
Abstract
Rapid advances over the past decade have uncovered the heterogeneous genomic and immunologic landscape of myelodysplastic syndromes (MDS). This has led to notable improvements in the accuracy and timing of diagnosis and prognostication of MDS, as well as the identification of possible novel targets for therapeutic intervention. For the practicing clinician, however, this increase in genomic, epigenomic, and immunologic knowledge needs consideration in a “real-world” context to aid diagnostic specificity. Although the 2016 revision to the World Health Organization classification for MDS is comprehensive and timely, certain limitations still exist for day-to-day clinical practice. In this review, we describe an up-to-date diagnostic approach to patients with suspected lower-risk MDS, including hypoplastic MDS, and demonstrate the requirement for an “integrated” diagnostic approach. Moreover, in the era of rapid access to massive parallel sequencing platforms for mutational screening, we suggest which patients should undergo such analyses, when such screening should be performed, and how those data should be interpreted. This is particularly relevant given the recent findings describing age-related clonal hematopoiesis.
Similar content being viewed by others

Introduction
Myelodysplastic syndromes (MDS) are a diverse group of clonal hematopoietic stem cell neoplasms characterized by ineffective hematopoiesis, peripheral blood cytopenias, and an inherent risk of progression to acute myeloid leukemia (AML) [1]. The incidence of MDS has been estimated at ~4.35 per 100 000 in the United States (age-adjusted incidence) [2] and 4.0 per 100 000 in Europe [3]. MDS is more common in males, with the exception of the MDS with isolated deletion 5q (del(5q) syndrome), which has a female predilection [4, 5]. However, the true incidence of MDS may actually be higher due to delayed presentation, inaccurate reporting, misinterpretation of subtle bone marrow (BM) morphologic findings, or misdiagnosis. MDS is predominantly a disease of the elderly, with a median age of onset in the seventh decade of life, although it may present much earlier [5]. Importantly, MDS may arise sporadically or be associated with an underlying germline predisposition syndrome, which may present at any age during childhood or adulthood [6]. In some cases, a precise diagnosis may be challenging, even in experienced centers. Conventionally, the prognostic scores utilized most frequently in daily clinical practice for MDS are the International Prognostic Scoring System (IPSS) and the revised IPSS (IPSS-R) [7, 8]. The IPSS, first described more than 20 years ago, encompasses the number of cytopenias, 3 cytogenetic subsets, and BM blast percentage. The IPSS-R has been refined to include the depth of cytopenias, 5 cytogenetic subsets, and further subdivides the BM blast percentage. Lower-risk (LR)-MDS is most commonly defined as those cases with Low-risk or Intermediate-1-risk MDS according to the IPSS [7]. It is extremely important to distinguish LR-MDS from other nonmalignant and non-clonal causes of cytopenias, such as vitamin deficiencies, side effects from drugs, autoimmune disorders, systemic infections such as human immunodeficiency virus (HIV) or inflammatory disorders, as well as from other myeloid neoplasms [1, 9].
Overview of disease pathogenesis
Over the past decade, major advances have been made in our understanding of how these diverse disorders arise. Cumulative cytogenetic, genomic, and immunologic data provide a fascinating insight into the varied pathogenetic mechanisms underlying disease development despite similar clinical and hematologic phenotypes. Advances in high-throughput DNA sequencing have led to the identification of multiple recurrent somatic mutations involved in disease initiation and progression. These include diverse genes involved in RNA splicing, DNA methylation, histone modification, signal transduction, transcription, and the cohesin complex [10]. The incidence of these mutations, prognostic significance, and associated disease characteristics are described in Table 1 [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. Furthermore, excessive DNA damage has been frequently reported in patients with MDS, and both intrinsic and extrinsic genotoxins have been implicated [31]. Defective DNA repair pathways have also been implicated in MDS pathogenesis although the evidence is less robust than that in AML [32].
Despite the established role of chronic inflammation in the pathogenesis of many malignancies, a potential role in MDS remained less clear until recently. One of the first reports recognizing an association between immunologic abnormalities and MDS was published by Mufti et al. [33], highlighting the higher incidence of abnormal serum immunoglobulins and autoantibodies in patients with MDS. Results from a collaborative study demonstrated that the presence of an autoimmune disorder was associated with an improvement in overall survival in MDS [34], most likely due to an initially “protective” adaptive immune response [35]. Further work has demonstrated modulation of both the adaptive and innate immune systems linked to disease initiation and progression [35]. An increase in the number of interleukin 17-producing T cells (Th17) in LR-MDS, whereby the ratio of Th17 cells to regulatory T cells (Tregs) was significantly higher in LR-MDS compared with higher-risk (HR) MDS, correlated with the presence of apoptosis [36]. Moreover, an immunosuppressive environment with increased Tregs in LR-MDS carries adverse prognostic significance [37]. The innate immune system also plays a key role in MDS pathogenesis via the upregulation of inflammatory cytokines through nuclear factor (NF)-κB activation and/or activation of the redox-sensitive NLRP3 inflammasome and β-catenin pathways leading to clonal propagation [38]. Lastly, it is becoming increasingly evident that the microenvironment plays a key role in disease propagation. Mesenchymal niche-induced genotoxic stress in hematopoietic stem/progenitor cells (HSPC) has been found to be predictive of leukemic evolution and progression-free survival in MDS [39]. Further refinements of the diagnostic and prognostic criteria for MDS will likely include these immunologic findings; however, it is important to stress that these immunologic findings need to be considered within both the clinical and hematologic context. In this review article, we describe an integrated approach to the diagnosis of patients with LR-MDS, taking into account the increasing “-omics” data concerning MDS pathogenesis and prognosis (Fig. 1).

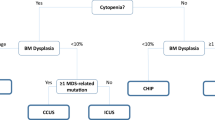
Differential diagnosis of ICUS, CCUS, CHIP, and MDS is important to informing prognosis and to guide treatment decisions (a), while accurate diagnosis of hypoMDS and AA can be challenging due to overlapping symptoms (b). The diagnosis of MDS has traditionally been achieved using morphology, measurement of blast count, and cytogenetic analyses. Newer diagnostic techniques, such as flow cytometry and mutational profiling, are becoming more widely used. These may facilitate diagnosis of hematopoietic disorders; however, the challenge will be to integrate these methods into a single diagnostic workflow that can be used for the differential diagnosis of each disease (c). AA aplastic anemia, aCGH array-based comparative genomic hybridization, CCUS clonal cytopenia of undetermined significance, CHIP clonal hematopoiesis of indeterminate potential, hMDS hypoplastic MDS, ICUS idiopathic cytopenia of undetermined significance, MDS myelodysplastic syndromes, PNH paroxysmal nocturnal hemoglobinuria, SNP single-nucleotide polymorphism, WHO World Health Organization
An up-to-date approach to the diagnosis of lower-risk MDS
History and physical examination
A detailed, focused, and systemic enquiry should be completed in the workup of any potential case of MDS to rule-out other reasons for cytopenia. This workup should include a detailed extended family history, occupational history, and comprehensive review of concomitant medications. Any history of other malignancies that may suggest an underlying germline predisposition syndrome, as well as any history of exposure to cytotoxic chemotherapy or radiotherapy, should be noted. The chronic nature of the cytopenias should be carefully evaluated and historical blood counts assessed where possible. Moreover, a thorough physical examination should be performed with particular reference to organomegaly, lymphadenopathy, stigmata of autoimmune disorders, and features suggestive of a constitutional BM failure disorder.
Morphologic examination of blood and BM
The diagnosis of MDS historically relied largely on morphologic findings of BM and blood [40]. In theory, morphologic examination is a technically simple and inexpensive method [41] and can be performed using well-prepared peripheral blood smears, BM aspirates, and BM trephine biopsies. However, as patients may present with hypocellular marrows or disease-related fibrosis, accurate morphologic assessment may be difficult. Moreover, differentiation between entities such as aplastic anemia (AA) and hypoplastic MDS may prove challenging due to the considerable overlap in morphologic findings (Fig. 1). In general, at least 200 cells in a blood film, 500 cells in a BM aspirate, and a minimum of 100 erythroblasts and 30 megakaryocytes should be evaluated where possible [42]. An accurate diagnosis in patients in the early stages of disease may be difficult. The 2016 revised World Health Organization (WHO) criteria for MDS rely heavily upon assessment of the degree of dysplasia and blast percentages rather than specific cytopenias. Suggested values for categorization of cytopenias in MDS remain hemoglobin <10 g/dl, platelet count <100 × 109/l, and absolute neutrophil count <1.8 × 109/l. When measuring neutrophil count, the ethnic origin of the patient and individual laboratory reference ranges should be considered, as some patient populations may have a lower minimal normal neutrophil count (<1.5 × 109/l). Importantly, MDS may initially present with anemia or thrombocytopenia above these arbitrary thresholds [43]. Although the threshold for defining dysplasia is 10% dysplastic cells in any 1 lineage, dysplasia in 1 cell lineage above 10% may occur in some healthy individuals and in other causes of cytopenia. The number of classical signs of dysplasia may also be low, depending on the case. Germing et al. [44] reported a median of six different dysplastic features in individual patients, highlighting the importance of testing both the peripheral blood and BM for signs of dysplasia. Although there is also inherent subjectivity in the classification of dysplasia, even among experienced hematopathologists [43], studies have shown moderate to substantial concordance among experts in assessing dysplasia in LR-MDS [45]. To estimate blast percentage, it is now recommended that all nucleated BM cells should be counted as the denominator, rather than just non-erythroid cells; [46, 47] this applies to all myeloid neoplasms [43]. Prussian blue staining of the BM is essential to determine if ring sideroblasts (RS; cells containing at least five siderotic granules surrounding the nucleus) are evident and how many are present. Most patients with RS are stratified into LR-MDS categories [48] (Table 2), and the revised WHO classification recommends a diagnosis of MDS-RS if an SF3B1 mutation (discussed in detail below) is present when RS comprise as few as 5% of the nucleated erythroid population. BM trephine biopsy can provide a more accurate assessment of the BM topography, cellularity, and presence or absence of fibrosis [49]. We recommend a trephine biopsy be performed at diagnosis and for follow-up assessments. Of note, the new 2016 WHO proposals for MDS classification were recently validated—and shown to be both pragmatic and feasible—in a large independent cohort [50].
Role of flow cytometry in MDS diagnosis: an overview
Over the past decade, flow cytometric advances have gained increasing importance in aiding the diagnosis of MDS. Multiparameter flow cytometry (MFC) may be used to detect the aberrant expression of differentiation-associated antigens in cells, as well as abnormal phenotypic patterns in maturing hematopoietic cells. However, as the underlying complexity and heterogeneity of the disease makes robust harmonization and reproducibility of testing difficult, immunophenotypic findings should always be considered in the context of other diagnostic results as part of an integrated diagnostic report (Fig. 1).
Attempts have been made to standardize immunophenotyping protocols, antibody selection, and interpretation of resultant diagnostic information. The European LeukemiaNet (ELN) collaborative group proposed minimal requirements for the standardization of flow cytometry in MDS, recommending the use of multicolor MFC to detect 4 key reproducible parameters: (i) percentage of CD34+ myeloid progenitors, (ii) frequency of B-cell progenitors within the CD34+ cell population, (iii) myeloid progenitor cell CD45 expression, and (iv) granulocyte side scatter value [51,52,53,54]. In a large “learning and validation” cohort study (797 patients; 417 with LR-MDS and 380 controls with non-clonal cytopenia) designed to develop and validate a flow cytometric score for MDS diagnosis, patients with MDS frequently displayed increased myeloid progenitor-related cluster size, decreased B-cell progenitor-related cluster size, reduced granulocyte side scatter, and aberrant CD45 expression. Overall, the diagnostic score had a sensitivity of 70% to correctly diagnose MDS. Cremers et al. [55] reported on the specificity of MFC in excluding MDS in 379 cytopenic patients with indeterminate cytomorphology or cytogenetic findings—the presence of normal MFC findings predicted a low probability of developing MDS within 1 year.
Prognostically, a higher flow cytometry score has been associated with multilineage dysplasia, severe cytopenias, red blood cell transfusion dependence, and poor-risk cytogenetics, leading to a higher revised International Prognostic Scoring System (IPSS-R) classification, and an increased risk of leukemic evolution [56]. More recently, Alhan et al. [57] analyzed the flow cytometric characteristics of BM aspirate samples from 109 individuals with MDS to derive an MDS Flow Cytometry Score (MFS); this was validated in a further 103 patients. This MFS incorporates three parameters; degree of sideward light scatter of myeloid progenitor cells, CD117 expression on myeloid progenitor cells, and CD13 expression on monocytes. A high MFS score was associated with significantly poorer outcomes versus patients with intermediate MFS scores. Of particular note, the MFS further refined prognostication within the IPSS-R low-risk group, whereby those with high MFS tended to have worse overall survival. A multinational collaborative group has suggested minimal diagnostic criteria for both MDS and pre-MDS states and importantly has included a focus on suggested FC panels [58].
While the above studies focused on the myeloid compartment, a recent study focused on the erythroid compartment with the aim of delineating dyserythropoiesis associated with MDS from non-clonal cytopenias, something that can be very difficult for even experienced morphologists. An erythroid flow cytometry marker incorporating CD36 and CD71 expression (expressed as a co-efficient of variation), combined with CD71 fluorescence intensity and the percentage of CD117+ erythroid progenitors formed a marker set with high specificity (92%; 95% confidence interval 86–97%) for discriminating between true MDS and other non-clonal cytopenias [59]. This approach was validated in a prospective clinical study of 106 patients with MDS [60].
Despite advances, several issues regarding the utility of FC remain: (1) the variable degree of sensitivity; (2) heterogeneous immunophenotypic findings dependent on antibody combination and gating strategies; (3) the lack of uniform standardization; and (4) reproducibility across platforms and users [61]. One area of interest should be the immunophenotypic signature of conditions, which may mimic LR-MDS, such as autoimmune and inflammatory-driven cytopenias. Where possible, laboratories should follow the guidance of the ELN working group for standardization of flow cytometry in MDS. Flow cytometry findings should be one facet of the integrated report, which should also include peripheral blood counts, a description of the BM aspirate and trephine morphology with any relevant immunohistochemistry, and the complete conventional karyotype and fluorescence in situ hybridization (FISH) data, as well as molecular data where available (Fig. 1).
Paroxysmal nocturnal hemoglobinuria (PNH) screening
The presence of paroxysmal nocturnal hemoglobinuria (PNH) in patients with MDS may have important implications for prognosis and treatment. Patients with MDS and increased PNH-type cells may have more severe thrombocytopenia but less pronounced blood cell-morphologic abnormality, lower rates of karyotypic abnormalities, and lower rates of progression to acute leukemia versus patients with MDS without increased PNH-type cells [62]. PNH clones can occur in both MDS and AA and can readily be detected by standard flow cytometric techniques showing lack of glycophosphatidylinositol-anchored proteins in the red cell, monocyte, and granulocyte compartments [63]. PNH clones have been reported in between 5.5% and 8% of patients with MDS [64, 65] and in 26.3% of patients with AA [64]. To detect PNH clones, samples should be analyzed within 24–48 h to ensure sensitivity at detecting PNH clones. Even when detected, the clone size can vary substantially and may often be clinically insignificant. If present at >1%, follow-up testing should generally be performed every 6 months [66] to determine whether clonal expansion has occurred. Patients with PNH and either AA or MDS may benefit from treatment with eculizumab, an anticomplement C5 monoclonal antibody that inhibits complement-mediated hemolysis of red blood cells [67].
Cytogenetic testing
Chromosome abnormalities are detected in approximately 50% of patients with newly diagnosed MDS and more than 80% of those with therapy-related MDS based on conventional G-banding karyotypic analysis, or FISH [68, 69]. Importantly, FISH analysis can be applied to both metaphase cell preparations and interphase cell nuclei. FISH may aid diagnosis in patients with poor-quality metaphases or submicroscopic alterations. As FISH has only limited ability to detect additional abnormalities that are undetectable by metaphase cytogenetics [70], it should primarily be used when adequate metaphases are unavailable for conventional cytogenetic analysis. FISH analysis of peripheral blood CD34+ cells may also be used when conventional chromosome banding analysis is not possible [71]. A suggested FISH panel for diagnostic laboratory use in MDS could include the following probes: (i) EGR1/D5S630/D5S21 probe set, to detect −5 and del(5q); (ii) D7S486 or alternative probe set, to detect −7 and del(7q); (iii) CEP 8 or alternative, to detect trisomy 8; (iv) D20S108/20qter probe set, to detect −20 and del(20q); (v) TP53 locus-specific probe, to detect del(17p13.1); (vi) RPN1/MECOM to detect t(3;3) and inv(3); (vii) MLL (KMT2A) to detect 11q23.1−q23.3 rearrangement; and (viii) D13S319/LAMP1 to detect del(13q).
As discussed, karyotype risk factors feature heavily in both the IPSS and IPSS-R prognostic stratification (Table 3) [7, 8] and can aid prediction of response to therapeutic intervention. For example, the presence of del(13q) is associated with a favorable response to immunosuppressive therapy [72]. In a comprehensive cytogenetic analysis on 2072 MDS patients, clonal abnormalities were found in 1084 (52%) [68]. A total of 684 different cytogenetic categories were identified, reflecting the marked karyotypic heterogeneity associated with MDS. The most frequent cytogenetic abnormalities in MDS are del(5q), monosomy 7/del(7q), trisomy 8, loss of Y, and complex karyotypes (conventionally defined as ≥3 chromosomal aberrations, including at least 1 structural aberration) [73] (Table 4) [8, 74]. In LR-MDS, the frequent findings are a normal karyotype, isolated del(5q), del(20q), and –Y [8, 74].
Array-based comparative genomic hybridization (aCGH) facilitates identification of small chromosomal abnormalities that may remain undetected with traditional cytogenetics [75]. These abnormalities may include deletion of the region of chromosome 4q24 containing the TET2 gene, small deletions on chromosome 5q31, and deletions of 7q22.1 and 21q22.12 [76]. A small study reported on the utility of high-resolution whole genome aCGH analysis of CD34+ progenitor cells isolated from the marrow of 44 LR-MDS patients, 25 of whom had no karyotypic aberration by conventional karyotyping. aCGH identified cryptic DNA alterations that were undetectable by conventional karyotyping and revealed copy number changes in 36 of 44 patients. Moreover, maintenance of genomic integrity (arbitrarily defined as a chromosomal disruption of <3 MB) was associated with lower risk of leukemic transformation and improved survival [77]. Similarly, single-nucleotide polymorphism arrays (SNP-A) carry significant technical advantages and can be used for high-resolution genotyping in MDS to identify additional aberrations, including measurement of gene copy number (hybridization signal intensity) and areas of loss of heterozygosity, which cannot be detected with conventional techniques [78]. In a study of SNP-A genotyping performed on 119 LR-MDS patients, the group from King’s College, London demonstrated the presence of uniparental disomy in 46%, deletions in 10%, and amplifications in 8% of patients [78]. SNP-A genotyping provides superior levels of resolution and is able to evaluate nondividing cells and detect acquired copy-neutral loss of heterozygosity [79]. Potential platforms include the Affymetrix (Santa Clara, CA, USA) SNP platform 6.0 version (with 1.8 million probes), and the Cytoscan HD platform (with 2.695 million probes). However, SNP-A does not detect balanced translocations or small clones.
Importantly for both diagnosis and follow-up, there is a high concordance between cytogenetic and genomic aberrations detectable in the BM and in the peripheral blood of MDS patients [80]. This study evaluated BM-derived “genetic markers” in peripheral blood and serum samples using SNP-A karyotyping, 454 parallel sequencing (454-PS), and Sanger sequencing of 22 genes frequently mutated in MDS: all exons of DNMT3A, RUNX1, CEBPα, TP53, EZH2, and ZRSR2 and mutation “hotspots” for NPM1, FLT3, ASXL1, IDH1, IDH2, MPL, JAK2, BRAF, cCBL, NRAS, KRAS, C-KIT, SF3B1, SRSF2, and U2AF35. TET2 was analyzed by Sanger sequencing. This study successfully demonstrated an excellent concordance for both SNP-A and mutation analyses between peripheral blood (not serum) and BM, albeit with a lower clonal burden in the peripheral blood. In practical terms, this means that sequential cytogenetic monitoring can be performed on the peripheral blood rather than subjecting patients, who are often elderly, to repeated marrow biopsies [80].
Genomic profiling: mutational landscape
The increasing availability of rapid sequencing has revolutionized the diagnostic mutational profiling of suspected MDS patients (Table 1) [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. Recurrent genetic mutations occur in diverse, pivotal cellular pathways. These include tyrosine kinases (FLT3, JAK2, MPL) and their downstream signaling pathways (RAS, CBL), transcription factors (RUNX1, NPM1, ETV6, GATA2), tumor suppressors (TP53, WT1), epigenetic modifiers (TET2, ASXL1, EZH2, DNMT3A, IDH), pre-mRNA splicing machinery (SF3B1, SRSF2, U2AF1, ZRSR2), and cohesion complex proteins (STAG2, RAD21, SMC3, SMC1A). Akin to cytogenetic profiling, the mutational landscape of MDS demonstrates great heterogeneity, although SF3B1 and TET2 remain the most commonly detected disease-associated mutations overall with incidences of 20–25% [12]. Furthermore, the DNA methylation-associated gene DNMT3A and the chromatin modification gene ASXL1 are mutated in more than 10% of MDS patients [81]. Della Porta et al. [82] reported on an association between the presence of severe granulocytic dysplasia and mutations in ASXL1, RUNX1, TP53, and SRSF2. The mean number of mutations is lower in LR-MDS compared with HR-MDS and, with the exception of SF3B1, DNMT3A, JAK2, and MPL, the majority of common mutations are more prevalent in high-risk subtypes [12]. Moreover, in an analysis of 288 patients with LR-MDS, 71% of the cohort had detectable mutations, most commonly involving TET2 (23% of samples), SF3B1 (22%), U2AF1 (16%), ASXL1 (15%), SRSF2 (15%), and DNMT3A (13%) [83].
Mutational profiling can also confer significant prognostic information and help predict response to therapy [20, 30]. As a result, mutational data are increasingly integrated into prognostic scoring systems and therapeutic treatment pathways. Mutations of ASXL1 are associated with poorer overall survival [25], while mutations of the tumor suppressor gene TP53 occur more frequently in patients with HR-MDS than LR-MDS [28] and are an independent predictor of decreased overall survival [25, 29]. Bejar et al. demonstrated that the presence of high abundance TET2 mutations were associated with an increased response rate to HMA therapy in a large MDS cohort, including cases of LR-MDS, particularly in the absence of ASXL1 [22]. Moreover, pre-allogeneic stem cell transplantation-targeted genomic profiling can aid prediction of transplantation outcomes in MDS [84].
In contrast to the majority of recurrently mutated genes in MDS that occur in HR phenotypes, SF3B1 mutation is more common in patients with LR-MDS and confers a favorable prognosis [11, 12, 29, 85]. Mutations of SF3B1 are strongly associated with favorable disease characteristics including the presence of RS [9,10,11,12,13, 42, 86] and normal cytogenetics [85]. Mian et al. [85] elegantly demonstrated that SF3B1 mutations in patients with MDS with RS arise early in rare hematopoietic stem cells; these may indeed be the initiating event and may propagate to myeloid progeny [85]. SF3B1 mutation was also shown to be an early event in MDS by Woll et al. [87] Of note, although there is a strong association between SF3B1 mutation and RS, the percentage of RS itself is not predictive of survival [86]. The PACE-MDS study, investigating the efficacy of the recombinant fusion protein luspatercept, also demonstrated that improvements in erythroid activity in anemic LR-MDS patients were associated with the presence of SF3B1 mutations [88].
Attempts to incorporate genetic mutation data into the existing IPSS-R are ongoing (Fig. 2) [27, 29]. The 2016 update to the WHO classification system also incorporates key mutational data into diagnostic criteria. The revised guidelines now include SF3B1 mutation as diagnostic of MDS with RS; patients with SF3B1 mutation and as few as 5% with RS can be classified as having MDS-RS [43].

Somatic mutations occurring in patients and their prognostic significance in the IPSS-Rm [25, 27]. aRemaining independent prognostic factors in a Cox proportional hazard model including age and IPSS-R score [25]. IPSS-R(m) revised International Prognostic Scoring System (molecular), MDS myelodysplastic syndromes, OS overall survival
The choice of platform utilized to establish the presence or absence of mutations is dependent on the institution and availability of technology. The number of genes on such panels is non-exhaustive and should be directed by local capabilities and clinical utility. One suggested comprehensive panel is highlighted in Table 5, including common and rare genes that may be mutated in MDS (with known hotspots) in addition to telomerase complex genes. This is solely the authors’ suggestion and is dependent upon the platform available, referral practice, and clinical population. The actual turnaround time for the resultant report is variable, ranging from 1 to 6 weeks in routine clinical practice. Moreover, there is ongoing debate about how detailed such a report should be; known pathogenetic mutations should be reported although debate currently exists concerning routine reporting of detected variants of unknown significance.
How to practically distinguish idiopathic cytopenia of undetermined significance, idiopathic dysplasia of undetermined significance, clonal cytopenia of undetermined significance, and clonal hematopoiesis of indeterminate potential from LR-MDS
Increasingly, the clinical community is utilizing targeted mutation testing in cases of cytopenia to aid both diagnostic and prognostic stratification; however, simply determining the presence of a myeloid disease-associated somatic mutation is not diagnostic per se of MDS or related disorders [58, 89].
It is well established that chronologic aging affects not only the hematopoietic stem cell compartment and progeny but also the supportive BM microenvironmental niche and the interacting immune system. Recently, Vas et al. [90] described how an aged niche might exert a distinct selection pressure on dominant hematopoietic progenitor clones. Furthermore, Jaiswal et al. [91] reported on whole exome sequencing (WES) data from 17 182 individuals unselected for hematologic phenotype in which the frequency of detectable somatic mutations rose appreciably with age. For individuals aged 70 to 79 years (n = 2229), 80 to 89 years (n = 317), and 90 to 108 years (n = 103), clonal mutations were observed in 9.5%, 11.7%, and 18.4%, respectively. The majority of the variants occurred in three genes: TET2, DNMT3A, and ASXL1. Somatic mutations were associated with an increased risk of hematologic malignancy, cardiovascular-related deaths, and all-cause mortality. Moreover, Genovese et al. [92] reported on an unselected cohort of 12 380 Swedish patients in which up to 10% of patients older than 65 years displayed clonal hematopoiesis with somatic mutations; in contrast, this feature was observed in only 1% of those younger than 50 years. Again, the most frequent somatic mutations were in TET2, DNMT3A, and ASXL1. The presence of this so-called age-related clonal hematopoiesis was a strong predictive factor for the subsequent development of a hematologic malignancy. Almost 42% of hematologic malignancies arose in patients with evidence of clonality at the time of sampling, at least 6 months before detectable disease [92]. However, not all patients with evidence of clonal hematopoiesis with MDS-type mutations occurring with age will subsequently develop a hematologic disorder, called clonal hematopoiesis of indeterminate potential (CHIP) (Fig. 1). The working definition requires an allele burden of ≥2% [58, 93]. Further longitudinal studies are required to correlate the presence of specific mutations and subsequent development of bona fide MDS [43].
Idiopathic cytopenia of undetermined significance (ICUS) is characterized by unexplained persistent cytopenia (≥4 months), in 1 or more lineages, which fails to meet the minimal diagnostic criteria for MDS and is not explained by other hematologic or non-hematologic disorders (Fig. 1) [58]. Arbitrary cutoff figures remain hemoglobin <11 g/dl, platelet count <100 × 109/l, and absolute neutrophil count <1.5 × 109/l [94]. Although ICUS may involve more than 1 cell lineage, the normal clinical situation is that of a severe unilineage cytopenia. Individuals with ICUS may be further divided into ICUS-A (anemia), ICUS-N (neutropenia), ICUS-T (thrombocytopenia), and ICUS-PAN (bi/pancytopenia [58]. The prevalence of ICUS and clonal cytopenia of undetermined significance (CCUS; in which a myeloid-disorder–associated mutation is detected in a cytopenic patient (≥4 months) in the absence of any other clonal BM neoplasm) is poorly understood and robust long-term follow-up studies are lacking [58]. Although not as frequent, it is important to recognize patients with idiopathic dysplasia of undetermined (unknown) significance (IDUS), who present with persistent peripheral blood or BM findings of dysplasia in >10% of cells but no persistent cytopenias and no other reason for dysplasia and who do not meet the minimal criteria for MDS. These individuals lack a detectable MDS-associated mutation. No specific management guidelines, beyond those recommended for a specific cytopenia in ICUS or CCUS are available, but ongoing observation is warranted overall.
Although the precise longer-term significance of ICUS, IDUS, CHIP, and CCUS requires further clarification, we are increasing our knowledge of factors that may predict progression to MDS. Kwok et al. [95] analyzed 144 patients with unexplained cytopenias. Based on cytomorphologic assessment, 15% were diagnosed with ICUS and some evidence of dysplasia, 69% with ICUS and no dysplasia, and 17% with MDS. Using a targeted 22-gene panel, mutations were identified in 71% of MDS patients, 62% of patients with ICUS and some dysplasia, and only 20% of ICUS patients with no dysplasia. This represents a higher rate of detection than would be expected even for age-related clonal hematopoiesis. Similar rates were found when these results were validated in a cohort of 91 patients with LR-MDS and 245 patients with ICUS [95]. More recently, Malcovati et al. [96] evaluated the significance of somatic mutations in patients with unexplained cytopenias. In a learning cohort of 683 patients, using a targeted panel of 40 genes, 64% of patients carried a somatic mutation in at least 1 of these genes. The presence of a somatic mutation with a variant allele frequency (VAF; a measurement of the mutational burden detected) ≥10% or having 2 or more detectable mutations had a positive predictive value of 0.86 and 0.88, respectively, for diagnosis of a myeloid disorder. Moreover, mutations in spliceosome genes or comutation of TET2 or DNMT3A with another mutation were strongly associated with high risk of progression to a myeloid neoplasm.
It is extremely important to consider the VAF of the mutation detected when using these panels and whether that may contribute to the presence of cytopenia or not. According to Steensma et al. [93], larger clones (VAF > 20%) may have more clinical significance than smaller clones (VAF < 10%); however, further evaluation of the dominant clonal architecture is needed. Future work may define patterns of mutations and VAFs that are predictive of a higher risk of progression to MDS. We suggest that follow-up and frequency of monitoring of patients with ICUS and CCUS is dependent on the degree of cytopenia, as well as the mutational burden in those with CCUS.
How to accurately distinguish hypoplastic mds from AA
Histologic differences between hypoplastic MDS (hMDS, which accounts for 10–15% of all MDS) and AA, can be subtle, and it can be extremely difficult, even for experienced histopathologists, to accurately discriminate between these two disorders. This is particularly true when the trephine cellularity is low and aspirates are pauciparticulate and markedly hypocellular [97]. AA and hMDS may have overlapping pathogenetic mechanisms, but clinically this distinction is highly relevant, as the therapeutic approach and prognosis will differ.
Dyserythropoiesis can be prominent in AA and is not specific to hMDS. More than 10% hypogranular neutrophils or pseudo–Pelger–Huet cells in the peripheral blood (in a sample of at least 100 cells), presence of dysmegakaryopoiesis and marrow granulocytic dysplasia, the presence of RS, fibrosis, abnormal localization of immature precursors, and increased blasts suggest hMDS rather than AA [97]. However, overlapping features are not uncommon and these criteria are not always accurate. In addition, it is important to note that clonality is not uncommon in AA. Yoshizato et al. [98] comprehensively described the presence of myeloid disease-associated somatic mutations in 156 of 439 (35.5%) AA patients as determined by targeted-capture deep sequencing. Moreover, in targeted exome sequencing on the BM of 150 patients with AA, 32 somatic mutations commonly identified in MDS were discovered in 19% of patients; mutations in DNMT3A, ASXL1, and BCOR were most common. The presence of these somatic mutations and disease duration of >6 months was associated with a 40% risk of transformation to MDS [99]. It is difficult therefore to accurately utilize a detected mutational pattern/clonal burden for discrimination; for example in both AA and MDS, mutations in DNMT3A and ASXL1 are common. In contrast, PIG1A and BCOR/BCOR1 mutations are overrepresented in AA [98, 99]. Collaborative approaches are required to revisit the criteria for hMDS incorporating morphologic, genomic, and immunologic features and are currently under way.
Delineating immune signatures in lr-mds
It is becoming increasingly accepted that the host background (e.g., human leukocyte antigen–type, DNA repair capability, and genomic characteristics), microenvironmental factors, and, importantly, the type of cellular immune response and immune checkpoints, may play a significant role in modulating clonal evolution in LR-MDS. As discussed previously, Kordasti et al. [36] were the first to identify an increased number of Th17 cells and increased Th17:Treg ratio in LR-MDS. Within the innate immune system, myeloid-derived suppressor cells (MDSCs) function as pivotal effectors of ineffective hematopoiesis and are markedly expanded in the BM of patients with LR-MDS. BM levels of the TLR4/CD33 ligand S100A9, which promotes both autocrine-reinforced MDSC activation and paracrine-mediated myeloid progenitor cell death, are also increased [100]. Utilization of findings such as these in routine diagnostics is as yet premature, but many advances are being made in this area.
When to consider inherited BM failure syndrome or germline predisposition syndromes in lr-mds
It is important to be alert to the possibility of inherited BM failure syndromes such as dyskeratosis congenita or even Fanconi anemia that may present later in life with an MDS phenotype. Moreover, integration of genomic analyses into diagnostic algorithms has led to an increasing recognition of underlying germline anomalies associated with an increased susceptibly to MDS. Recognition of such syndromes is essential not only for accurate diagnostic classification but also genetic counseling and psychological support for other family members. A detailed analysis of diagnostic approaches is outside the remit of this article, but several key points will be highlighted. An accurate and extended family history, with particular focus on hematologic disorders and solid organ tumors, should be obtained, and the clinician should be alert to any findings suggestive of a constitutional BM failure disorder/germline predisposition that may present as LR-MDS. Symptoms, signs, and laboratory findings may be highly variable but may include a personal or family history of cutaneous and nail anomalies, short stature, premature graying, thrombocytopenia, hemorrhagic phenomena, and limb or tooth anomalies [101]. Imaging may reveal the presence of cardiac abnormalities, liver fibrosis, or pulmonary fibrosis [101]. Where the clinical situation warrants telomere assessment, telomere length analysis should be considered either by reverse transcription polymerase chain reaction or Flow-FISH [102] (a technique combining flow cytometry with FISH), depending on local expertise. Mutations in TERC (encoding the RNA component of telomerase) or TERT (encoding the telomerase reverse transcriptase enzyme) can also be evaluated, and comprehensive telomerase gene complex targeted sequencing panels are under development [103, 104]. Regulator of telomere elongation helicase 1 (RTEL1) is a DNA helicase critical to telomere maintenance and stability and contributes to DNA repair. It plays a pivotal role in dismantling T loops and counteracts telomeric G4-DNA [105]. Biallelic germline mutations have been identified that clinically manifest with the dyskeratosis congenita phenotype and Hoyeraal–Hreidarsson syndrome [106]. Of relevance to this diagnostic workup, Marsh et al. [107] have recently reported that heterozygous RTEL1 variants classified as likely pathogenic can be associated with unexplained cytopenias, AA, and hMDS, seen both at an early age and in adulthood. These variants had variable penetrance, and it was noted that telomere length analysis alone may not detect all primary telomere defects, as RTEL1 variants were detected that were associated with eroded 3′ overhangs only. For Fanconi anemia, although late presentation is more unusual, presence of a positive chromosomal breakage test (using agents such as mitomycin C or diepoxybutane) remains the gold standard for diagnosis [108]. FANC gene mutational analyses can also be performed. Analyses of GATA2 mutational status are also highly relevant given the well-documented predisposition to MDS [109,110,111]. Lastly, a range of other germline mutations may present with late-onset MDS and these should always be considered dependent on phenotype and history (Table 6) [103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121].
Conclusions
The diagnosis of LR-MDS can be complex due to the difficulty of distinguishing true MDS from age-related or nonmalignant causes of cytopenia. There is also increasing recognition of cases that represent ICUS, IDUS, or CCUS rather than true MDS. However, as patient management is currently determined by accurate disease classification and prognostication, a delayed or incorrect diagnosis may delay treatment and adversely affect outcomes. As new tools are being developed to improve diagnostic/prognostic assessment, the challenge is to incorporate these tools into a streamlined, standardized diagnostic protocol. An up-to-date diagnostic approach, such as the one described here, will improve diagnostic accuracy and permit therapeutic stratification where required. An example of an integrated report of a LR-MDS case is shown in Fig. 3.

An integrated diagnostic approach for lower-risk MDS. aCGH array comparative genomic hybridization, BM bone marrow, BMF bone marrow failure, LR-MDS lower-risk MDS, MDS myelodysplastic syndromes, MDS-RS myelodysplastic syndromes-ring sideroblasts, MFS MDS flow cytometry score, PB peripheral blood, PNH paroxysmal Nocturnal Hemoglobinuria, RS ring sideroblasts, SNP-A single-nucleotide polymorphism-A, SSC side scatter, T-LGL T cell large granular lymphocyte leukemia, TNC total nucleated cells, VAF variant allele frequency
In conclusion, over the past decade, we have gained understanding of how genomic characteristics, microenvironmental factors, host background, type of adaptive/innate immune response, and immune checkpoints may play a significant role in modulating clonal evolution in MDS and response to therapy. We hypothesize that future developments will incorporate these findings into diagnostic and prognostic models that will permit stratified therapeutic intervention and improved outcomes.
Case study 1: a patient with delayed presentation of a constitutional BM failure syndrome
A 55-year-old man with a known diagnosis of pulmonary fibrosis for the past 3 years was referred to hematology due to progressive pancytopenia. He had no other significant past medical history or family history of note. He was on maintenance steroids for his pulmonary fibrosis. A physical examination revealed that he had a cushingoid facies with telangiectasia and evidence of vitiligo on his eyelids. His liver edge was palpable at 2 cm and his spleen at 3 cm below the right and left costal margins, respectively. A routine workup demonstrated a hemoglobin level of 88 g/l, a white blood cell count 2.5 × 109/l, neutrophils 1.9 × 109/l, and platelets 75 × 109/l. His mean corpuscular volume was raised at 102 fl. The patient had a normal reticulocyte count, and normal B12 and folate levels. His ferritin level was 1945 g/l. His renal function was normal. His bilirubin level was normal at 14 µmol/l, alkaline phosphatase 109 IU/l, aspartate aminotransferase 78 U/l, and gamma-glutamyl transpeptidase 402 U/l. His lactate dehydrogenase level was normal. An abdominal ultrasound scan revealed hepatomegaly, the liver demonstrated a diffuse coarse texture on imaging, no focal lesions, and an enlarged spleen (17 cm). Subsequent BM aspiration and trephine biopsy results revealed a hypercellular marrow, trilineage dysplasia, and blasts were evident at 2%. Reticulin deposition was increased at grade 2, CD34+ 1%, and CD117 2%. BM cytogenetics revealed 46 XY, deletion (20)(q11q13) in 30 metaphases. Overall diagnosis was consistent with MDS, subtype MDS with Multilineage Dysplasia (MDS-MLD). A targeted gene panel incorporating commonly mutated telomerase genes in addition to the most frequent MDS-associated anomalies was performed, and this revealed that the patient was heterozygous for a known pathogenetic telomerase RNA component (TERC) gene mutation only. This case highlights the importance of always considering a constitutional BM failure syndrome dependent upon the clinical presentation.
Case study 2: a patient with pancytopenia and a hypocellular BM
A 59-year-old woman presented in June 2013 with new-onset severe pancytopenia and demonstrated a hemoglobin level of 76 g/l, a white blood cell count 1.8 × 109/l, neutrophils 0.4 × 109/l, and platelets 2 × 109/l. Her mean corpuscular volume was normal. Her symptoms were limited to fatigue and intermittent epistaxis. No other significant comorbidities were recorded. A peripheral blood film showed normal red cell morphology and confirmed leukopenia and thrombocytopenia. There were no circulating blasts. Flow cytometry revealed a small population of cells with a PNH phenotype: glyscosylphosphatidylinositol (GPI)-deficient CD15 + neutrophils 12%; GPI-deficient CD64 + monocytes 11.2%; and GPI-deficient red cells 0.5% only. Her lactate dehydrogenase level and hemolytic markers were normal. An autoimmune screen and direct antiglobulin test results were negative. No T cell large granular lymphocyte population was identified on peripheral blood flow cytometry. BM aspiration and flow cytometry revealed an aparticulate and severely hypocellular sample, predominantly scattered non-clonal plasma cells and lymphocytes. There was marked dyserthropoiesis and, in assessable areas, mild granulocytic dysplasia. Conventional karyotyping failed and a single-nucleotide polymorphism array (SNP-A) was normal. One hypolobated megakaryocyte was seen. A targeted gene panel analysis revealed the presence of an ASXL1 mutation (c.1934dupG) with a variant allele frequency of 45%. A BM trephine biopsy was severely hypocellular (10%), with evidence of sparse yet hypolobated megakaryocytes, scattered lymphocytes and plasma cells, and marked reduction in erythropoiesis and granulocytic activity. There were no excess blasts. Reticulin deposition was patchy yet grade 2 in some regions. Based on these cumulative findings, she was diagnosed with hypoplastic MDS with a small PNH clone rather than AA, and commenced immunosuppressive therapy with cyclosporine A to which she had mounted a response by 3 months. Unrelated donors for a future allograft were identified.
References
Bejar R, Steensma D. Recent developments in myelodysplastic syndromes. Blood. 2014;124:2793–803.
Surveillance, Epidemiology, and End Results Program (SEER) Cancer Statistics 2000–2013. Available at: https://seer.cancer.gov/faststats/selections.php?#Output (accessed 19 January 2017).
Dinmohamed AG, Visser O, van Norden Y, Huijgens PC, Sonneveld P, van de Loosdrecht AA, et al. Trends in incidence, initial treatment and survival in myelodysplastic syndromes: a population-based study of 5144 patients diagnosed in the Netherlands from 2001 to 2010. Eur J Cancer. 2014;50:1004–12.
Neukirchen J, Schoonen WM, Strupp C, Gattermann N, Aul C, Haas R, et al. Incidence and prevalence of myelodysplastic syndromes: data from the Düsseldorf MDS-registry. Leuk Res. 2011;35:1591–6.
Ma X. Epidemiology of myelodysplastic syndromes. Am J Med. 2012;125(7 Suppl):S2–5.
Babushok DV, Bessler M. Genetic predisposition syndromes: when should they be considered in the work-up of MDS? Best Pract Res Clin Haematol. 2015;28:55–68.
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Steensma D, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16.
Pellagatti A, Boultwood J. The molecular pathogenesis of the myelodysplastic syndromes. Eur J Hematol. 2015;95:3–15.
Malcovati L, Karini M, Papaemmanuil E, Ambaglio I, Jädersten M, Jansson M, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126:233–41.
Haferlach T, Nagata Y, Grossman V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7.
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–95.
Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–10.
Thol F, Kade S, Schlamann C, Löffeld P, Morgan M, Krauter J, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119:3578–84.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9.
Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2011;44:53–7.
Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122:4021–34.
Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17:5–19.
Bejar R, Stevenson KE, Caughey B, Lindsley RC, Mar BG, Stojanov P, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol. 2014;32:2691–8.
Santamaria C, Ramos F, Puig N, Barragán E, de Paz R, Pedro C, et al. Simultaneous analysis of the expression of 14 genes with individual prognostic value in myelodysplastic syndrome patients at diagnosis: WT1 detection in peripheral blood adversely affects survival. Ann Hematol. 2012;91:1887–95.
Bejar R, Lord A, Stevenson K, Bar-Natan M, Pérez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705–12.
Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25:1153–8.
DiNardo CN, Jabbour E, Ravandi F, Takahashi K, Daver N, Routbort M, et al. IDH1 and IDH2 mutations in myelodysplastic syndromes and role in disease progression. Leukemia. 2016;30:980–4.
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506.
Chen TC, Hou HA, Chou WC, Tang JL, Kuo YY, Chen CY, et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 2014;4:e177.
Nazha A, Narkhede MS, Radivoyevitch T, Kalaycio M, Patel BJ, Gerds AT, et al. The revised International Prognostic Scoring System “molecular” (IPSS-Rm), a validated and dynamic model in treated patients with myelodysplastic syndromes (MDS). Blood. 2015;126:607.
Harada H, Harada Y. Recent advances in myelodysplastic syndromes: molecular pathogenesis and its implications for targeted therapies. Cancer Sci. 2015;106:329–36.
Bejar R, Papaemmanuil E, Haferlach T, Garcia-Manero G, Maciejewski JP, Sekeres MA, et al. Somatic mutations in MDS patients are associated with clinical features and predict prognosis independent of the IPSS-R: analysis of combined datasets from the International Working Group for Prognosis in MDS – Molecular Committee. Blood 2015; 126: Abstract 907.
Mallo M, del Rey M, Ibáñez M, Calasanz MJ, Arenillas L, Larráyoz MJ, et al. Response to lenalidomide in myelodysplastic syndromes with del(5q): influence of cytogenetics and mutations. Br J Haematol. 2013;162:74–86.
Zhou T, Chen P, Gu J, Bishop AJ, Scott LM, Hasty P, et al. Potential relationship between inadequate response to DNA damage and development of myelodysplastic syndrome. Int J Mol Sci. 2015;16:966–89.
Valka J, Vesela J, Votavova H, Jonasova A, Cermak J, Belickova M. Differential expression of the homologous recombination DNA repair genes in early and advanced stages of myelodysplastic syndrome. Blood 2016; 128: Abstract 5513.
Mufti GJ, Figes A, Hamblin TJ, Oscier DG, Copplestone JA. Immunological abnormalities in myelodysplastic syndromes. I. Serum immunoglobulins and autoantibodies. Br J Haematol. 1986;63:143–7.
Komrokji RS, Kulasekaraj A, Al Ali NH, Kordasti S, Bart-Smith E, Craig BM, et al. Autoimmune diseases and myelodysplastic syndromes. Am J Hematol. 2016;91:E280–3.
Aggarwal S, van de Loosdrecht AA, Alhan C, Ossenkoppele GJ, Westers TM, Bontkes HJ. Role of immune responses in the pathogenesis of low-risk and high-risk MDS: implications for immunotherapy. Br J Haematol. 2011;153:568–581.
Kordasti SY, Afzali B, Lim Z, Ingram W, Hayden J, Barber L, et al. IL-17-producing CD4(+) T cells, pro-inflammatory cytokines and apoptosis are increased in low risk myelodysplastic syndrome. Br J Haematol. 2009;145:64–72.
Kahn JD, Chamuleau MED, Westers TM, Van de Ven PM, van Dreunen L, van Spronsen M, et al. Regulatory T cells and progenitor B cells are independent prognostic predictors in lower risk myelodysplastic syndromes. Haematologica. 2015;100:e220–2.
Sallman DA, Cluzeau T, Basiorka AA, List A. Unraveling the pathogenesis of MDS: The NLRP3 inflammasome and pyroptosis drive the MDS phenotype. Front Oncol. 2016;6:151.
Zambetti NA, Ping Z, Chen S, Kenswil KJ, Mylona MA, Sanders MA, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19:613–27.
Mufti GJ, Bennett JM, Goasguen J, Bain BJ, Baumann I, Brunning R, et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica. 2008;93:1712–7.
Invernizzi R, Quaglia F, Porta MG. Importance of classical morphology in the diagnosis of myelodysplastic syndrome. Mediterr J Hematol Infect Dis. 2015;7:e2015035.
Malcovati L, Hellström-Lindberg E, Bowen D, Adès L, Cermak J, Del Cañizo C, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122:2943–64.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Germing U, Strupp C, Giagounidis A, Haas R, Gattermann N, Starke C, et al. Evaluation of dysplasia through detailed cytomorphology in 3156 patients from the Düsseldorf Registry on myelodysplastic syndromes. Leuk Res. 2012;36:727–34.
de Swart L, Smith A, MacKenzie M, Symeonidis A, Neukirchen J, Mikulenková D, et al. Cytomorphology review of 100 newly diagnosed lower-risk MDS patients in the European LeukemiaNet MDS (EUMDS) registry reveals a high inter-observer concordance. Ann Hematol. 2017;96:1105–12.
Bennett JM, Tuechler H, Aul C, Strupp C, Germing U. Dysplastic erythroid precursors in the myelodysplastic syndromes and the acute myeloid leukemias: is there biologic significance? (How should blasts be counted?). Leuk Res. 2016;47:63–69.
Wang SA, Patel KP, Pozdnyakova O, Peng J, Zuo Z, Dal Cin P, et al. Acute erythroid leukemia with <20% bone marrow blasts is clinically and biologically similar to myelodysplastic syndrome with excess blasts. Mod Pathol. 2016;29:1221–31.
Patnaik MM, Tefferi A. Refractory anemia with ring sideroblasts and RARS with thrombocytosis. Am J Hematol. 2015;90:549–59.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51.
Strupp C, Nachtkamp K, Hildebrandt B, Giagounidis A, Haas R, Gattermann N, et al. New proposals of the WHO working group (2016) for the diagnosis of myelodysplastic syndromes (MDS): characteristics of refined MDS types. Leuk Res. 2017;57:78–84.
Ogata K, Della Porta MG, Malcovati L, Picone C, Yokose N, Matsuda A, et al. Diagnostic utility of flow cytometry in low-grade myelodysplastic syndromes: a prospective validation study. Haematologica. 2009;94:1066–74.
van de Loosdrecht AA, Alhan C, Béné MC, Della Porta MG, Dräger AM, Feuillard J, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica. 2009;94:1124–34.
Westers TM, Ireland R, Kern W, Alhan C, Balleisen JS, Bettelheim P, et al. Standardization of flow cytometry in myelodysplastic syndromes: a report from an international consortium and the European LeukemiaNet Working Group. Leukemia. 2012;26:1730–41.
Porwit A, van de Loosdrecht AA, Bettelheim P, Brodersen LE, Burbury K, Cremers E, et al. Revisiting guidelines for integration of flow cytometry results in the WHO classification of myelodysplastic syndromes-proposal from the International/European LeukemiaNet Working Group for Flow Cytometry in MDS. Leukemia. 2014;28:1793–8.
Cremers EM, Westers TM, Alhan C, Cali C, Wondergem MJ, Poddighe PJ, et al. Multiparameter flow cytometry is instrumental to distinguish myelodysplastic syndromes from non-neoplastic cytopenias. Eur J Cancer. 2016;54:49–56.
Della Porta MG, Picone C, Pascutto C, Malcovati L, Tamura H, Handa H, et al. Multicenter validation of a reproducible flow cytometric score for the diagnosis of low-grade myelodysplastic syndromes: results of a European LeukemiaNET study. Haematologica. 2012;97:1209–17.
Alhan C, Westers TM, Cremers EMP, Cali C, Witte BI, Ossenkoppele GJ, et al. The myelodysplastic syndromes flow cytometric score: a three-parameter prognostic flow cytometric scoring system. Leukemia. 2016;30:658–65.
Valent P, Orazi A, Steensma DP, Ebert BL, Haase D, Malcovati L, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget. 2017;8:73483–500.
Westers TM, Cremers EMP, Oelschlaegel U, Johansson U, Bettelheim P, Matarraz S, et al. Immunophenotypic analysis of erythroid dysplasia in myelodysplastic syndromes. A report from the IMDSFlow working group. Haematologica. 2017;102:308–19.
Cremers EMP, Westers TM, Alhan C, Cali C, Visser-Wisselaar HA, Chitu DA, et al. Implementation of erythroid lineage analysis by flow cytometry in diagnostic models for myelodysplastic syndromes. Haematologica. 2017;102:320–6.
Aanei CM, Picot T, Tavernier E, Guyotat D, Campos Catafal L. Diagnostic utility of flow cytometry in myelodysplastic syndromes. Front Oncol. 2016;6:161.
Wang H, Chuhjo T, Yasue S, Omine M, Nakao S. Clinical significance of a minor population of paroxysmal nocturnal hemoglobinuria–type cells in bone marrow failure syndrome. Blood. 2002;100:3897–902.
Höchsmann B, Rojewski M, Schrezenmeier H. Paroxysmal nocturnal hemoglobinuria (PNH): higher sensitivity and validity in diagnosis and serial monitoring by flow cytometric analysis of reticulocytes. Ann Hematol. 2011;90:887–99.
Movalia MK, Weitz IC, Lim SH, Illingworth A. Incidence of PNH clones by diagnostic code utilizing high sensitivity flow cytometry. Blood 2011; 118: Abstract 1033.
Wang SA, Pozdnyakova O, Jorgensen JL, Medeiros LJ, Stachurski D, Anderson M, et al. Detection of paroxysmal nocturnal hemoglobinuria clones in patients with myelodysplastic syndromes and related bone marrow diseases, with emphasis on diagnostic pitfalls and caveats. Haematologica. 2009;94:29–37.
Borowitz MJ, Craig FE, DiGiuseppe JA, Illingworth AJ, Rosse W, Sutherland DR, et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom. 2010;78:211–30.
Al-Ani F, Chin-Yee I, Lazo-Langner A. Eculizumab in the management of paroxysmal nocturnal hemoglobinuria: patient selection and special considerations. Ther Clin Risk Manag. 2016;12:1161–70.
Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110:4385–95.
Godley LA, Larson RA. Therapy-related myeloid leukemia. Semin Oncol. 2008;35:418–429.
Pitchford CW, Hettinga AC, Reichard KK. Fluorescence in situ hybridization testing for -5/5q, -7/7q,+8, and del(20q) in primary myelodysplastic syndrome correlates with conventional cytogenetics in the setting of an adequate study. Am J Clin Pathol. 2010;133:260–4.
Braulke F, Platzbecker U, Müller-Thomas C, Götze K, Germing U, Brümmendorf TH, et al. Validation of cytogenetic risk groups according to International Prognostic Scoring Systems by peripheral blood CD34+FISH: results from a German diagnostic study in comparison with an international control group. Haematologica. 2015;100:205–13.
Hosokawa K, Katagiri T, Sugimori N, Ishiyama K, Sasaki Y, Seiki Y, et al. Favorable outcome of patients who have 13q deletion: a suggestion for revision of the WHO ‘MDS-U’ designation. Haematologica. 2012;97:1845–9.
Bacher U, Schanz J, Brauke F, Haase D. Rare cytogenetic abnormalities in myelodysplastic syndromes. Mediterr J Hematol Infect Dis. 2015;7:e2015034.
Haase D. Cytogenetic features in myelodysplastic syndromes. Ann Hematol. 2008;87:515–26.
Stevens-Kroef MJ, Olde Weghuis D, Elldrissi-Zaynoun N, van der Reijden B, Cremers EMP, Alhan C, et al. Genomic array as compared to karyotyping in myelodysplastic syndromes in a prospective clinical trial. Genes Chromosomes Cancer. 2017;56:524–34.
Thiel A, Beier M, Ingenhag D, Servan K, Hein M, Moeller V, et al. Comprehensive array CGH of normal karyotype myelodysplastic syndromes reveals hidden recurrent with individual genomic copy number alterations with prognostic relevance. Leukemia. 2011;25:387–99.
Starczynowski DT, Vercauteren S, Telenius A, Sung S, Tohyama K, Brooks-Wilson A, et al. High-resolution whole genome tiling path array CGH analysis of CD34+ cells from patients with low-risk myelodysplastic syndromes reveals cryptic copy number alterations and predicts overall and leukemia-free survival. Blood. 2008;112:3412–24.
Mohamedali A, Gäken J, Twine NA, Ingram W, Westwood N, Lea NC, et al. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood. 2007;110:3365–73.
Tiu RV, Gondek LP, O’Keefe CL, Elson P, Huh J, Mohamedali A, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011;117:4552–60.
Mohamedali AM, Alkhatabi H, Kulasekararaj A, Shinde S, Mian S, Malik F, et al. Utility of peripheral blood for cytogenetic and mutation analysis in myelodysplastic syndrome. Blood. 2013;122:567–570.
Zhang L, Padron E, Lancet J. The molecular basis and clinical significance of genetic mutations identified in myelodysplastic syndromes. Leuk Res. 2015;39:6–17.
Della Porta MG, Travaglino E, Boveri E, Ponzoni M, Malcovati L, Papaemmanuil E, et al. Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes. Leukemia. 2015;29:66–75.
Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30:3376–82.
Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376:536–47.
Mian SA, Smith AE, Kulasekararaj AG, Kizilors A, Mohamedali AM, Lea NC, et al. Spliceosome mutations exhibit specific associations with epigenetic modifiers and proto-oncogenes mutated in myelodysplastic syndrome. Haematologica. 2013;98:1058–66.
Patnaik MM, Hanson CA, Sulai NH, Hodnefield JM, Knudson RA, Ketterling RP, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119:5674–7.
Woll PS, Kjällquist U, Chowdhury O, Doolittle H, Wedge DC, Thongiuea S, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. 2014;25:794–808.
Platzbecker U, Germing U, Götze KS, Kiewe P, Mayer K, Chromik J, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18:1338–47.
Bejar R. Myelodysplastic syndromes diagnosis: what is the role of molecular testing? Curr Hematol Malig Rep. 2015;10:282–91.
Vas V, Wandhoff C, Dörr K, Niebel A, Geiger H. Contribution of an aged microenvironment to aging-associated myeloproliferative disease. PLoS ONE. 2012;7:e31523.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Steensma DP. Myelodysplastic syndromes: diagnosis and treatment. Mayo Clin Proc. 2015;90:969–83.
Valent P, Horny HP, Bennett JM, Fonatsch C, Germing U, Greenberg P, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: consensus statements and report from a working conference. Leuk Res. 2007;31:727–36.
Kwok B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. 2015;126:2355–61.
Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129:3371–8.
Bennett JM, Orazi A. Diagnostic criteria to distinguish hypocellular acute myeloid leukemia from hypocellular myelodysplastic syndromes and aplastic anemia: recommendations for a standardized approach. Haematologica. 2009;94:264–8.
Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35–47.
Kulasekararaj AG, Jang J, Smith AE, Mohamedali AM, Mian S, Gandhi S, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124:2698–704.
Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N, et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest. 2013;123:4595–611.
Chirnomas SD, Kupfer GM. The inherited bone marrow failure syndromes. Pediatr Clin North Am. 2013;60:1291–310.
Montpetit AJ, Alhareeri AA, Montpetit M, Starkweather AR, Elmore LW, Filler K, et al. Telomere length: a review of methods for measurement. Nurs Res. 2014;63:289–99.
Ghemlas I, Li H, Zlateska B, Klaassen R, Fernandez CV, Yanofsky RA, et al. Improving diagnostic precision, care and syndrome definitions using comprehensive next-generation sequencing for the inherited bone marrow failure syndromes. J Med Genet. 2015;52:575–84.
Thota S, McMahon S, Przychodzen B, LaFramboise T, Makishima H, Sekeres MA et al. Comprehensive identification of germline alterations in telomerase complex genes by whole exome sequencing of MDS and related myeloid neoplasms. Blood 2013; 122: Abstract 522.
Vannier J-B, Pavicic-Kaltenbrunner V, Retalcorin MIR, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149:795–806.
Ballew BJ, Joseph V, De S, Sarek G, Vannier JB, Stracker T, et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 2013;9:e1003695.
Marsh JC, Gutierrez-Rodrigues F, Cooper J, Jiang J, Gandhi SA, Kajigaya S, et al. Heterozygous RTEL1 variants in bone marrow failure and myeloid neoplasms. Blood Adv. 2018;2(1):36–48.
Oostra AB, Nieuwint AWM, Joenje H, de Winter JP. Diagnosis of Fanconi anemia: chromosomal breakage analysis. Anemia. 2012;2012:238731.
Rodrigues NP, Janzen V, Forkert R, Dombkowski DM, Boyd AS, Orkin SH, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106:477–84.
Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–7.
Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119:1283–91.
Lewinsohn M, Brown AL, Weinel LM, Phung C, Rafidi G, Lee MK, et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood. 2016;127:1017–23.
Li R, Sobreira N, Witmer PD, Pratz KW, Braunstein EM. Two novel germline DDX41 mutations in a family with inherited myelodysplasia/acute myeloid leukemia. Haematologica. 2016;101:e228–31.
Polprasert C, Schulze I, Sekeres MA, Makishima H, Przychodzen B, Hosono N, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell. 2015;27:658–70.
Sakurai M, Kunimoto H, Watanabe N, Fukuchi Y, Yuasa S, Yamazaki S, et al. Impaired hematopoietic differentiation of RUNX1-mutated induced pluripotent stem cells derived from FPD/AML patients. Leukemia. 2014;28:2344–54.
Churpek JE, Pyrtel K, Kanchi KL, Shao J, Koboldt D, Miller CA, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015;126:2484–90.
Pippucci T, Savoia A, Perrotta S, Pujol-Moix N, Noris P, Castegnaro G, et al. Mutations in the 5′ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Human Genet. 2011;88:115–20.
Noris P, Biino G, Pecci A, Civaschi E, Savoia A, Seri M, et al. Platelet diameters in inherited thrombocytopenias: analysis of 376 patients with all known disorders. Blood. 2014;124:e4–10.
Bannon SA, DiNardo CD. Hereditary predispositions to myelodysplastic syndrome. Int J Mol Sci. 2016;17:838.
Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48:792–7.
Schwartz JR, Wang S, Ma J, Lamprecht T, Walsh M, Song G, et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia. 2017;31:1827–30.
Acknowledgements
The authors received editorial and writing support provided by Victoria Edwards, PhD, of Excerpta Medica, supported by Celgene Corporation. The authors are fully responsible for content and editorial decisions for this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
GJM has received honoraria for speakers bureau/advisory boards for Celgene Corporation and Novartis. DPM has received honoraria for speakers bureau/advisory boards for JAZZ Pharmaceuticals, Novartis, and Gilead. AAvdL has received honoraria for participating in advisory boards for Janssen, Amgen, Celgene Corporation, and Novartis, as well as research support from Celgene Corporation and Alexion. UG has received speakers honoraria from Celgene Corporation, Janssen, and Novartis and institutional research support from Celgene Corporation and Novartis. RPH has received an honorarium for consulting for Celgene Corporation.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mufti, G.J., McLornan, D.P., van de Loosdrecht, A.A. et al. Diagnostic algorithm for lower-risk myelodysplastic syndromes. Leukemia 32, 1679–1696 (2018). https://doi.org/10.1038/s41375-018-0173-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0173-2
This article is cited by
-
Significant improvement of bone marrow-derived MSC expansion from MDS patients by defined xeno-free medium
Stem Cell Research & Therapy (2023)
-
Management of patients with lower-risk myelodysplastic syndromes
Blood Cancer Journal (2022)
-
RUNX1 mutations contribute to the progression of MDS due to disruption of antitumor cellular defense: a study on patients with lower-risk MDS
Leukemia (2022)
-
A novel automated image analysis system using deep convolutional neural networks can assist to differentiate MDS and AA
Scientific Reports (2019)
