
Abstract
Despite worldwide promising clinical outcome of CD19 CAR-T therapy, relapse after this therapy is associated with poor prognosis and has become an urgent problem to be solved. We conducted a CD22 CAR T-cell therapy in 34 relapsed or refractory (r/r) B-ALL pediatric and adult patients who failed from previous CD19 CAR T-cell therapy. Complete remission (CR) or CR with incomplete count recovery (CRi) was achieved in 24 of 30 patients (80%) that could be evaluated on day 30 after infusion, which accounted for 70.5% of all 34 enrolled patients. Most patients only experienced mild cytokine-release syndrome and neurotoxicity. Seven CR patients received no further treatment, and 3 of them remained in remission at 6, 6.6, and 14 months after infusion. Eleven CR patients were promptly bridged to transplantation, and 8 of them remained in remission at 4.6 to 13.3 months after transplantation, resulted in 1-year leukemia-free survival rate of 71.6% (95% CI, 44.2–99.0). CD22 antigen loss or mutation was not observed to be associated with relapsed patients. Our study demonstrated that our CD22 CAR T-cells was highly effective in inducing remission in r/r B-ALL patients, and also provided a precious window for subsequent transplantation to achieve durable remission.
Similar content being viewed by others

Introduction
B-cell acute lymphoblastic leukemia (B-ALL), is usually treated with chemotherapy and allogeneic hematopoietic stem cell transplantation (allo-HCT). Patients who are refractory or relapsed (r/r) after these conventional therapies have extremely poor prognosis [1,2,3]. Recently, clinical trials with CD19 chimeric antigen receptor (CAR) T-cell therapy have shown 70 to 90% complete remission (CR) rate in patients with r/r B-ALL [4,5,6]. However, some patients display no response [7, 8], and a large proportion (43–55%) of CR patients relapsed within 1 year [9, 10]. Loss or mutation of CD19 are frequently observed, which hamper the secondary application of CD19 CAR T-cell therapy [4, 10,11,12,13,14]. Even in some treatment-failure cases which preserves high CD19 expression on leukemia cells, a secondary infusion of CD19 CAR T-cells has not been reproducibly successful, which may be partly due to immune-mediated clearance of murine-derived CARs [15,16,17]. Therefore, new therapeutic methods are required for r/r patients who have failed previous CD19 CAR T-cell therapy.
CD22 represents one of the candidates for replacing CD19 as targeting antigen by CAR T cells, due to its high expression on leukemic cells from most r/r B-ALL patients and the restricted expression on normal B cells [18,19,20,21]. A recent study showed that CD22 CAR T-cell therapy in 21 r/r B-ALL patients resulting in a 74% CR rate, but most patients relapsed after CD22 CAR T-cell infusion [22], raising the question of whether CD22 CAR T-cell therapy could benefit these patients. Furthermore, whether the CAR T manufacture process, characteristics of the patients, and clinical protocol play roles in the efficacy, safety and long-term outcome of the r/r B-ALL patients receiving CD22 CAR T-cell therapy remains largely unknown.
Here we report a study on the therapy with CD22 CAR T-cells in 34 r/r B-ALL patients that have mostly failed previous CD19 CAR T-cell therapy and were considered to be incurable, showing the efficacy, adverse effect and long-term outcome of this therapy. CD22 CAR T-cell therapy resulted in high CR rate with mild or moderate cytokine-release syndrome and neurotoxicity in most patients. In the long-term follow-up, only a minority of the patients without further therapy remained in remission, while patients who were bridged to a subsequent transplantation mostly remained in remission. We also examined CD22 expression on leukemia cells in some relapsed patients, and did not observed an association between relapse and loss/mutation of CD22 antigen.
Methods
Study design and patients
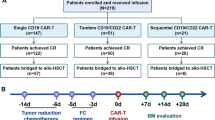
We conducted CD22 CAR T-cell therapy in 34 r/r B-ALL patients in Beijing Boren Hospital, Beijing, China. The study was approved by the institutional review board of Beijing Boren Hospital, and informed consent was obtained in accordance with the Declaration of Helsinki. This study included 14 patients who were treated in the proof of concept (POC) procedure before trial registration, and 20 patients who were enrolled after registration on Chinese Clinical Trial Registry/WHO International Clinical Trial Registry (ClinicalTrials#: ChiCTR-OIC-17013523). Patients were eligible if they were r/r CD22+B-ALL without curative treatment options including CD19 CAR-T therapy (the details of inclusion and exclusion (I/E) criteria are shown in Supplementary Methods). The baseline disease status was assessed immediately before enrollment. After enrollment, there was no further bridging chemotherapy before lymphodepleting procedure. The 14 POC patients were consistent with those of the 20 patients enrolled after trial registration in I/E criteria, study protocol, follow-up strategy, and adverse effect management principle (Detailed clinical trial protocol had been on ChiCTR website). Enrolled patients received CD22 CAR T-cell infusion between July 6, 2017 and May 9, 2018, and were evaluated for responses and adverse effects. After CAR T-cell infusion, non-transplanted patients were mostly bridged to subsequent transplantation. Clinical outcomes including leukemia-free survival (LFS), relapse, and treatment-related mortality (TRM) were evaluated up to date as of October 17th, 2018.
Design, construction and cytotoxicity validation of CAR
The antigen recognition domain of this CD22 specific CAR was obtained from a human antibody phage display library. A lentiviral vector carrying a CD22 CAR with 4-1BB costimulatory domain and a CD3-zeta signaling domain was constructed as previously described [4,5,6]. The activity of CAR T-cells based on this construct was evaluated in vitro by a cytotoxicity assay against CD22+ Nalm6 cells (the details had been in Supplementary Methods).
Manufacture of CAR T-cells
CD22 CAR T-cells were manufactured with T cells, obtained through leukapheresis, and transduced with the lentiviral vector expressing the anti-CD22 CAR with the same protocol of the production of our CD19 CAR T-cells as previously described [4], and the details were in Supplementary Methods. Leukemia burden of ≤30% in peripheral blood (PB) was required for leukapheresis in non-transplanted patients, while both patients and donors were allowed as cell sources for previously transplanted patients. When previously transplanted patients had >30% blasts in PB or low WBC counts (<1 × 109/L), T cells were obtained from donors. The agent was administered at dosage of ≤4 × 106 per kilogram (/kg) of body weight in non-transplanted patients and at dosage of ≤1 × 106/kg for previously transplanted patients (lower dosages of CAR T-cells derived from donor lymphocytes were applied for reducing risk of severe adverse effects [23, 24]).
Clinical procedures
After leukapheresis, patients received lymphodepleting chemotherapy composing of Fludarabine 30 mg/m2/day and Cyclophosphamide 250 mg/m2/day on days—5–3 before CD22 CAR T-cells infusion (day 0). All the patients underwent bone marrow (BM) biopsy examination and radiology studies on day 30 to determine the response and remission status. Complete remission (CR) or CR with incomplete count recovery (CR/CRi), relapse and minimal residual disease (MRD) were defined in accordance with the National Comprehensive Cancer Network (NCCN) guidelines, version 1.2016 [25]. FCM-MRD− was defined as the absence of leukemia cells in BM determined by FCM. The sensitivity of the FCM-MRD analyses was 0.01%. TRM were calculated from date of allo-HCT to date of death. LFS was calculated from date of allo-HCT to date of relapse or death, or the last follow-up.
Assessment and management of adverse effect
CRS were graded according to the adopted CRS scoring system by Lee et al. [26], and individual organ toxicities and neurotoxicity were graded through the National Cancer Institute Common Terminology Criteria for Adverse Events, CTCAE Version 4.03 [27], The details of CRS managements and supportive care had been shown in Supplementary Methods. Steroids (2 to 15 mg/kg/d methylprednisolone) were given by intravenous injection (IV) in severe CRS patients (grade ≥ 3). Mannitol (2.5 ml/kg/dose IV), furosemide (1 mg/kg/dose IV) and dexamethasone (2–5 mg, intrathecal injection) were used in patients with neurotoxicity (grade ≥ 2).
Flow cytometry and cell sorting
Specimens were analyzed by FCM according to our previously described protocol [4]. Cell sorting was processed on BD Aria II, the leukemia cell staining and gating strategy were determined by clinical diagnosis standard, which is detailed in Supplementary Methods.
RT-PCR, Sanger sequencing, and transcriptome sequencing
The leukemic and normal B cells were sorted from the bone marrow samples of patients. The total RNA was extracted from sorted cells and subjected to reverse transcription-polymerase chain reaction (RT-PCR), Sanger sequencing, or global transcriptome sequencing. Details were in Supplementary Methods.
Statistical analysis
Exact methods (Welch’s ANOVA test, Clopper-Pearson, 95% confidence intervals and Fisher’s exact tests) were used for categorical variables. Difference between two groups was analyzed by unpaired two-tailed Student’s t-test and χ2 test. Spearman correlation coefficient was used for evaluating the correlation between in vivo expansion of CAR T-cells and different serum cytokines. The Kaplan-Meier approach was performed to estimate time-to-event analyses. All statistical analyses were performed using SPSS 25, and all the P-values are two-sided.
Results
Design and in vitro validation of a CD22 CAR construct
The efficacy of CAR T-cells can be affected by expression level, affinity and binding epitope of the antigen recognition domain toward target antigen [28]. To evaluate the expression and cytotoxicity capability of our CAR, we compared our product with another CAR using a different antigen recognition domain derived from anti-CD22 antibody, m971, that has been well used by others for CAR T-cell therapy [20, 22, 29]. The CAR T-cells using our construct (YK-CD22BB-002) out-performed m971-based CAR (m971BB-002), with the same costimulatory and signaling domain, both in CAR expression as well as in cytotoxicity against Nalm6 cells (Fig. 1).

Preclinical evaluation of CD22 CAR T cells. a Flow cytometry analysis of CAR expression in T cells following lentiviral transduction. Left, un-transduced T cells; middle, T cells transduced with m971BB-002; right, T cells transduced with YK-CD22BB-002. b Cytotoxicity of CAR-T cells against target cells carrying luciferase reporter gene evaluated by luminescent assay, after co-culturing with Nalm6 cells for 4 h at the indicated E:T ratios, with m971BB-002 CAR-T cells and YK-CD22BB-002 CAR-T cells respectively. Statistical analysis was performed using one-way ANOVA: #, not statistically significant; *P < 0.05
Patient enrollment and clinical characteristics
The characteristics of all enrolled patients were shown in Table 1. The median age was 10 (range, 1–55). Thirty one/34 patients (91%) had failed previous CD19 CAR T-cell therapy, and among them 11 (35%) had CD19 or CD19dim relapse, and 20 (65%) were CD19bright patients that displayed no response to primary or secondary CD19 CAR T-cells infusion. The other 3 patients (9%) had CD19 weak expression initially, and therefore did not receive CD19 CAR-T therapy. Thirteen of 34 patients (38%) had previously undergone allo-HCT. Extramedullary diseases (EMDs) were commonly observed in 11 patients (32%) including 4 (12%) with exclusive EMDs. Twenty-five patients (74%) had hematological relapse (HR) with a median marrow leukemia burden of 63.8% (rang, 5% to 97%) blasts. Five patients (15%) had FCM-MRD+ with a median marrow blasts of 3% (range, 0.3% to 5%) by FCM. Complex chromosome aberrations, high-risk fusion and mutated genes had been commonly detected. There are no significant differences in patient characteristics between POC and clinical trial groups (Supplementary Table 1). Thirty-two patients had been evaluated to have positive CD22 expression on blasts by FCM before enrollment (30 patients had CD22+ blasts in BM and 2 patients had CD22+ blasts in their exclusive EMDs, Supplementary Fig. 1). The other 2 exclusive EMDs patients had positive CD22 expression on blasts in BM when they were initially diagnosed. They were diagnosed as EMDs by immunohistochemistry instead of FCM in other hospital and were not further evaluated for CD22 expression in EMDs in our hospital due to sustained thrombocytopenia. None of our enrolled patients received any CD22 directed therapy prior to CD22 CAR T-cell infusion.
Before CD22 CAR T-cell infusion, a total of 34 patients received lymphodepleting chemotherapy. Non-transplanted patients received a median dose of 7.5 × 105/kg (range, 0.3–34.7 × 105/kg) CAR T-cells; while transplanted patients received a lower median dose of 1 × 105/kg (range, 0.2 to 10 × 105/kg) CAR T-cells (P = 0.001, Supplementary Fig. 2). Six of 13 previously transplanted patients received CAR T-cells manufactured from donors.
Response rate
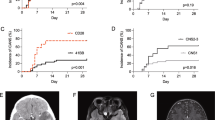
Four patients died within 30 days during CAR T-cell treatment: 2 died on day 10 without evaluation; 1 achieved CRi on day 15 while the other did not. Of the 30 patients who survived for 30 days or longer and were evaluated, 24 (80%) achieved CR/CRi (Fig. 2a), while the total CR rate in 34 enrolled patients was 70.5% on day 30. Of the 21 patients with hematological relapse at baseline, 19 (90%) achieved CR/CRi, and 18 (86%) achieved FCM-MRD‒ (Fig. 2a). Of the 5 FCM-MRD+ patients, 3 (60%) converted to FCM-MRD- (Fig. 2a). Of the 4 patients with exclusive EMDs, 2 (50%) achieved CR, and 1 had a partial reduction in his EMD (mass size: from 10.0 × 5.3 × 5.9 cm to 6.2 × 2.3 × 3.1 cm by ultrasonography). (Fig. 2a). The detailed response of all patients (both in POC and clinical trial) after CAR T-cell therapy are in Supplementary Table 3. There was no significant difference in response rate between patients in POC and clinical trials (Supplementary Table 2).

Response Rate of the 30 Patients. a The rate of MRD-negative, MRD-positive complete remission, partial remission and no response among all evaluated patients (n = 30), hematological relapse patients (n = 21), FCM-MRD+ patients (n = 5), and only EMDs patients (n = 4). Numbers of patients in different groups are indicated in brackets, and numbers of patients with different response are indicated in bars. b The correlation between CAR T-cell expansion and the occurrence of objective response (P = 0.048). c The rate of complete remission according to clinical characteristics of the patients at baseline. Diamonds indicate the observed proportions of complete remission according to different reference category, and lines extended from the diamonds indicate the 95% confidence intervals. The values of confidence intervals are also presented to the right of the plot. Reference categories are identified by the absence of 95% confidence intervals. MRD denotes minimal residual disease, and EMDs extramedullary diseases
The CD22 CAR T-cells were readily detectable in the PB from most patients, indicating the high in vivo expansion rate. The expansion peaked from days 12 to 15 after infusion (1.8-2200 fold). The median duration of in vivo persistence of CAR T-cell detectable by FCM was 28 days (range, 0 to 96) (Supplementary Fig. 3). Serum cytokine markers of systemic inflammation including tumor necrosis factor-α, interleukin-6, interlekin-10, interleukin-2, and interferon-γ were increased during CAR T-cell treatment. While the increase of interleukin-2 and tumor necrosis factor-α, but not interleukin-6, interlekin-10, or interferon-γ were significantly correlated with the expansion kinetics of CAR T-cells (Spearman r = 0.59 and 0.43, Supplementary Fig. 4). The cytokine profile was different from other reports of CAR T therapy [30], which might be due to the distinct targeting antigen, feature of ScFv, and expanding feature of our CAR T-cells.
The response is significantly associated with in vivo expansion of CAR T-cells (P = 0.048), and 3 of the 5 patients without response displayed no expansion of CAR T-cells in vivo (Fig. 2b). CR rate was not significantly different between subgroups categorized according to previous transplantation, disease burden, high risk gene mutations, and fusion genes (Fig. 2c).
Adverse effects
Cytokine-release syndrome (CRS) and neurotoxicity have been reported to be the most common CAR T-cell related toxic effects, while adverse events that are unrelated to CAR T-cell toxicity may also occur [26]. The median onset of CRS, as manifested by fever, decreased appetite, nausea, vomiting, fatigue, edema, rash, headache, hypotension, and transaminitis, occurred on day 7 (range, 0 to 17), with median resolution on day 15 after CAR T-cell infusion. CRS occurred in 31 of 34 patients (91.2%) (Fig. 3a) and was mild or moderate (grade 1 or 2) in 30 patients (Fig. 3a, and Supplementary Fig. 5) who received supportive care alone and resolved fully within 1 week. The median onset of neurotoxicity, occurred on day 8 (range, 1 to 17), with median resolution on day 15 (range, 5 to 17) after CAR T-cell infusion. Grade 1 neurotoxicity, as manifested by headache and mental status changes, occurred in 5 of 34 patients (Fig. 3a). One patient developed grade 2 neurotoxicity on day 7 after CAR T-cell infusion as manifested by brief generalized seizure, and was immediately managed with mannitol and furosemide to reduce intracranial pressure, and dexamethasone to alleviate inflammation, and completely resolved on day 10. The duration of treatment lasted for 1 week.

Cytokine release syndrome and neurotoxic effects. a Left: The grade of cytokine release syndrome and neurotoxic effects in all patients, and the horizontal line indicates the median; Right: the grade of each symptom of CRS in all patients. The horizontal line indicates the median. b The correlation of peak CAR T-cell expansion in peripheral blood, as determined by the percentage of CAR T cells among CD3+ cells, with the occurrence of severe adverse events which led to death in 4 patients (One grade 5 CRS, two infection, and one VOD patients) and neurotoxic effects (P = 0.361 and P = 0.518, respectively). Horizontal lines indicate medians. c The rate of severe adverse events according to clinical characteristics of the patients at baseline. Diamonds indicate the observed proportions severe cytokine release syndrome according to different reference category, and the lines extended from the diamonds indicate the 95% confidence intervals. The values of confidence intervals are also presented to the right of the plot. Reference categories are identified by the absence of the 95% confidence intervals
Four patients died during CAR T-cell treatment. One patient previously received a DLI 20 days before CAR T-cell treatment, and she developed grade 5 CRS and died of severe hepatotoxicity and disseminated intravascular coagulation (DIC) on day 10 after infusion despite the treatment with steroids and plasma exchange (BM not evaluated). Since there was only minimal CAR T-cell expansion (peak level of 0.5% among CD3+ cells) post-infusion, the severe adverse effect in this patient was suspected to be contributed by both complications of DLI and CRS. One transplanted patient received donor-derived CAR T-cells and experienced fever and mild transaminitis without elevated bilirubin (grade 2) during day 6 to 16. After achieving MRD- on day 15, he had rapidly elevated bilirubin, transaminitis and ascites, and developed veno-occlusive disease (VOD) on day 19 and died on day 29. Both secondary engraftment of donor stem cells and CRS may contribute to his death. Other two patients died of complications unrelated to CAR T-cell toxicity. These two patients with a pre-enrollment history of infection developed severe Klebsiella sepsis and Aspergillus pneumonia shortly after lymphodepletion procedure, and died of septic shock and hemoptysis on day 10 (BM not evaluated) and 24 (not remission on day 15) respectively, and they only exhibited mild symptoms of CAR T-cell related CRS or neurotoxicity.
The peak CAR T-cell expansion values were not significantly different between the patients with no/mild and those with sever adverse events, and there was also no difference with regard to the severity of neurotoxicity (Fig. 3b). We also analyzed the incidence of severe adverse events in patients according to different subgroups, and found that most clinical features including age, leukemia burden, and EMDs were unrelated with the incidence of severe adverse effect (Fig. 3c).
Long-term follow up
The patients were followed up after CAR T-cell infusion during extended observation time. The only-EMD patient who displayed partially reduced EMD lesions after CAR T-cell infusion had his EMD progressed and relapse in BM on day 90. Of the 24 patients who attained CR after infusion, 4 withdrew from the study to receive chemotherapies due to the willingness of themselves, 2 were lost of follow-up after being discharged, 11 were bridged to a subsequent allo-HCT (most of them had haploidentical donors as described in Supplementary Table 3 and details of donor types, conditioning and stem cell source were listed in Supplementary Table 4) at a median time of 55 days (range, 40 to 70) after CAR T-cell infusion. To the observation endpoint, 3 of the 7 CR patients without further treatment remained in remission at 6, 6.6, and 14 months, but 4 had a relapse at 1.7 to 6 months (median, 3.4 months) after CAR T-cell infusion. Of the 11 CR patients who received allo-HCT, 2 died of TRM at 1.5 and 6 months, 1 had a relapse at 1.1 months, and 8 remained in remission at 4.6 to 13.3 months after HCT, resulted in 1-year LFS rate of 71.6% (95% CI, 44.2–99.0) (Fig. 4a) and a 1-year relapse rate of 9.1% (95% CI, 0–26.2) (Fig. 4b). One-year LFS rate of all 24 CR patients was 58.1% (95% CI, 35.2–81.0) (Fig. 4c). The detailed outcome of these 25 patients (both in POC and clinical trial) responded to CAR T-cell therapy are listed in Supplementary Table 3.

Long-term outcome of CR patients. a LFS among the patients who have received a subsequent transplantation after CD22 CAR T-cell therapy. b Relapse and TRM of patients who have received a subsequent transplantation after CD22 CAR T-cell therapy. c LFS among the patients who have achieved CR after CD22 CAR T-cell infusion. In A and C, dashed lines indicate 95% confidence intervals. In all panels, tick marks indicate the time of data censored at the last follow-up
Leukemic CD22 expression and normal B cells in relapsed patients
Relapse after CAR T-cell therapy might be due to the loss, mutation or preferential expression of alternatively spliced isoform of targeted antigen. In patients 11, 12, 14, and 31 we compared the leukemic cell surface CD22 protein levels between the time point before CAR T-cell infusion and that of relapse in patients 11, 13, 27, and 33, with standard FCM protocol which had also been used to detect antigen expression by other researchers [11, 31]. Indeed, CD22 was still expressed on most leukemic cells in patients 11, 27, and 33 who had a relapse at 2, 2.6, and 4 months, taking non B lineage cells as the control for negativity for CD22 expression in the same plot of each patient (Fig. 5a). However, since a relative, instead of absolute, quantification FCM method was used in our study, it is possible that the absolute CD22 expression level might have changed across different time points, which could not be detected by our method. Dramatic attenuation of CD22 expression by the blasts was observed in patient 13 who had a relapse at 6 months after infusion, but we still can induce this patient into CR again by a secondary infusion, suggesting that decreased CD22 expression did not necessarily prevent leukemic cells from elimination by CAR T-cells. Patient 13 finally relapsed. We also performed semi-quantitative RT-PCR to analyze the expression of CD22 mRNA splicing isoforms in the pre-treatment and relapsed leukemia cells from patient 27, and another relapsed leukemia sample from patient 11, whose matched pre-treatment sample was not available. In all the samples, the two longer CD22 transcript variants NM_001771 and NM_001185100, which share the same extracellular region encoding sequence, were dominantly expressed, and no significant variation of CD22 isoforms was detected between these samples, so as compared to normal B cells and leukemia cells from CR patients (Fig. 5b). Furthermore, Sanger sequencing of CD22 RT-PCR products in relapsed samples from patient 27 displayed no mutation of the CD22 coding sequence (Supplementary Fig. 6). To further identify possible genetic or transcriptomic mechanisms underlying relapse, leukemia samples before CAR T-cell infusion and after relapse were subjected to transcriptome sequencing, and no RNA-splicing variations, exon deletions and point mutations in the CD22 mRNA were detected; CD22 mRNA levels only slightly decreased after CAR T-cell treatment (Fig. 5c). Lineage switch, a reported mechanism of relapse with CD19 CAR therapy [14], did not occur in relapsed leukemic cells (Fig. 5c). Despite the number of relapsed cases was small in this study, these results suggest that a loss or mutation of CD22 antigen may not be the only mechanism of relapse.

CD22 expression in relapsed patients. a Change of CD22 protein level on tumor cells in patients before CAR T-cell infusion and after relapse, presented and compared by the relevant dot plots and flow cytometric histograms of CD22. A population of CD22 negative cells with similar size to the leukemia blasts was used as control for CD22 level analysis. b The CD22 splicing variants in leukemic cells of indicated patients. The total 5 splicing variants of CD22 recorded by NCBI GeneBank were listed. CD22 extracellular domain coding region was marked by gray color. Paired primers F1 and R1, F2, R2 were designed to detect various CD22 isoforms. The results were presented as DNA electrophoresis from semi-quantitative RT-PCR. NB, normal B cells from healthy donor; CR, B-ALL cells from a complete remission patient before CAR T-cell treatment; Pt27-Pre and Pt27-R indicate samples from patient 27, before CART-cell infusion and after relapse; Pt11-R indicates samples from Patient11 after relapse. c The transcriptome sequencing analysis demonstrated no altered splicing or expression of CD22 mRNA between leukemic cells from patients 27 before CART-cell infusion and after relapse. Expression of CD22 mRNA was presented as FPKM (fragments per kilobase of exon per million fragments mapped) value. Heat map shows the transcript levels of lineage marker genes before CAR T-cell infusion (pre) and after relapse (R). d Percentages of normal B cells and leukemic cells in CCR and relapse patients at the time points of day 0, day 30 and the follow up endpoint date during CAR T therapy
B cell aplasia can be used as a pharmacodynamic measure of CAR T-cell function.6Though normal B cells were absent in all patients with continued remission, normal B cells were present in all the relapsed patients evaluated (Fig. 5d and Supplementary Fig. 7), suggesting persistence of CD22 CAR T-cell surveillance is critical for preventing relapse.
Discussion
A considerable proportion of patients had relapsed after CD19 CAR T-cell therapy, and their prognosis was extremely poor due to the lack of additional therapeutic option. A recent work recently conducted by Fry et al., as the first in human trial toward 21 r/r B-ALL patients most of whom had failed from previous CD19 CAR T-cell therapy, has demonstrated the efficacy of CD22 CAR T-cells in inducing CR in a considerable proportion of such patients. However, its long-term term efficacy and applicable value of CD22 CAR T-cells still warrants further investigation. In our study involving 34 r/r B-ALL patients who have failed previous CD19 CAR T-cell therapy, we demonstrated that therapy with relatively low dosage of T-cells transduced with our CAR vector induce CR in 80% (24/30) patients that could be evaluated on day 30 (76% attained MRD-), and in 70.5% (24/34) of all enrolled patients. Regarding different disease categories at baseline, CR was achieved in 90% (19/21) evaluated patients with hematological relapse (86% attained MRD-), in 60% (3/5) patients with FCM-MRD+ status, and in 50% (2/4) patients with excluding EMDs. Our CD22 CAR T-cell therapy thus demonstrates high efficacy in the induction of remission for these CD19 CAR-refractory r/r B-ALL patients.
Low grades of CRS and neurotoxicity were observed in most patients, which are self-limiting and need no specific treatment, contrasting to the higher CRS in CD19 CAR T-cell treated patients [4,5,6,7,8]. Only 1 patient had suffered moderate neurotoxicity (grade 2), and recovered from interventions. Four patients died early after CAR T-cell infusion, but were most likely attributed to severe infection (2 pts), or the combined effect of CRS with other complications (DLI and stem cell engraftment). These results suggest that CD22 CAR T-cell related toxicity is generally low, but the complications with other medical conditions in these heavily-treated patients may increase the risk of severe adverse effect, which need to be taken into consideration in the future.
In the long-term follow up, 4 patients had withdrawn for personal willingness and maintained with chemotherapies, and 2 patients were lost of follow up after discharged. 4 of the 7 CR patients without further therapy had a relapse, suggesting that administration of only CD22 CAR T-cells was accompanied by a high relapse rate. However, among the 11 CR patients who were successfully bridged to subsequent allo-HCT, only 1 had a relapse, indicating that allo-HCT after CD22 CAR T-cell therapy could favor long-term leukemia-free survival. In consistent with this, our previous study also demonstrated allo-HCT promoted durable remission after CD19 CAR T-cell therapy [4].
Relapse after CAR T-cell therapy might be attributed to the loss or mutation of targeted antigen [11]. In our study, leukemic cells from 4 relapsed patients still have CD22 expression on most leukemic cells. Furthermore, alternative CD22 mRNA splicing or CD22 gene mutation is not detected in relapsed cases. These results suggest that loss or mutation of CD22 antigen might not essentially occurred in relapsed cases. Nevertheless, since only a very limited number of relapsed cases were available for evaluation in our study, we can not exclude the possibility of CD22 antigen loss or mutation in relapsed patients in our future study. Fry et al. have demonstrated that CD22 down-regulation in relapsed patients. In our study, we could not exclude the possibility that CD22 expression was in some degree down-regulated but could not be detected by our relatively quantification method across different time points. Indeed, one of our relapsed patients exhibited drastic down-regulation of CD22 expression. However, this patient could still be re-induced to CR by a secondary CD22 CAR T-cell infusion, implicating that down-regulation of CD22 may not essentially prevent leukemic cells from being killed.
The duration of CAR T-cell persistence has been indicated to be critical for preventing relapse of leukemia [6]. Although CAR T-cells were only detectable by FCM in our patients within 3 months after infusion, the patients in sustained remission had no normal B cells, which has been considered as a reliable measure of CAR T-cell function [6], suggesting that functional CAR T-cells were present in them for a relatively long period despite that the cell numbers are under FCM detection threshold. In contrast, normal B cells are detectable in all the 4 patients with a relapse, suggesting that loss of CAR T-cell surveillance is likely an underlying mechanism of relapse. Therefore, future effort to understand the mechanisms that affect the duration of CAR T-cell persistence and to further improve CAR T-cells persistence might be important to ensuring long-term CR after CAR T-cell infusion.
In conclusion, our study demonstrated that our CD22 CAR T-cells are capable of inducing a high remission rate in B-ALL patients who are refractory or relapsed after chemotherapy, transplantation, and even CD19 CAR T-cell therapy. Although relapse rate was high after CD22 CAR T-cell therapy without additional treatment, subsequent bridging to allo-HCT after CD22 CAR T-cell therapy can reduce the relapse rate. Our results also suggest that persistent CAR T-cell surveillance may be important for durable remission, and future optimization of CAR technique and clinical protocol may further improve therapeutic efficacy.
References
Sun W, Malvar J, Sposto R, Verma A, Wilkes JJ, Dennis R et al. Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia. 2018. https://doi.org/10.1038/s41375-018-0094-0.
Pulsipher MA, Langholz B, Wall DA, Schultz KR, Bunin N, Carroll W, et al. Risk factors and timing of relapse after allogeneic transplantation in pediatric ALL: for whom and when should interventions be tested? Bone Marrow Transpl. 2015;50:1173–9.
Sellar RS, Rowntree C, Vora AJ, Furness CL, Goulden N, Mitchell C, et al. Relapse in teenage and young adult patients treated on a paediatric minimal residual disease stratified ALL treatment protocol is associated with a poor outcome: results from UKALL2003. Br J Haemato. 2018;181:515–22.
Pan J, Yang JF, Deng BP, Zhao XJ, Zhang X, Lin YH, et al. High efficacy and safety of low dose CD19 directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia. 2017;31:2587–93.
Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Lee DW, Stetler-Stevenson M, Yuan CM, Fry TJ, Shah NN, Delbrook C, et al. Safety and response of incorporating CD19 chimeric antigen receptor T cell therapy in typical salvage regimens for children and young adults with acute lymphoblastic leukemia. Blood. 2015;126:684.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR-T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25.
Kenderian SS, Porter DL, Gill S. Chimeric antigen receptor T cells and hematopoietic cell transplantation: how not to put the CART before the horse. Biol Blood Marrow Transpl. 2016;23:235–46.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28.
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+and CD4+CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116.
Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Disco. 2015;5:1282–95.
Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017–23.
Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–10.
Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–38.
Jain MD, Davila ML. Concise review: emerging principles from the clinical application of chimeric antigen receptor T cell therapies for B cell malignancies. Stem Cells. 2018;36:36–44.
Cao J, Wang G, Cheng H, Wei C, Qi K, Sang W, et al. Potent anti-leukemia activities of humanized CD19-targeted Chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am J Hematol. 2018;93:851–8.
Uy N, Nadeau M, Stahl M, Zeidan AM. Inotuzumabozogamicin in the treatment of relapsed/refractory acute B cell lymphoblastic leukemia. J Blood Med. 2018;9:67–74.
Raponi S, De Propris MS, Intoppa S, Milani ML, Vitale A, Elia L, et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma. 2011;52:1098–107.
Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–74.
Shah NN, Stevenson MS, Yuan CM, Richards K, Delbrook C, Kreitman RJ, et al. Characterization of CD22 expression in acute lymphoblastic leukemia. Pedia Blood Cancer. 2015;62:964–9.
Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24:20–8.
Yu WJ, Mo XD, Zhang XH, Xu LP, Wang Y, Yan CH et al. Occurrence and severity of donor lymphocyte infusion-associated chronic graft-versus-host disease influence the clinical outcomes in relapsed acute leukemia after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2018. https://doi.org/10.1016/j.bbmt.2018.11.024.
Jaiswal SR, Bhakuni P, Joy A, Kaushal S, Chakrabarti A, Chakrabarti S. CTLA4Ig primed donor lymphocyte infusion: a novel approach to immunotherapy after haploidentical transplantation for advanced leukemia. Biol Blood Marrow Transpl. 2019. https://doi.org/10.1016/j.bbmt.2018.12.836.
Alvarnas JC, Brown PA, Advani A, Aoun P, Ballen KK, Barta SK et al. NCCN clinical practice guidelines in oncology: acute lymphoblastic leukemia, version 1.2016. Accessed date: 6 April 2016. https://www.nccn.org/professionals/physician_gls/pdf/all.pdf.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95.
US Department of Health and Human Services. Common terminology criteria for adverse events. V4.03. Accessable date: 14 June 2010. Publication: 2009. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE4.032010-06-14.
Long AH, Haso WM, Orentas RJ. Lessons learned from a highly-active CD22-specific chimeric antigen receptor. Oncoimmunology. 2013;2:e23621.
Xiao X, Ho M, Zhu Z, Pastan I, Dimitrov DS. Identification and characterization of fully human anti-CD22 monoclonal antibodies. MAbs. 2009;1:297–303.
Circosta P, Elia AR, Landra I, Machiorlatti R, Todaro M, Aliberti S, et al. Tailoring CD19xCD3-DART exposure enhances T-cells to eradication of B-cell neoplasms. Oncoimmunology. 2018;7:e1341032.
Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24:1504–6.
Acknowledgements
This work was supported by the National Key Basic Research Program of China (No. 2016YFC1303403, and No. 2015CB964400), National Natural Science Foundation of China (No. 81272325, and No. 81670107), National Natural Science Foundation of China Youth Project (No. 31501082), and the CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-1-003 and 2017-I2M-1-015). The Tianjin Science Funds for Distinguished Young Scholars. We thank Professor Chen Dong, Division of Hematopathology, Mayo Clinic for critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
AHC is also a founding member of Shanghai YaKe Biotechnology Ltd., a biotechnology company focused on research and development of tumor cellular immunotherapy. YZ and SL are also employees of Shanghai YaKe Biotechnoloy Ltd. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pan, J., Niu, Q., Deng, B. et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 33, 2854–2866 (2019). https://doi.org/10.1038/s41375-019-0488-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-019-0488-7
This article is cited by
-
Myeloid leukemia-derived galectin-1 downregulates CAR expression to hinder cytotoxicity of CAR T cells
Journal of Translational Medicine (2024)
-
Safety and efficacy of co-administration of CD19 and CD22 CAR-T cells in children with B-ALL relapse after CD19 CAR-T therapy
Journal of Translational Medicine (2023)
-
Complete spectrum of adverse events associated with chimeric antigen receptor (CAR)-T cell therapies
Journal of Biomedical Science (2023)
-
Targeting CD22 for B-cell hematologic malignancies
Experimental Hematology & Oncology (2023)
-
CD22 is a potential target of CAR-NK cell therapy for esophageal squamous cell carcinoma
Journal of Translational Medicine (2023)
