/blood.2019003062.
Montalban-Bravo G, Benton CB, Wang SA, Ravandi F, Kadia T, Cortes J, et al. More than 1 TP53 abnormality is a dominant characteristic of pure erythroid leukemia. Blood. 2017;129:2584–7.
Almeida AM, Prebet T, Itzykson R, Ramos F, Al-Ali H, Shammo J, et al. Clinical outcomes of 217 patients with acute erythroleukemia according to treatment type and line: A retrospective multinational study. Int J Mol Sci. 2017;18:837.
Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med. 2016;375:2023–36.
Acknowledgements
This work was supported by a grant from the Italian Association for Cancer Research (AIRC: n. 23604) and Fondation ARC (Leopold Griffuel Award) to B.F. This work was supported by the Edward P. Evans Foundation and the National Institutes of Health (DK092883, CA183252, AG036695, and CA125123) and by the Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program.
Author information
Authors and Affiliations
Contributions
PS, DS, MAG, and BF, conceived the study. AS, SS, AM, LB, BD, RR, GP, CB, GV, and AS performed the experiments and analyzed the data. DS, FMA, and LB performed cytometric analysis. AGG, JMR, SS, and AS performed molecular analysis. AM, FMA, DS, and AS carried out histological analysis. VT and GS constructed analytical and visualization tools and databases. MPM provided logistical support and performed critical review of the manuscript. PS, DS, MAG, and BF wrote the manuscript. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sportoletti, P., Sorcini, D., Guzman, A.G. et al. Bcor deficiency perturbs erythro-megakaryopoiesis and cooperates with Dnmt3a loss in acute erythroid leukemia onset in mice. Leukemia 35, 1949–1963 (2021). https://doi.org/10.1038/s41375-020-01075-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-01075-3
This article is cited by
-
Functional and epigenetic phenotypes of humans and mice with DNMT3A Overgrowth Syndrome
Nature Communications (2021)
