
Abstract
Peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS) is a diagnosis of exclusion, being the most common entity in mature T-cell neoplasms, and its molecular pathogenesis remains significantly understudied. Here, combining whole-exome and targeted-capture sequencing, gene-expression profiling, and immunohistochemical analysis of tumor samples from 133 cases, we have delineated the entire landscape of somatic alterations, and discovered frequently affected driver pathways in PTCL, NOS, with and without a T-follicular helper (TFH) cell phenotype. In addition to previously reported mutational targets, we identified a number of novel recurrently altered genes, such as KMT2C, SETD1B, YTHDF2, and PDCD1. We integrated these genetic drivers using hierarchical clustering and identified a previously undescribed molecular subtype characterized by TP53 and/or CDKN2A mutations and deletions in non-TFH PTCL, NOS. This subtype exhibited different prognosis and unique genetic features associated with extensive chromosomal instability, which preferentially affected molecules involved in immune escape and transcriptional regulation, such as HLA-A/B and IKZF2. Taken together, our findings provide novel insights into the molecular pathogenesis of PTCL, NOS by highlighting their genetic heterogeneity. These results should help to devise a novel molecular classification of PTCLs and to exploit a new therapeutic strategy for this group of aggressive malignancies.
Similar content being viewed by others


Introduction
PTCLs represent a clinically, histologically, and molecularly heterogeneous group of non-Hodgkin lymphomas (NHLs) derived from mature post-thymic T cells [1, 2]. Among them, the most common entity is PTCL, NOS, accounting for ~30% of all PTCLs [3]. Patients with PTCL, NOS generally demonstrate aggressive clinical course and are often refractory to standard therapy. By definition, PTCL, NOS includes cases that do not meet the criteria for any specific PTCL subtypes and has been considered a “wastebasket” category.
It has been recognized that a subset of PTCLs classified as PTCL, NOS has a T-follicular helper (TFH) cell phenotype (i.e., positive for CD4, PD-1, CD10, CXCL13, BCL6, and so on) and some pathological features of angioimmunoblastic T-cell lymphoma (AITL) [4,5,6]. In addition, recent genetic studies revealed that these cases share some of the recurrent genetic alterations found in AITL, such as mutations affecting TET2, DNMT3A, and RHOA [7,8,9,10,11]. Among these, the RHOA G17V mutation is highly specific to both PTCL subtypes and, when expressed in mouse T cells, induces TFH-cell specification and, together with TET2 loss, results in the development of AITL-like tumors [12]. On the basis of these findings, the revised World Health Organization (WHO) classification of hematological malignancies recommended that this subset of PTCL, NOS should be classified as PTCL with a TFH-cell phenotype as a provisional entity (referred to as “TFH PTCL, NOS”) [5]. However, the molecular pathogenesis of the remaining cases in the PTCL, NOS category is still poorly understood. The currently available genetic data from several small series reported different recurrent mutations and copy-number alterations (CNAs) [13,14,15,16], which preclude a solid conclusion as to the genomic landscape of the tumor. Systematic characterization of genetic alterations should significantly contribute to refining the molecular classification, improving prognostication, and identifying candidate therapeutic targets in this entity, as demonstrated in other lymphomas [6, 17].
Here, we conducted a comprehensive genetic analysis to determine the spectrum of mutations, CNAs, and structural variations (SVs) in PTCL, NOS with and without TFH-cell phenotype. In particular, our efforts focused on genetically dissecting the molecular pathogenesis and identifying a new molecular subgroup of PTCL, NOS, showing unique genetic and clinicopathological features.
Materials and methods
Patient samples
A total of 142 patients diagnosed with PTCL, NOS at six institutions were enrolled in this study according to the protocols approved by the Institutional Review Boards. This study was approved by the institutional ethics committees of the Graduate School of Medicine, Kyoto University and other participating institutes. All cases were reviewed, and a consensus diagnosis was made by expert hematopathologists according to the criteria of the 2008 WHO classification [18], of which 94 cases examined for tumor content. HTLV-1 infection was examined by anti-HTLV-1 antibody detection and/or southern blotting for HTLV-1 proviral DNA. HTLV-1-positive cases were considered as adult T-cell leukemia/lymphoma (ATL) and excluded from this study before analysis. Based on the recent revision of the WHO classification [5], TFH markers, including PD-1, CD10, CXCL13, and BCL6, were evaluated, and PTCL, NOS cases positive for at least two TFH markers were diagnosed as TFH PTCL, NOS. Because the minimum criteria for assignment of TFH phenotype is not well established, we considered PTCL, NOS cases expressing only one TFH marker as unclassifiable PTCL, NOS. Age, sex, and other clinical characteristics are summarized in Supplementary Table S1. Genomic DNA was extracted from fresh frozen tumor tissues or buccal swabs (as normal control) using the QIAamp DNA Mini kit (QIAGEN, Hilden, Germany) or commercially prepared by SRL Inc. (Tokyo, Japan). RNA was extracted from fresh frozen or formalin-fixed paraffin-embedded (FFPE) tumor tissues with RNeasy Mini Kit (QIAGEN).
Whole-exome sequencing (WES)
SureSelect Human All Exon v5 kits (Agilent Technologies, Santa Clara, CA, USA) were used for exome capture according to the manufacturer’s instructions. Sequencing data were generated using the Illumina HiSeq 2500 platform with a standard 125-bp paired-end read protocol, as previously described [19]. WES data for 3 PTCL, NOS, 3 AITL, and 81 ATL cases were described in our previous reports [10, 19]. Publicly available WES data for PTCL, NOS [accession number phs000689.v1.p1 [9]], AITL [phs000689.v1.p1 [9] and SRP029591 [20]], anaplastic large cell lymphoma [ALCL, SRP044708 [21]], and extranodal NK/T-cell lymphoma [ENKTL, SRP057085 [22]] were obtained from the National Center for Biotechnology Information Sequence Read Archive. Sequence alignment and mutation calling were performed using the Genomon pipeline (https://github.com/Genomon-Project), as previously described [19, 23], with minor modifications. Putative somatic mutations with (i) Fisher’s exact P-value <0.01; (ii) >4 variant reads in tumor; (iii) allele frequency in tumor >0.025; and (iv) sequencing depth in tumor ≥30 were adopted and filtered by excluding (i) synonymous single-nucleotide variants (SNVs); (ii) variants only present in unidirectional reads; and (iii) variants occurring in repetitive genomic regions. These candidate mutations were further filtered by removing known variants listed in the 1000 Genomes Project (October 2014 release), NCBI dbSNP build 131, National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) 6500, the Human Genome Variation Database (version 2.0), the Exome Aggregation Consortium (ExAC), or our in-house single-nucleotide polymorphism (SNP) database, unless they were listed in the COSMIC database (v70). Moreover, recurrently altered genes, including RHOA, TET2, IDH2, DNMT3A, and TP53, were manually reviewed for additional mutations. Finally, mapping errors were removed by visual inspection with Integrative Genomics Viewer (IGV).
Targeted-capture sequencing
Targeted-capture sequencing was performed using a custom SureSelect library (Agilent Technologies), for which 140 genes (Supplementary Table S2) reported to be recurrently mutated in PTCL, NOS, AITL, ATL, ALCL, ENKTL, cutaneous T-cell Lymphoma, and major subtypes of B-cell lymphomas (Supplementary Table S3; refs [5, 6, 9, 10, 17, 19,20,21,22, 24,25,26,27,28,29,30,31]). Additional probes for 1999 SNPs were included to calculate genomic copy numbers [32]. Mutation calling was performed with Empirical Bayesian Mutation Calling (EBCall) [33]. Candidate mutations were filtered in the same manner as for WES analysis, except for the inclusion of (i) P-value <10−4; (ii) >4 variant reads in tumor; and (iii) allele frequency in tumor >0.025, and (iv) the exclusion of missense SNVs with allele frequency of 0.35–0.65 in copy-neutral regions, unless they were listed in the COSMIC database (v70).
RNA sequencing (RNA-seq)
Libraries for RNA-seq were prepared from the total RNA extracted from fresh frozen tumor tissues using the NEBNext Ultra RNA Library Prep kit for Illumina (New England BioLabs, Beverly, MA, USA), and subjected to sequencing using the HiSeq 2500 instrument with a standard 125-bp paired-end read protocol. The sequencing reads were aligned to the human reference genome (hg19) using STAR (v2.5.3) [34]. The mapped reads per gene were counted with featureCounts (v1.5.3) from the R-package “Rsubread”, and normalized to counts per million (CPM) (R-package “edgeR”) [35]. To identify significantly enriched pathways in each group, Gene Set Enrichment Analysis (GSEA: v2.2.4) with the Molecular Signatures Database-curated gene sets (hallmark and C2: v6.1) was performed for genes expressed at >1 CPM in two or more samples.
nCounter gene-expression assay
Details of the nCounter assay (NanoString Technologies, Seattle, WA, USA) have been reported previously [36]. Briefly, the GATA3, TBX21, and 20 housekeeping gene probes (NanoString Technologies) were hybridized to 300 ng of the total RNA for 16 h at 65 °C, and applied to the nCounter Preparation Station for automated removal of excess probe and immobilization of probe–transcript complexes on a streptavidin-coated cartridge. The data were analyzed by using the nSolver 4.0 software (NanoString). To test the validity of nCounter analysis, a linear regression analysis was performed between normalized counts and CPM for GATA3 and TBX21 expressions, respectively.
SV and CNA analysis
SVs and CNAs were detected using the Genomon pipeline and the CNACS algorithm, respectively, as previously described [19, 32, 37]. Putative SVs were manually curated and further filtered by removing those (i) with Fisher’s exact P-value >0.01; (ii) with ≥6 supporting reads in tumor; (iii) with allele frequency in tumor <0.02; or (iv) present in any of control samples. SV breakpoints were visually inspected using IGV. Candidate focal CNAs (shorter than half a chromosome arm, except for 17 p deletions involving TP53) were assessed in genomic regions where sequencing coverage was sufficient in unmatched control samples, and then manually reviewed and further filtered by removing those with <3 probes. Frequency of focal CNAs were calculated for 49 genes (i) with recurrent mutations or SVs (found in ≥3 cases) in our cohort (47 genes) and/or (ii) with focal homozygous deletions or high-level (copy number ≥4) amplifications in at least two samples (2 genes: CDKN2A and ARID2). To confirm CNAs detected by the CNACS algorithm, we conducted SNP array karyotyping for 24 samples using the Affymetrix GeneChip Human Mapping 250 K NspI array (Affymetrix, Santa Clara, CA, USA), as previously described [19, 23]. Microarray data were analyzed to estimate the total and allele-specific copy numbers using CNAG/AsCNAR algorithms. Significantly recurrent arm-level CNAs were identified using a binomial distribution test, as previously described [38].
Mutation analysis
Pairwise correlations between alterations (present in ≥10 cases) were assessed by Fisher’s exact test with Benjamini–Hochberg correction (q < 0.1). Mutational signature was determined by pmsignature (version 0.2.1), as previously described [39]. The RHOA G17V mutations were separately analyzed, because they behaved differently from other RHOA mutations.
Hierarchical clustering
Unsupervised hierarchical clustering of recurrent somatic alterations, including 49 genes (affected by mutations and/or focal CNAs) and 14 arm-level CNAs, was performed with Spearman’s rank correlation and Ward’s linkage algorithm (R-package “heatmap.2”).
Detection of HTLV-1 genome
For the detection of HTLV-1 sequence, after sequencing reads were mapped to the HTLV-1 genome (AB513134), the number of the HTLV-1-aligned reads were enumerated and divided by the number of the total reads mapped to the human reference genome (GRCh37). Then, the obtained ratio was evaluated for the cutoff value of 0.01%, which was determined so that all confirmed ATL cases were included (data not shown).
Survival analysis
Survival data were available for 46 patients with PTCL, NOS. Observations were censored at the last follow-up. The median follow-up was 22.6 months in surviving patients, and 24 patients were alive at the last follow-up. The Kaplan–Meier method was used to estimate overall survival, and the log-rank test was used to assess differences in overall survival between patient groups (R-package “survival”).
Immunohistochemical analysis
Immunohistochemical analysis (IHC) for PD-1, CD10, CXCL13, BCL6, GATA3, and TBX21 was performed on FFPE tissue sections using antibodies directed against PD-1 (NAT105, Abcam, Cambridge, UK), CD10 (56C6, Leica Biosystems, Newcastle, UK), CXCL13 (polyclonal, R&D Systems, Minneapolis, MN, USA), BCL6 (EP529Y, Abcam; PG-B6P, Dako, Glostrup, Denmark; and LN22, Novocastra, Newcastle, UK), GATA3 (L50–823, Nichirei Bioscience, Tokyo, Japan), and TBX21 (4B10, Abcam). The antigen–antibody complexes were visualized with Histofine Simple Stain MAX PO (Nichirei Bioscience), Bond polymer Refine Detection kit (Leica Biosystems), or the REAL EnVision Detection system (Dako).
CRISPR-mediated gene targeting
Human IRF2BP2 sgRNA-targeted sites were designed manually and checked in silico. The pSpCas9(BB)-2A-GFP (pX458) vector expressing Cas9 (Addgene plasmid 48138) was digested with BbsI and ligated to annealed and phosphorylated sgRNA oligonucleotides. Jurkat cells, obtained from the RIKEN Cell Bank, were transfected with indicated vectors using the Amaxa Nucleofector system (Lonza, Bazel, Switzerland) according to the manufacturer’s instructions. CRISPR/Cas9-mediated targeting was confirmed by PCR-based deep-sequencing and expression analysis by real-time quantitative PCR, as previously described [37]. The sgRNA sequences and PCR primers are listed in Supplementary Table S4.
Real-time quantitative PCR
cDNA synthesis from the total RNA was performed with ReverTra Ace qPCR RT Kit (TOYOBO, Tokyo, Japan), and subjected to quantitative reverse transcription PCR with SYBR Premix Ex TaqII (Tli RNaseH Plus) (TaKaRa, Shiga, Japan) and LightCycler 480 System (Roche, Basel, Switzerland), according to the manufacturer’s instructions. All assays were performed in three technical replicates for each biological replicate, and relative expression was normalized for 18S rRNA.
Immunoblot analysis
Cells were lysed, subjected to SDS–PAGE, and transferred to a PVDF membrane (Millipore, Bedford, MA, USA). The blot was incubated with the antibodies listed in Supplementary Table S5, and visualized by Immobilon Western Chemiluminescent HRP Substrate (Millipore).
Luciferase assay
Jurkat cells were collected 48 h after transfection with pX458 and pGL4.30 (luc2P/NFAT-RE/Hygro, Promega, Madison, WI, USA) vectors and assayed for NFAT luciferase activity using the Dual-Luciferase Reporter Assay System (Promega) and Wallac ARVO SX 1420 Multilabel Counter (PerkinElmer, Waltham, MA, USA). Firefly luciferase activity was normalized by Renilla luciferase activity (phRL-TK vector, Promega) in each sample, and is presented with a logarithmic scale relative to the activity in mock-transfected cells.
Cell proliferation assay
Five thousand cells transduced with indicated vectors were inoculated into 96-well culture plates, and their growth was monitored using Cell Counting Kit-8 (DOJINDO LABORATORIES, Kumamoto, Japan), according to the manufacturer’s protocol.
Statistical analysis
Statistical analyses were performed with R 3.1.3 software (The R Foundation for Statistical Computing). Comparisons between groups were based on the Wilcoxon rank-sum test for continuous data with Bonferroni correction (if necessary), and the Fisher’s exact test with Benjamini–Hochberg correction (if necessary) for categorical data. For functional assays, normality of data distribution and homogeneity of variance were assessed by the Shapiro–Wilk’s test and F-test, respectively. Student’s two-tailed t test was used to compare two groups, and a Welch’s correction was applied when comparing groups with unequal variance (F-test, P < 0.05). In box plots, the center line and lower and upper hinges correspond to the median, and the first and third quartiles (25 and 75 percentiles), respectively. The upper and lower whiskers extend from the upper and lower hinges to the largest or smallest values, no further than 1.5× interquartile range from the hinges.
Results
WES of patients with PTCL, NOS
To delineate the entire picture of genetic alterations in PTCL, NOS, we initially performed WES analysis of tumor and normal samples from 20 PTCL, NOS patients (Supplementary Fig. 1A, B), including seven TFH, three unclassifiable, and four non-TFH PTCL, NOS cases from our cohort. In total, we detected 1068 somatic mutations (1.5 mutations/Mb/sample), including 971 SNVs and 97 insertions and deletions (indels), as well as 42 SVs (Fig. 1a; Supplementary Tables S6–S8). These mutations mainly consisted of age-related C > T transitions at CpG sites, followed by C > A substitutions at the CpCpT context, whose etiology has been unknown (Fig. 1b). Approximately a half of patients exhibited a low overall mutation frequency (<0.5 mutations/Mb), while there were four samples showing a moderate-to-high mutation rate (2–10 mutations/Mb) (Fig. 1a). Additional targeted sequencing not only validated the somatic mutations detected by WES sequencing but also captured multiple previously reported mutations with low allele frequencies, such as those involving TET2 and RHOA (colored in red in Fig. 1c) [7,8,9,10,11], suggesting a possibility that some driver mutations overlooked with WES analysis. In addition to TFH-related mutations, the observed alterations included recurrent mutations and deletions of TP53 (n = 7), a well-known tumor suppressor gene [40], which were associated with a higher tumor mutation burden (Fig. 1d). Moreover, a number of mutations frequently observed in other subtypes of lymphomas, such as those in CDKN2A, VAV1, and TBL1XR1 [5, 17, 19, 41], were also detected (Fig. 1a, c; Supplementary Fig. S1C; Supplementary Table S6). These results suggest a potential role of lymphoma-associated mutations, particularly those affecting TP53, as driver alterations in the molecular pathogenesis of PTCL, NOS.

WES analysis for 20 PTCL, NOS cases. a The number of coding mutations (top), frequency of mutational signature (middle), and a heatmap showing the distribution of mutations in TET2, RHOA, IDH2, TP53, and CDKN2A alterations, TFH markers, sample source, and molecular subtype are depicted. All panels are aligned, with the vertical tracks representing 20 PTCL, NOS cases. b Two mutational signatures identified by pmsignature algorithm in PTCL, NOS. Signature 1 was predominated by age-related C > T transitions at CpG dinucleotides, while signature 2 (of unknown etiology) consisted of C > A substitutions at CpCpT context. c Hierarchy of somatic mutations is shown with their allele frequencies in four representative PTCL, NOS cases. Lymphoma-associated alterations are shown in green, and mutations that were undetectable by WES but were identified later by targeted-capture sequencing are shown in red. Mutations located in non-amplified regions and TP53/CDKN2A deletions are depicted. d Box plots showing the number of somatic mutations identified by WES in cases with or without TP53 alterations. **P < 0.005, Wilcoxon rank-sum test
Overview of PTCL, NOS genomes revealed by deep-targeted-capture sequencing
On the basis of these results, we then carried out deep-targeted-capture sequencing that covered 140 lymphoma-associated genes (Supplementary Table S2) in a cohort of 142 patients with PTCL, NOS (including 11 WES cases), with a mean depth of 627× (range, 399–830×) (Supplementary Fig. S2A, B). Unexpectedly, 18 cases had a substantial number of sequencing reads mapped to the HTLV-1 proviral genome (Supplementary Fig. S2C). After excluding these cases, who were considered to have ATL, we analyzed the remaining 124 cases and identified 438 non-silent somatic mutations (333 SNVs and 105 indels), with a median of three per sample (range 0–13) (Supplementary Table S9). These included numerous driver mutations which did not appear to be readily identifiable by WES due to low allele frequency (Supplementary Fig. S2D), suggesting that deep-targeted sequencing would be required to delineate the entire landscape of driver alterations in PTCL, NOS.
When the results from targeted-capture sequencing and WES were combined, a total of 41 genes were found to be recurrently mutated (in ≥3 cases), of which 12 (such as TET2, TP53, RHOA, and DNMT3A) were affected in more than 5% of a total of 133 cases (Supplementary Fig. S2E). Copy-number analysis based on the sequencing method identified 222 focal CNAs in 41 recurrently altered genes (6 amplified and 35 deleted genes) (Supplementary Fig. S2F; Supplementary Table S10). Among them, ten (such as TP53, CDKN2A, CD28, HLA-B, and IKZF2) were affected in more than 5% of the cases, some of which showed high-level amplifications or homozygous deletions (Supplementary Figs. S2F, S3A, B). In addition, 251 arm-level CNAs were detected in 14 significantly altered chromosome arms (seven gains and seven losses) (Supplementary Fig. S3C). SNP array karyotyping was also performed for 24 samples, in which 25 out of 27 focal CNAs (93%) and 136 of 141 arm-level CNAs (96%) were confirmed (Supplementary Fig. S3D; Supplementary Table S10).
We also identified 32 SVs (21 deletions, 5 inversions, 5 tandem duplications, and 1 translocation) in recurrently affected genes, including IKZF2 and CD274 (Supplementary Fig. S2F; Supplementary Table S11). Overall, 107 (80%) and 69 (52%) of 133 PTCL, NOS patients carried at least one driver mutation and CNA/SV, respectively, which belong to a wide spectrum of T-cell-related biological processes (Figs. 2, 3). When evaluated together, 116 (87%) patients harbored at least one somatic alteration, and 49 genes, including 25 previously unreported genes (HLA-A/B, KMT2C, NOTCH1, ARID1A, and so on), were recurrently affected (in ≥3 cases), including 10 genes affected in more than 10% of the cases (Fig. 2). Among these 133 cases, three or more TFH makers were evaluated by IHC in 98 cases, of which 37, 25, and 36 were considered to have TFH, unclassifiable, and non-TFH PTCL, NOS, respectively (Supplementary Table S1).

Landscape of somatic alterations in PTCL, NOS. a Frequency and type of somatic alterations in 49 recurrently altered genes (found in ≥3 cases) for 133 PTCL, NOS cases, including 127 cases from our series and 6 cases from a previous study. Genes not previously reported as altered in PTCL, NOS are shown in red. b Co-mutation plot showing the spectrum of somatic alterations in recurrently altered genes (n = 49) and chromosome arms (n = 14) across 133 PTCL, NOS cases. Samples were organized by hierarchical clustering with Spearman’s rank correlation and Ward’s linkage algorithm. Molecular subtype, experimental platform, IHC (TFH markers, GATA3, and TBX21) as well as related functional pathways (right) are also shown. Other RHOA mutations (three and two cases in group 2 and 3, respectively) are shown in a different color from G17V mutation

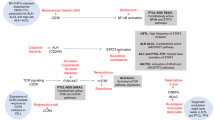
Commonly affected functional pathways in PTCL, NOS. Driver alterations, including mutations, CNAs, and SVs, are summarized according to their functionalities. Frequencies of mutations (left) and CNAs/SVs (right) are expressed as the percentage of altered cases in 133 PTCL, NOS cases. Major determinants of the molecular classification are highlighted by green (group 1) or red (group 2) boxes. Gain-of-function mutations and activating CNAs/SVs shown in red, and loss-of-function mutations and disrupting CNAs/SVs are shown in blue
RHOA G17V and IDH2 R172 mutations are highly specific for TFH PTCL, NOS
In accordance with previous reports [7,8,9,10,11], TET2 (44%), RHOA (26%), and DNMT3A (12%) were frequently altered in this cohort (Figs. 2, 3; Supplementary Fig. S4A). Although IDH2 mutations had not previously been reported in PTCL, NOS, including that with TFH phenotype [42, 43], 10 (8%) cases harbored IDH2 mutations, mostly consisting of R172 substitutions (Fig. 2; Supplementary Fig. S4A). Immunohistochemical evaluation revealed significant associations of TET2 and RHOA mutations with the expression of TFH markers, such as PD-1, CD10, CXCL13, and BCL6 (Fig. 4a). Although DNMT3A and IDH2 mutations tended to occur more frequently in TFH PTCL, NOS, many types of alterations, such as CD28 mutations and amplifications, were present irrespective of TFH marker status, suggesting partially overlapping genetic mechanisms involved in TFH and non-TFH PTCL, NOS (Fig. 4a; Supplementary Fig. S4B). Interestingly, at least one TFH marker was positive in all cases with RHOA G17V mutations, which were almost invariably accompanied by TET2 mutations with higher variant allele frequencies (Figs. 2b, 4a; Supplementary Fig. S4C, D). By contrast, none of five cases with other RHOA mutations were TFH PTCL, NOS, suggesting that the G17V substitution is pathognomonic for TFH-related PTCL. As expected, unclassifiable PTCL, NOS cases exhibited a genetic feature intermediate between TFH and non-TFH PTCL, NOS, suggesting this entity consisted of a mixed population of TFH and non-TFH PTCL, NOS cases (Supplementary Fig. S4B, C).

Co-occurring and mutually exclusive associations define two molecular subtypes in PTCL, NOS. a Comparison of frequencies of recurrent somatic alterations between patients with 37 TFH and 36 non-TFH PTCL, NOS. Recurrently altered genes (n = 19) present in ≥10 cases (7%) in the entire cohort are shown. *q < 0.1, **q < 0.01, Fisher’s exact test with Benjamini–Hochberg correction. b Pairwise associations among 19 recurrently altered genes found in ≥10 cases (7%) in the entire cohort. Only significant correlations (OR > 10 and q < 0.1, Fisher’s exact test with Benjamini–Hochberg correction) are shown with their odds ratios (OR). Orange colors depict gene pairs that are co-mutated more than expected by chance, and blue colors depict mutually exclusive gene pairs. c Two molecular subtypes [group 1 (TFH-related) and group 2 (TP53/CDKN2A)] and their major determinants in PTCL, NOS. Orange and blue lines represent co-occurring and mutually exclusive associations, respectively. The line width is proportional to the statistical significance (q value) of the association. Genes showing at least two significant associations in (b) are shown
A distinct molecular subtype characterized by TP53 and CDKN2A alterations in non-TFH PTCL, NOS
Although rarely reported in the previous literatures [13, 14, 44, 45], TP53 mutations and deletions were found in as many as 37 cases (28%) with PTCL, NOS, where the majority (51%) had a biallelic lesion (Figs. 2, 3; Supplementary Fig. S4A, E). CDKN2A, another tumor suppressor, was focally deleted in 17 cases (13%), of which 11 had homozygous deletions (Figs. 2, 3; Supplementary Fig. S3A). Remarkably, TP53 and CDKN2A represented two leading targets of genetic alterations in non-TFH PTCL, NOS, and their alterations negatively correlated with TFH marker expression (Fig. 4a). Prompted by the inverse correlation between TP53 and CDKN2A and TFH-related alterations, we investigated co-occurrence and exclusion between somatic alterations (Fig. 4b). TFH-related abnormalities, including TET2 alterations and RHOA G17V and IDH2 mutations, showed a strong tendency to co-occur, whereas the RHOA G17V mutations were mutually exclusive with TP53 and CDKN2A alterations (Fig. 4b). Moreover, the latter two alterations significantly co-occurred with somatic aberrations, involving the HLA-A, HLA-B, CD58, and IKZF2 genes (Fig. 4b). Taken together, these observations clearly depicted two molecular subtypes in PTCL, NOS: subtypes characterized by TFH-related alterations and TP53 and CDKN2A alterations, respectively (Fig. 4c).
TP53/CDKN2A-altered PTCL, NOS shows marked chromosome instability
In consistent with these findings, hierarchical clustering of recurrent somatic alterations revealed three molecular subtypes with discrete genetic features: those with TFH-related alterations (TET2, RHOA G17V, and IDH2) (group 1), those with TP53/CDKN2A alterations (group 2) and, those lacking any of the above alterations (group 3) (Fig. 2). While group 1 shows similar immunophenotype and genetic alterations to TFH PTCL, NOS [5], group 2 is supposed to represent a novel molecular subtype in PTCL, NOS (Fig. 5a). As revealed by genome-wide copy-number profiling, almost all group 2 cases exhibited extensive chromosomal abnormalities, which were rarely seen in other groups (Fig. 5b), pointing to a discrete genetic feature of group 2 tumors. This difference was quantitatively substantiated by a higher number of abnormal genomic segments in TP53- or CDKN2A-altered cases than those harboring TFH-related or other alterations (Fig. 5c; Supplementary Fig. S5A, B). Although group 3 showed a lower number of genetic alterations, ATM mutations and deletions were detected in a subset of group 3, some of which had extensive CNAs, similar to group 2 cases (Figs. 2b, 5b; Supplementary Fig. S5A, B). Given that ATM regulates the ARF-TP53 tumor suppressor pathway in response to DNA damage [46] (Fig. 3), ATM-altered tumors may exploit a shared oncogenic mechanism with group 2, which is characterized by tumor suppressor inactivation.

TP53/CDKN2A-altered PTCL, NOS shows a distinct genetic features characterized by chromosome instability and cell cycle dysregulation. a Comparison of frequencies of TFH marker positivity among molecular subtypes. Fisher’s exact test. b The heatmap shows somatic CNA segments (copy-number gains/amplifications, losses/deletions, and uniparental disomies (UPD) with ≥10 probes) in each sample (horizontal axis) plotted by chromosomal location (vertical axis). Samples are vertically aligned in the same order as in Fig. 2b. c The number of abnormal chromosomal segments identified in cases with indicated alterations, regardless of presence or absence of other alterations. Each dot represents a single case. **P < 0.005, ***P < 0.0005, Wilcoxon rank-sum test with Bonferroni correction. d Significantly enriched gene signatures for group 1 (left) and 2 (right) in GSEA analysis with RNA-seq data from 16 cases (four, six, and six cases in group 1, 2, and 3, respectively) using the hallmark (top) and C2 (bottom) gene sets. e Dot plots of normalized counts of GATA3 (left) and TBX21 (right) expressions measured by nCounter analysis in each group (10, 13, and 11 cases in group 1, 2, and 3, respectively). Wilcoxon rank-sum test
GSEA analysis with RNA-seq data from 16 fresh frozen tumor tissues using curated gene sets showed that AITL-related genes were the second most enriched signature in group 1, confirming the validity and reliability of expression analysis (Fig. 5d; Supplementary Table S12). Among hallmark gene sets, genes associated with stromal response and inflammation were enriched in group 1, whereas cell cycle-related genes were overrepresented in group 2 (Fig. 5d; Supplementary Table S12), suggesting that differences of genetic features among molecular subtypes are reflected in gene-expression profiles. Although it has been reported that PTCL, NOS can be classified into two subgroups by GATA3 and TBX21 expressions [47], these expressions measured by RNA-seq, nCounter analysis, or IHC were similar among subtypes (Fig. 5e; Supplementary Fig. S5C–F).
Histologically, group 2 showed a higher tumor content than group 1 (Fig. 6a), which is consistent with gene-expression profiling data showing strong immune or stromal cell-related signatures in group 1. With regard to clinical outcome, group 2 showed the worst survival, followed by group 1, whereas group 3 had an excellent outcome (Fig. 6b), suggesting that TP53/CDKN2A alterations and associated chromosomal instability confer an adverse prognostic impact. When compared with other PTCL subtypes, group 1, corresponding to TFH PTCL, NOS, showed a similar pattern of mutations and CNAs to AITL (Fig. 6c; Supplementary Fig. S6; Supplementary Tables S13–S21), consistent with previous reports [9,10,11]. By contrast, group 2 showed a unique profile of somatic alterations, although it shared a number of aberrations with other PTCL subtypes (Fig. 6c). These findings indicate that TP53/CDKN2A-altered cases have a molecular pathogenesis distinct from other PTCL subtypes, which may underlie their different clinical behavior.

Clinical and genetic differences among three molecular subtypes of PTCL, NOS. a Comparison of tumor cell fraction among three molecular subtypes of PTCL, NOS. Fisher’s exact test. b Kaplan–Meier survival curves of overall survival of 46 PTCL, NOS cases stratified by molecular subtype. The prognostic impact on survival was evaluated by log-rank test. c Comparison of frequencies of somatic alterations among the entire cohort (n = 133) and each molecular subtype (n = 50, 42, and 41 for group 1, 2, and 3, respectively) of PTCL, NOS, AITL (n = 26), ATL (n = 81) [19], ALCL (n = 23) [21], and ENKTL (n = 25) [22]. Diagonal lines represent no data available. d Proportion of the number of somatic alterations belonging to each functional pathway among three molecular subtypes of PTCL, NOS. **q < 0.01, ***q < 0.001, Fisher’s exact test with Benjamini–Hochberg correction
Frequent genetic alterations associated with immune evasion in TP53/CDKN2A-altered PTCL, NOS
Recurrent alterations in different PTCL, NOS subtypes affected a number of discrete functional pathways. Among these, uniquely overrepresented in group 2 was the pathways involved in immune surveillance (Fig. 6d; Supplementary Fig. S7), which include the components of the class I major histocompatibility complex (MHC) (HLA-A and HLA-B), the transactivator of MHC class II (CIITA), immune checkpoints (CD274), and molecules engaged in cell adhesion (CD58) and death signaling (FAS) (Figs. 2, 3). In group 2, most of these genes were affected by loss-of-function alterations, particularly by focal deletions (Fig. 7a; Supplementary Fig. S8A, B), suggesting a possible link to genomic instability characteristic of this subgroup. Intriguingly, we identified recurrent loss-of-function mutations involving the PDCD1 gene (3% in the entire cohort), the gene encoding an inhibitory receptor, PD-1 (Fig. 7a). In addition to focal deletions found in ATL and other T-cell lymphomas, which were recently reported to induce T-cell malignancies in mice [48], frameshift and nonsense mutations of PDCD1 were observed in PTCL, NOS, suggesting that loss of PD-1 function takes place through multiple mechanisms.

Genetic alterations in molecules associated with immune evasion and transcriptional regulation. a Positions and types of somatic mutations encoded in HLA-A (NM_002116), PDCD1 (NM_005018), IRF2BP2 (NM_001077397), and YTHDF2 (NM_001173128) detected in 133 PTCL, NOS cases. IRF2BP2 mutations observed in 81 ATL cases are also shown. b Validation of CRISPR/Cas9-mediated targeting of IRF2BP2 gene by amplicon sequencing. Representative sequencing data for mock- (left) and sgIRF2BP2-1- (right) transfected samples were visualized with IGV. c Relative expression of IRF2BP2 mRNA in Jurkat cells transfected with the indicated sgRNA vectors (n = 4). Expression values were normalized to the mock-transfected control. The data represent means ± s.d., ***P < 0.0005, Student’s t test with Welch’s correction. d Immunoblot analysis of IRF2BP2 in Jurkat cells transfected with the indicated sgRNA vectors. The representative result of two independent experiments. e Luciferase assays of NFAT transcriptional activity in Jurkat cells transfected with the indicated sgRNA vectors with or without NFAT1 expression vector, in the presence or absence of CD3/CD28 stimulation (n = 3–5 biological replicates, respectively). RLU, relative luminometer units. The data represent means ± s.d. *P < 0.05, **P < 0.005, Student’s t test with Welch’s correction
Enrichment of somatic lesions in transcriptional and post-transcriptional regulators in TP53/CDKN2A-altered PTCL, NOS
Another significant finding was the enrichment of somatic alterations in transcriptional and post-transcriptional regulators in group 2 (Fig. 6d). These regulators included transcription factors (IKZF2, PRDM1, and ETV6), transcriptional co-repressors (TBL1XR1 and IRF2BP2), and RNA-binding proteins (DDX3X and YTHDF2) (Figs. 2, 3). IKZF2, also known as HELIOS, is one of the major regulators of T-cell development and affected in 29% of group 2 tumors exclusively through SV/CNAs, such as intragenic deletions, duplications, and inversions, most likely leading to dominant-negative spliced variants [49] (Supplementary Figs. S7, S8C).
IRF2BP2, which encodes an IRF2-dependent transcriptional co-repressor [50, 51], was another common genetic target in group 2 tumors (Supplementary Fig. S7) and also affected in other lymphoma types [19, 52], in which frequent nonsense or frameshift mutations are thought to lead to loss of function of IRF2BP2 (Fig. 7a). To assess the functional consequence of IRF2BP2 mutations on T-cell lymphomagenesis, we evaluated the effect of IRF2BP2 disruption on cellular growth and the transcriptional activity of NFAT, a major downstream target of T-cell receptor (TCR) signaling, using CRISPR/Cas9-mediated gene editing (Fig. 7b–d). Although IRF2BP2 disruption did not affect cell proliferation, it caused an enhanced transcription from an NFAT response element in a human T-cell line (Jurkat), regardless of co-transfection with NFAT1 or CD3/CD28 stimulation, suggesting that loss-of-function alterations of IRF2BP2 lead to TCR signaling activation (Fig. 7e; Supplementary Fig. S8D). By contrast, recurrent deteriorating deletions and mutations in YTHDF2 were detected in both groups 1 and 2 (8% of the entire cohort) (Fig. 7a; Supplementary Fig. S7). This gene encodes a reader protein that recognizes N6-methyladenosine, the most abundant internal modification in mammalian mRNA, and reduces the stability of target transcripts [53], which may suggest the functional importance of deregulated mRNA stability in the pathogenesis of T-cell lymphoma.
Other commonly affected pathways and molecules in TP53/CDKN2A-altered PTCL, NOS
Signal transduction molecules were also common mutational targets in group 2 tumors, including NOTCH1 and SOCS1 (Fig. 6d; Supplementary Fig. S7). Although activating mutations in genes related to TCR signaling are reported in TFH-cell-derived lymphomas [11], in our cohort, more than two-thirds of both groups 1 and 2 cases harbored somatic changes in the components of TCR–NF-κB signaling and their downstream pathways (Fig. 6d). However, the spectrum of target genes substantially differed between group 1 and 2 tumors. In group 1, RHOA mutations represented by far the most predominant alterations. By contrast, the alterations in group 2 involved a broader spectrum of genes than those in group 1, such as CD28, PLCG1, CARD11, TNFAIP3, and PTPRC, which were frequently affected by focal CNAs, including high-level amplifications or homozygous deletions, rather than missense mutations (Supplementary Figs. S3B, S7).
In addition to TFH-related mutations, such as those affecting TET2, IDH2, and DNMT3A, recurrent mutations and CNAs/SVs were also present in a variety of epigenetic regulators, including histone modifiers (KMT2C (MLL3), KMT2D (MLL2), SETD1B, SETD2, and CREBBP) and SWI/SNF-mediated chromatin remodelers (ARID1A and ARID2), in our entire cohort (Figs. 2, 3). Among these, two histone 3 lysine 4 methyltransferases, KMT2C and SETD1B, were frequently inactivated by loss-of-function mutations or focal deletions in group 2 (Supplementary Figs. S7, S8E). The remaining group of molecules affected in PTCL, NOS were G protein-coupled receptors involved in T-cell trafficking, such as CCR4 and CCR7, which are also commonly mutated in other T-cell neoplasms (Figs. 2, 3).
Discussion
Through extensive genetic analyses using high-throughput sequencing, we have delineated a comprehensive registry of genetic alterations in PTCL, NOS. It includes not only known mutational targets in PTCL, NOS and other lymphoma subtypes but also novel recurrently altered genes previously unreported in this tumor type, such as KMT2C, SETD1B, YTHDF2, and PDCD1. As expected from a highly variable clinical presentation and prognosis, as well as pathological findings, PTCL, NOS is shown to be a heterogeneous entity in terms of genetic profile [3]. However, it should be underscored that PTCL, NOS does not represent a mere waste basket category, but comprises several discrete subtypes of mature T-cell neoplasms on the basis of unique genetic profiles.
Group 1 tumors, characterized by TFH-related mutations, such as TET2, RHOA G17V, and IDH2 mutations, correspond to a provisional entity of TFH PTCL, NOS, according to the revised WHO classification [5]. These tumors also exhibit a variety of somatic alterations at low frequencies, such as VAV1, CD28, and YTHDF2, most of which are shared by other PTCL, NOS subtypes, suggesting overlapping mechanisms of lymphomagenesis. Group 2 tumors are a previously unrecognized molecular subtype, which harbors frequent TP53 and/or CDKN2A alterations. This subtype shows the unique genetic features characterized by an increased burden of CNAs, which preferentially target molecules involved in immune surveillance and transcriptional regulation, including HLA-A/B and IKZF2. The high prevalence of TP53/CDKN2A alterations demonstrates the biological relevance of tumor suppressor inactivation and resultant genomic instability during T-cell lymphomagenesis, which is supported by the fact that T-cell lymphoma is one of the most common malignancies observed in p53-deficeint mice [40]. Except for ATM alterations in their subset, group 3 tumors lack a subtype-defining alteration, suggesting the necessity for further molecular investigation in this subtype.
Many efforts have been undertaken to further molecularly characterize and subdivide the heterogeneous group of tumors classified as this category. Microarray analysis of gene expression identified a biologically distinct entity showing a proliferation signature associated with a shorter survival [54]. More recently, large-scale gene-expression profiling enabled the characterization of two different molecular subgroups related to high expression of either TBX21 or GATA3 [47]. However, the molecular categorization of PTCL, NOS still remains controversial due to the inadequate understanding of the genetic landscape of the tumor. Therefore, the identification of the TP53/CDKN2A-altered molecular subtype with different genetic features, and prognosis can offer a clue to understand the genetic heterogeneity of PTCL, NOS and provide novel insights into its molecular classification and patient stratification, hopefully leading to the improvement of diagnostic and therapeutic strategy for this deadly disease.
References
Vose J, Armitage J, Weisenburger D. International TCLP. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol: Off J Am Soc Clin Oncol. 2008;26:4124–30.
Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117:6756–67.
Broccoli A, Zinzani PL. Peripheral T-cell lymphoma, not otherwise specified. Blood. 2017;129:1103–12.
de Leval L, Rickman DS, Thielen C, Reynies A, Huang YL, Delsol G, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109:4952–63.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. France: IARC Press; 2017.
Moffitt AB, Dave SS. Clinical applications of the genomic landscape of aggressive non-Hodgkin lymphoma. J Clin Oncol: Off J Am Soc Clin Oncol. 2017;35:955–62.
Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. New Engl J Med. 2012;366:95–6.
Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120:1466–9.
Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166–70.
Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171–5.
Vallois D, Dobay MP, Morin RD, Lemonnier F, Missiaglia E, Juilland M, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood. 2016;128:1490–502.
Cortes JR, Ambesi-Impiombato A, Couronne L, Quinn SA, Kim CS, da Silva Almeida AC, et al. RHOA G17V induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell. 2018;33:259–73 e257.
Schatz JH, Horwitz SM, Teruya-Feldstein J, Lunning MA, Viale A, Huberman K, et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia. 2015;29:237–41.
Simpson HM, Khan RZ, Song C, Sharma D, Sadashivaiah K, Furusawa A, et al. Concurrent mutations in ATM and genes associated with common gamma chain signaling in peripheral T cell lymphoma. PLoS ONE. 2015;10:e0141906.
Fujiwara SI, Yamashita Y, Nakamura N, Choi YL, Ueno T, Watanabe H, et al. High-resolution analysis of chromosome copy number alterations in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified, with single nucleotide polymorphism-typing microarrays. Leukemia. 2008;22:1891–8.
Hartmann S, Gesk S, Scholtysik R, Kreuz M, Bug S, Vater I, et al. High resolution SNP array genomic profiling of peripheral T cell lymphomas, not otherwise specified, identifies a subgroup with chromosomal aberrations affecting the REL locus. Br J Haematol. 2010;148:402–12.
Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171:481–94 e415.
Swerdllow S, Campo E, Harris NL. WHO classification of tumours of haematopoietic and lymphoid tissues. France: IARC Press; 2008.
Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47:1304–15.
Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:371–5.
Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27:516–32.
Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47:1061–6.
Kataoka K, Iwanaga M, Yasunaga JI, Nagata Y, Kitanaka A, Kameda T, et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood. 2018;131:215–25.
Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293–6.
da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat Genet. 2015;47:1465–70.
Kiel MJ, Sahasrabuddhe AA, Rolland DC, Velusamy T, Chung F, Schaller M, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat Commun. 2015;6:8470.
Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47:1011–9.
de Miranda NF, Georgiou K, Chen L, Wu C, Gao Z, Zaravinos A, et al. Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood. 2014;124:2544–53.
Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–81.
Okosun J, Wolfson RL, Wang J, Araf S, Wilkins L, Castellano BM, et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat Genet. 2016;48:183–8.
Mansouri L, Noerenberg D, Young E, Mylonas E, Abdulla M, Frick M, et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood. 2016;128:2666–70.
Yoshizato T, Nannya Y, Atsuta Y, Shiozawa Y, Iijima-Yamashita Y, Yoshida K, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129:2347–58.
Shiraishi Y, Sato Y, Chiba K, Okuno Y, Nagata Y, Yoshida K, et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41:e89.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30.
Kulkarni MM. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr Protoc Mol Biol 2011; Chapter 25: Unit25B 10.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016;534:402–6.
Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15.
Shiraishi Y, Tremmel G, Miyano S, Stephens M. A simple model-based approach to inferring and visualizing cancer mutation signatures. PLoS Genet. 2015;11:e1005657.
Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–70.
Abate F, da Silva-Almeida AC, Zairis S, Robles-Valero J, Couronne L, Khiabanian H, et al. Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T-cell lymphomas. Proc Natl Acad Sci USA. 2017;114:764–9.
Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood. 2015;126:1741–52.
Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–3.
Vasmatzis G, Johnson SH, Knudson RA, Ketterling RP, Braggio E, Fonseca R, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012;120:2280–9.
Matsushima AY, Cesarman E, Chadburn A, Knowles DM. Post-thymic T cell lymphomas frequently overexpress p53 protein but infrequently exhibit p53 gene mutations. Am J Pathol. 1994;144:573–84.
Sherr CJ. Divorcing ARF andp53: an unsettled case. Nat Rev Cancer. 2006;6:663–73.
Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123:2915–23.
Wartewig T, Kurgyis Z, Keppler S, Pechloff K, Hameister E, Ollinger R, et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature. 2017;552:121–5.
Asanuma S, Yamagishi M, Kawanami K, Nakano K, Sato-Otsubo A, Muto S, et al. Adult T-cell leukemia cells are characterized by abnormalities of Helios expression that promote T cell growth. Cancer Sci. 2013;104:1097–106.
Childs KS, Goodbourn S. Identification of novel co-repressor molecules for interferon regulatory factor-2. Nucleic Acids Res. 2003;31:3016–26.
Carneiro FR, Ramalho-Oliveira R, Mognol GP, Viola JP. Interferon regulatory factor 2 binding protein 2 is a new NFAT1 partner and represses its transcriptional activity. Mol Cell Biol. 2011;31:2889–901.
Bruno A, Boisselier B, Labreche K, Marie Y, Polivka M, Jouvet A, et al. Mutational analysis of primary central nervous system lymphoma. Oncotarget. 2014;5:5065–75.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Cuadros M, Dave SS, Jaffe ES, Honrado E, Milne R, Alves J, et al. Identification of a proliferation signature related to survival in nodal peripheral T-cell lymphomas. J Clin Oncol: Off J Am Soc Clin Oncol. 2007;25:3321–9.
Acknowledgements
This work was supported by Grant-in-Aid from the Japan Agency for Medical Research and Development [Medical Research and Development Programs Focused on Technology Transfer (15im0210102h0001) and Project for Cancer Research and Therapeutic Evolution (16cm0106501h0001)] and the Naito Foundation. We thank Miki Sagou and Tsuyoshi Shirahari for technical assistance. The supercomputing resources were provided by the Human Genome Center, the Institute of Medical Science, The University of Tokyo.
Author contributions
WY and KK designed the study, analyzed the data, and wrote the paper; Y Shiraishi, KC, H Tanaka, and S Miyano developed sequence data processing pipelines; Y Nagata, YO, YT, JT, HU, Y Kogure, Y Shiozawa, NK, T Yoshizato, M Nakagawa, Y Nanya, KY, H Makishima, MS, and SO performed sequencing data analysis; Y Sato, KN, YG, H Miyoshi, KS, LZ, MS-Y, M Noguchi, RM, SC, NH, TI, NN, JK, TM, Y kito, H Tsurumi, KS, KT, KO, and T Yoshino managed patients and prepared samples; and all authors reviewed the paper during its preparation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Watatani, Y., Sato, Y., Miyoshi, H. et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia 33, 2867–2883 (2019). https://doi.org/10.1038/s41375-019-0473-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-019-0473-1
This article is cited by
-
SWI/SNF complexes in hematological malignancies: biological implications and therapeutic opportunities
Molecular Cancer (2023)
-
Deciphering the spectrum of cutaneous lymphomas expressing TFH markers
Scientific Reports (2023)
-
N6-methyladenosine reader YTHDF2 promotes multiple myeloma cell proliferation through EGR1/p21cip1/waf1/CDK2-Cyclin E1 axis-mediated cell cycle transition
Oncogene (2023)
-
TET2 guards against unchecked BATF3-induced CAR T cell expansion
Nature (2023)
-
Moving towards biologically informed treatment strategies for T-cell lymphomas
International Journal of Hematology (2023)
