
Abstract
Pharmacological mobilization of hematopoietic stem progenitor cells (HSPCs) from bone marrow (BM) into peripheral blood (PB) is a result of mobilizing agent-induced “sterile inflammation” in the BM microenvironment due to complement cascade (ComC) activation. Here we provide evidence that ATP, as an extracellular nucleotide secreted in a pannexin-1-dependent manner from BM cells, triggers activation of the ComC and initiates the mobilization process. This process is augmented in a P2X7 receptor-dependent manner, and P2X7-KO mice are poor mobilizers. Furthermore, after its release into the extracellular space, ATP is processed by ectonucleotidases: CD39 converts ATP to AMP, and CD73 converts AMP to adenosine. We observed that CD73-deficient mice mobilize more HSPCs than do wild-type mice due to a decrease in adenosine concentration in the extracellular space, indicating a negative role for adenosine in the mobilization process. This finding has been confirmed by injecting mice with adenosine along with pro-mobilizing agents. In sum, we demonstrate for the first time that purinergic signaling involving ATP and its metabolite adenosine regulate the mobilization of HSPCs. Although ATP triggers and promotes this process, adenosine has an inhibitory effect. Thus, administration of ATP together with G-CSF or AMD3100 or inhibition of CD73 by small molecule antagonists may provide the basis for more efficient mobilization strategies.
Similar content being viewed by others


Introduction
Hematopoietic stem/progenitor cells (HSPCs) circulate under steady-state conditions in peripheral blood (PB), and their number increases during inflammation, tissue or organ injuries, and after administration of pro-mobilizing drugs, such as granulocyte colony-stimulating factor (G-CSF) or an antagonist of the CXCR4 receptor, AMD3100 (plerixafor) [1,2,3,4,5,6,7]. One of the problems with clinical mobilization of patients as donors of HSPCs for transplantation is the fact that a significant percentage of patients are poor mobilizers, and more efficient mobilization strategies are needed. Therefore, to develop better mobilization strategies, we have to better understand the mobilization process at the molecular and cellular levels.
During mobilization, HSPCs are released from their bone marrow (BM) niches and migrate across the BM–PB endothelial barrier in BM sinusoids. This process is regulated by several redundant mechanisms, but, as we have proposed, activation of the complement cascade (ComC) through the mannan-binding lectin (MBL)-dependent pathway has a crucial role [8, 9]. Here we suggest the novel view that mobilization is due to the release from cells of extracellular nucleotides (EXNs), mainly ATP, that activate the ComC and purinergic signaling receptors in the BM microenvironment [1]. As ATP is an important danger-associated molecular pattern (DAMP) molecule recognized by MBL, it provides an important link between purinergic signaling and ComC activation as a trigger of “sterile inflammation” in the BM microenvironment [8, 10].
Purinergic signaling is a form of extracellular signaling mediated mainly by ATP and its metabolite adenosine. The purinergic signaling system has been found in bacteria, yeast, insects, and vertebrates, and purinergic receptors, represented by the P1, P2X, and P2Y receptor families, are among the most abundant receptors in living organisms [11]. EXNs, and in particular ATP and adenosine, have been reported to promote proliferation of HSPCs and the trafficking of granulocytes and monocytes and inhibit proliferation and migration of leukemic cells [12,13,14,15]. Interestingly, a related member of the EXN family and a metabolite in glycogen synthesis, UDP-glucose, has been reported to induce mobilization of HSPCs [9, 16].
Hematopoietic stem cells express several receptors for nucleotide- and nucleoside-based EXNs, which belong to two different purinergic receptor families, P2 and P1 [11, 17, 18]. The P2 family includes eight receptors that have been identified so far (P2Y1, 2, 4, 6, 11, 12, 13, and 14), which are G protein-coupled receptors that respond to stimulation by ATP, ADP, UTP, or UDP, depending on the receptor subtype. The P2X ionotropic channel receptor family consists of seven members (P2X1, 2, 3, 4, 5, 6, and 7), which are activated by ATP [11, 18, 19]. The P1 receptor family consists of four G protein-coupled receptor subtypes, A1, A2A, A2B, and A3, which are activated by adenosine [18,19,20].
We recently became interested in the role of ATP in the mobilization of HSPCs. Our interest was prompted by discovery of the role of ATP as a DAMP molecule in activation of mannan-binding lectin (MBL) pathway activation of the ComC but also as a major mediator of purinergic signaling within the BM microenvironment [1, 8, 9]. We demonstrate for first time that purinergic signaling involving ATP and its metabolite adenosine have an important role in the egress of HSPCs from BM niches into PB. Moreover, these mediators have opposite effects on the mobilization of HSPCs. Although ATP triggers this process, adenosine inhibits it by acting as a negative feedback molecule in the process of ATP degradation by the ectonucleotidases CD39 and CD73. Thus, administration of ATP or inhibition of CD73 by small molecule antagonists of this cell-surface-expressed enzyme may provide the basis for new and more efficient mobilization strategies.
Material and methods
Animals
Pathogen-free, 4–6-week-old C57BL/6J wild-type (WT), B6.129P2-P2rx7tm1Gab/J (P2X7–/–), and B6.129S1-Nt5etm1Lft/J (CD73–/–) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) at least 2 weeks prior to experiments. Animal studies were approved by the Animal Care and Use Committee of the University of Louisville (Louisville, KY, USA).
Detection of PANX1 by western blot
Murine cells were isolated from pathogen-free C57BL/6 mice, suspended in BD Pharm Lyse buffer (BD Biosciences, San Jose, CA, USA) to remove RBCs, and washed and resuspended in RPMI medium. An aliquot of mononuclear cells (MNCs, 1.5 × 106) were used for protein extraction. The remainder of the MNCs was expanded to grow stroma. After the cells reached confluence, they were trypsinized, and 1 × 106 stromal cells (SCs) were used for protein extraction. Cord blood cells were enriched for light-density MNCs by Ficoll–Paque centrifugation. The MNCs (1.5 × 106) were enriched for CD34+ cells by magnetic separation on an autoMACS separator according to the manufacturer’s protocol (Miltenyi Biotec, Auburn, CA, USA) and used for protein extraction. Briefly, cells were lysed for 30 min on ice in RIPA lysis buffer (Santa Cruz Biotechnology, Dallas, TX, USA) containing protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO, USA). Equal amounts of protein (15 μg) were separated on a 12% SDS-PAGE gel and transferred to a PVDF membrane. The PANX1 level was detected by incubation with primary mouse monoclonal anti-PANX1 antibody (R&D systems, Minneapolis, MN, USA), followed by incubation with an HRP-conjugated goat anti-mouse secondary antibody (Cell Signaling, Danvers, MA, USA). Equal loading in the lanes was evaluated by stripping the blots and reprobing with primary rabbit anti-β-actin antibody (Novus Biologicals, Littleton, CO, USA), followed by incubation with HRP-conjugated goat anti-rabbit secondary antibody (Cell Signaling). The membranes were developed with enhanced chemiluminescence (ECL) reagent (Amersham Life Sciences, Arlington Heights, IL, USA) and subsequently exposed to film (Hyperfilm; Amersham Life Sciences).
Isolation of Gr-1+ cells
Gr-1+ cells were isolated from the BM of adult mice as described [21, 22]. Briefly, BM was flushed from femurs, and the population of total nucleated cells was obtained after lysis of red blood cells (RBCs) using 1×BD Pharm Lyse buffer (BD Pharmingen, San Jose, CA, USA). The cells were subsequently stained with phycoerythrin (PE)–anti-Gr-1 antibody (anti-Ly-6G and Ly-6C, clone RB6-8C5) for 30 min in medium containing 2% fetal bovine serum (FBS). The cells were then washed, resuspended in RPMI-1640 medium and sorted using a Moflo XDP cell sorter (Beckman Coulter, Indianapolis, IN, USA) as populations of granulocytes (SSChighGr-1+).
Measuring ATP levels
Gr-1+ cells were resuspended in RPMI-1640 medium plus 0.5% bovine serum albumin (BSA, 2 ml cells/400 μl medium) and incubated overnight at 37 °C. Subsequently, cells were stimulated by adding G-CSF (100 ng/ml), AMD3100 (3 μM), or G-CSF + probenecid (100 µM) and incubated for 12 h at 37 °C. The cells were centrifuged, and conditioned media (CM) were collected. The ATP levels were measured using the ATP Colorimetric/Fluorometric Assay kit and the Deproteinizing Sample Preparation kit (BioVision, Milpitas, CA, USA), according to the manufacturer’s protocol. Fluorescence analysis was performed with Ex/Em set at 535/585 nm.
In vivo mobilization studies
Mice were mobilized with G-CSF (Amgen, Thousand Oaks, CA, USA) for 3 days (short mobilization) or 6 days (long mobilization) at 100 μg/kg/day by subcutaneous injection (SC); with AMD3100 (Sigma-Aldrich, St. Louis, MO, USA) for 1 day at 5 mg/kg by intraperitoneal injection (IP); or with UDP-glucose (Sigma-Aldrich) for 6 days at 200 mg/kg by SC. In some cases mice received ATP (3 days, 15 mg/kg, IP), probenecid (4 days, 200 mg/kg, IP), adenosine (3 days, 5 mg/kg, IP), or Brilliant Blue G (3 days, 50 mg/kg, IP). At 6 h after the last G-CSF injection, 1 h after AMD3100 injection, or 4 h after the last UDP-glucose injection, the mice were bled from the retro-orbital plexus for plasma and hematology analysis, and PB was obtained from the vena cava (with a 25-gauge needle and 1-ml syringe containing 250 U heparin). MNCs were obtained by hypotonic lysis of RBCs in BD Pharm Lyse buffer (BD Biosciences) as described [23,24,25,26,27,28].
Fluorescence-activated cell sorting (FACS) analysis
For staining of Lin−/Sca-1+/c-Kit+ (SKL) cells and Lin−/Sca-1+/CD45+ hematopoietic stem cells (HSCs), the following monoclonal antibodies were used: FITC–anti-CD117 (also known as c-Kit, clone 2B8; BioLegend, San Diego, CA, USA) and PE–Cy5–anti-mouse Ly-6 A/E (also known as Sca-1, clone D7; eBioscience, San Diego, CA, USA). All anti-mouse lineage marker antibodies were purchased from BD Biosciences and conjugated with PE [29], including: anti-CD45R (also known as B220, clone RA3-6B2), anti-Ter-119 (clone TER-119), anti-CD11b (clone M1/70), anti-T cell receptor β (clone H57-597), anti-Gr-1 (clone RB6-8C5), anti-TCRγδ (clone GL3), and anti-CD45 (clone 30-F11) [28, 30]. Staining was performed in RPMI-1640 medium containing 2% FBS. All monoclonal antibodies were added at saturating concentrations, and the cells were incubated for 30 min on ice, washed twice, and analyzed with an LSR II flow cytometer (BD Biosciences).
Evaluation of HSPC mobilization
For evaluation of circulating colony-forming unit-granulocyte/macrophage (CFU-GM) and SKL cells, the following formulas were used: (number of white blood cells [WBCs] × number of CFU-GM colonies)/number of WBCs plated = number of CFU-GM per ml of PB; and (number of WBCs × number of SKL cells)/number of gated WBCs = number of SKL cells per μl of PB [23, 24, 26, 27].
PB parameter counts
To obtain white and red blood cell counts, 50 μl of PB was taken from the retro-orbital plexus of mice into microvette EDTA-coated tubes (Sarstedt Inc., Newton, NC, USA) and run on a HemaVet 950FS hematology analyzer (Drew Scientific Inc., Oxford, CT, USA) within 2 h of collection [23, 25].
Generation of hematopoietic chimeras
Recipient mice were irradiated with a lethal dose of irradiation (1000cGy), and 24 h later they were transplanted through the retro-orbital plexus with 5 × 106 BM-MNCs from donor mice. Animals that had undergone transplantation were allowed to recover for 9 weeks before experiments.
Transwell migration assay
Medium (650 μl of RPMI-1640 medium plus 0.5% BSA) containing SDF-1 (10 ng/ml; Pepro Tech, Rocky Hill, NJ, USA), sphingosine-1-phosphate (S1P; 0.1 μM; Cayman Chemical Company, Ann Arbor, MI, USA), ceramide-1-phosphate (C1P; 100 µM; Sigma-Aldrich), or ATP (0.25 ng/ml; Sigma-Aldrich) was added to the lower chambers of a Costar Transwell 24-well plate (Corning Costar, Cambridge, MA, USA). Aliquots of murine BM-MNCs resuspended in assay medium (1 × 106 cells/100 μl) were loaded onto the upper chambers with 5-μm-pore filters and then incubated for 3 h (37 °C, 5% CO2). An aliquot of cells from the lower chambers was harvested and counted by FACS analysis. The cells were gated according to their forward scatter (FSC) and side scatter (SSC) parameters and counted during a 30-s acquisition at a high flow rate. The remaining cells were resuspended in human methylcellulose base medium provided by the manufacturer (R&D Systems), supplemented with murine GM-CSF (25 ng/ml) and IL-3 (10 ng/ml), for determining the number of CFU-GM colonies. Cultures were incubated for 7 days (37 °C, 95% humidity, and 5% CO2), at which time the colonies were counted under an inverted microscope [26, 27, 30].
Fibronectin cell-adhesion assay
Human and murine cell lines at a density of 5 × 104/100 µl were made quiescent overnight or for 3 h at 37 °C in RPMI-1640 medium containing 0.5% BSA. Subsequently, the cells were incubated with either probenecid (P8761, Sigma-Aldrich) or the pannexin-1 peptide inhibitor 10panx (WRQAAFVDSY, 200 µM; Bio-Techne) for 1 h at 37 °C. The samples was then incubated for an additional hour at 37 °C with either ATP (250 ng/ml) or adenosine (250 ng/ml). The cells were then added directly to 96-well plates that had been previously incubated overnight at 4°C with fibronectin (10 µg/ml) and later blocked with medium containing 0.05% BSA for 2 h. After a 5 min incubation at room temperature, non-adherent cells were washed from the wells, and the remaining cells counted using an inverted microscope [30].
Real-time quantitative reverse-transcription PCR
Total RNA of murine BM-MNCs was isolated with the RNeasy Kit (Qiagen, Valencia, CA, USA). The RNA was reverse-transcribed with MultiScribe reverse transcriptase and oligo-dT primers (Applied Biosystems, Foster City, CA, USA). Quantitative assessment of mRNA levels was done by real-time RT-PCR using an ABI 7500 instrument with Power SYBR Green PCR Master Mix reagent. PCR conditions were as follows: 95 °C (15 s), 40 cycles at 95 °C (15 s), and 60 °C (1 min). According to melting point analysis, only one PCR product was amplified under these conditions. The relative quantity of a target, normalized to the endogenous β2 microglobulin gene as control and relative to a calibrator, is expressed as 2–DDCt (fold difference), where Ct is the threshold cycle, DCt = (Ct of target genes) − (Ct of the endogenous control gene, β-microglobulin), and DDCt = (DCt of samples for the target gene) − (DCt of the calibrator for the target gene). The following primer pair was used for analysis of heme oxygenase-1 (HO-1) expression: 5′-AGGTACACATCCAAGCCGAGAA-3′ and 5′-CTCTGGACACTGACCCTTCTG-3′.
Statistical analysis
All results are presented as mean ± SD. Statistical analysis of the data was done using Student’s t-test for unpaired samples (Excel, Microsoft Corp., Redmond, WA, USA) with a value of p ≤ 0.05 considered significant.
Results
ATP is released through the pannexin-1 channel, acting as a DAMP molecule to trigger mobilization of HSPCs in a ComC activation-dependent manner
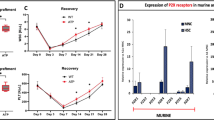
ATP is the most important DAMP molecule, and, as we reported in our previous work, ATP and other DAMPs are secreted from granulocytes and monocytes in the BM microenvironment when stimulated by G-CSF or AMD3100, activating the ComC through the mannan-binding lectin (MBL)–mannan-associated serum protease (MASP)-dependent pathway to trigger the mobilization process [1, 8, 9]. ATP is released from cells, mainly through pannexin channels [31], and we first evaluated the expression of pannexin-1 (PANX1) on the surface of murine and human mononuclear cells and cells enriched for HSPCs as well as the level of free ATP in conditioned media from BM cells stimulated by G-CSF or AMD3100 (Fig. 1a). We observed expression of PANX1 by western blot of the studied cell types as well as the presence of ATP in media conditioned by BM cells. Furthermore, secretion of ATP was inhibited after exposure to the PANX1-blocking agent probenecid [32] (Fig. 1a).

The impact of ATP on HSPC mobilization. a The expression of PANX1 in human cord blood and murine bone marrow cells was evaluated at the protein level by western blotting, and representative blots are shown. The same membranes were reprobed with β-actin to confirm equal loading of total protein. The ATP level was evaluated in conditioned medium from Gr-1+ cells from WT mice stimulated with G-CSF, AMD3100, or G-CSF + probenecid. Results are shown as the percentage of control cells. For mobilization studies, mononuclear cells were isolated from WT mice treated with ATP after 3 days of administration with G-CSF (b) or AMD3100 (c) or after 3 days of G-CSF together with probenecid (d). The numbers of WBCs, SKL (Sca-1+/c-kit+/Lin−) cells, HSCs (Sca-1+/CD45+/Lin−), and CFU-GM clonogenic progenitors were evaluated in PB. Results from two independent experiments are pooled together. *p ≤ 0.05
Next, we performed mobilization studies with induction by G-CSF + ATP (Fig. 1b) and AMD3100 + ATP (Fig. 1c) in normal wild-type (WT) mice and found that exogenous ATP additionally enhances the egress of HSPCs from BM into PB. This effect was significantly inhibited in mice treated with probenecid that is PANX1-blocking agent (Fig. 1d). In Supplementary Figure 1, it can be seen that a PANX1-specific blocking peptide as well as probenecid enhance adhesion of murine and human HSPCs and at the same time decrease migration not only in response to ATP but also to other important HSPC chemottractants, such as SDF-1, S1P, and C1P [33, 34]. This result suggests an autocrine involvement of ATP in the sensitization of HSPCs to chemoattractants, which requires further study.
ATP-mediated P2X7 receptor signaling is crucial for egress of HSPCs from BM into PB
P2X7 is a member of a purinergic ionotropic channel receptor family and is activated by extracellular ATP [35]. Interestingly, it has been postulated that the P2X7 receptor enhances calcium influx in response to ATP stimulation and thus co-operates with PANX1 in cell migration [31, 34]. To address the role of P2X7 in the mobilization process, we induced P2X7-KO mice with G-CSF for 3 or 6 days (Fig. 2a, b) or by AMD3100 (Fig. 2c). We found that these mice have a defect in mobilization in response to G-CSF but not AMD3100, which suggests a different involvement of P2X7 receptor in mobilization induced by these two different mobilizing agents. In fact we observed similar phenomenon in the past during mobilization of C2fB-deficent mice [25].

The impact of P2X7 signaling on HSPC mobilization. Mononuclear cells (MNCs) were isolated from WT and P2X7–/– mice after 3 days (a) or 6 days (b) of G-CSF mobilization or AMD3100 mobilization (c) or after 3 days of G-CSF mobilization performed on chimeric mice (d). The numbers of WBCs, SKL (Sca-1+/c-kit+/Lin−) cells, HSCs (Sca-1+/CD45+/Lin−), and CFU-GM clonogenic progenitors were evaluated in PB. Results from two independent experiments are pooled together. *p ≤ 0.05
Defective mobilization of HSPCs has also been observed in another type of experiment in which we blocked expression of P2X7 on the surface of hematopoietic cells with the P2X7 receptor blocking agent Brilliant Blue G (Supplementary Figure 2).
To address whether this poor G-CSF-mobilizing effect in P2X7-KO mice is due to the microenvironment or a hematopoietic stem cell-mediated defect, we created chimeric WT animals reconstituted with P2X7-KO bone marrow cells and P2X7-KO mice reconstituted with BM from WT mice (Fig. 2d). The poor mobilization effect was reproduced in WT mice reconstituted with P2X7 bone marrow cells, which demonstrates that the defect is dependent on defective expression of P2X7 receptors on the surface of hematopoietic cells.
To learn more about the role of P2X7 signaling in the mobilization of HSPCs, we measured the levels of extracellular ATP in conditioned media from the BM of mobilized WT and P2X7 mice and found that mobilizing agents increase secretion of ATP from BM cells (Fig. 3a). This finding suggests that interaction between the P2X7 and PANX1 receptors is more complex, most likely at the intracellular calcium signaling level [31], and, as is shown in Fig. 3b, BM-MNCs as well as CFU-GM progenitor cells from P2X7-KO mice show defective migration in response to crucial HSPC chemoattractants. This again suggests interplay between ATP and the P2X7 receptor in autocrine modulation of HSPC migration and requires further study. This phenomenon was reproduced by blocking the P2X7 receptor with Brilliant Blue G (Supplementary Figure 3).

The role of the P2X7 receptor in mobilization of HSPCs. a The ATP level was evaluated in conditioned medium from Gr-1+ cells from P2X7–/– mice stimulated with G-CSF, AMD3100, or G-CSF + probenecid. Results are shown as a percentage of control WT cells. b The chemotactic responsiveness of WT and P2X7–/–-derived BM-MNCs to SDF-1, S1P, C1P, and ATP gradients evaluated by FACS (left panel) and clonogenic CFU-GM progenitors (right panel). Results are combined from three independent experiments. *p ≤ 0.05. c For mobilization studies, MNCs were isolated from WT and P2X7–/– mice after 6 days of UDP-glucose mobilization. The numbers of WBCs, SKL (Sca-1+/c-kit+/Lin−) cells, HSCs (Sca-1+/CD45+/Lin−), and CFU-GM clonogenic progenitors were evaluated in PB. Results from two independent experiments are pooled together. *p ≤ 0.05
As mentioned in the introduction, it has been reported that infusion of UDP-glucose, which is a related member of the EXN family and a metabolite in glycogen synthesis, induces mobilization of HSPCs [16]. This has been postulated to be dependent on a specific interaction of UDP-glucose with a G protein-coupled member of the P2Y purinergic receptor family, P2Y14 [36]. To address the potential involvement of the P2X7 receptor in this phenomenon, we mobilized WT mice and P2X7-KO mice with UDP-glucose and found that UDP-glucose alone significantly increases the number of circulating HSPCs, both in WT and P2X7-KO mice (Fig. 3c). This finding supports the notion that UDP-glucose-mediated mobilization does not depend on the P2X7 receptor but involves the P2Y14 G protein-coupled receptor, as previously proposed [36].
Interestingly, despite the important role of P2X7 in mobilization, P2X7-KO mice have normal hematopoietic parameters in PB as well as normal numbers of clonogenic progenitors in BM under steady-state conditions (Supplementary Figure 4).
The ATP metabolite adenosine is a negative regulator of HSPC mobilization
On the basis of the facts that mobilization of HSPCs is the result of ComC-mediated sterile inflammation in BM [1, 9] and that adenosine has a well-known anti-inflammatory effect [37, 38], we became interested in the role of adenosine as an EXN in the mobilization process. First, we employed CD73-KO mice, which do not process extracellular ATP to adenosine [39, 40] and thus have low levels of this EXN in biological fluids. As shown (Fig. 4a), CD73-KO mice have significantly enhanced mobilization in response to G-CSF and AMD3100 (Fig. 4b). Furthermore, the reciprocal model of creating irradiation chimeras between CD73-KO and WT mice revealed that the observed CD73 defect depends on defective expression of CD73 on the surface of hematopoietic cells and not on the BM microenvironment (Fig. 4c). Finally, in direct mobilization experiments in WT animals we observed that mice injected with adenosine show defective HSPC mobilization by G-CSF (Fig. 5a) and AMD3100 (Fig. 5b).

The impact of CD73 ectonucleotidase on mobilization. Mononuclear cells (MNCs) were isolated from WT and CD73–/– mice after 3 days of G-CSF (a) or AMD3100 (b) mobilization or after 3 days of G-CSF mobilization performed on chimeric mice (c). The numbers of WBCs, SKL (Sca-1+/c-kit+/Lin−) cells, HSCs (Sca-1+/CD45+/Lin−), and CFU-GM clonogenic progenitors were evaluated in PB. Results from two independent experiments are pooled together. *p ≤ 0.05

Adenosine as a negative regulator of HSPC mobilization. Mononuclear cells (MNCs) were isolated from WT mice treated with adenosine after 3 days of G-CSF (a) or AMD3100 (b) mobilization. The numbers of WBCs, SKL (Sca-1+/c-kit+/Lin−) cells, HSCs (Sca-1+/CD45+/Lin−), and CFU-GM clonogenic progenitors were evaluated in PB. Results from two independent experiments are pooled together. c HO-1 expression was evaluated at the mRNA level by real-time PCR. Results from three independent experiments are pooled together. *p ≤ 0.05. d A proposed ATP-mediated, MBL-induced model for triggering sterile inflammation in BM. A mobilizing agent (e.g., G-CSF) induces secretion of DAMP molecules (e.g., ATP) and free radicals (ROS) from Gr-1+ cells (granulocytes or monocytes). ATP directly and ROS indirectly (by exposing neoepitope antigens in the BM microenvironment that are recognized by naturally occurring antibodies [Nabs] to form neoepitope–NAbs complexes) are recognized by MBL, which initiates activation of the ComC in a MASP-dependent manner. In parallel, ATP activates the coagulation cascade (CoaC). ATP is also metabolized to adenosine by the ectonucleotidases CD39 and CD73 as well as by E-NPP, which has the opposite effect and inhibits both sterile inflammation in BM as well as egress of HSPCs into PB. Egress of HSPCs is also inhibited by HO-1, which is an anti-inflammatory enzyme that inhibits the ComC
Interestingly, despite the important role of CD73 in mobilization, CD73-KO mice, as demonstrated in Supplementary Figure 5, have normal hematopoietic parameters in PB as well as normal numbers of clonogenic progenitors in BM under steady-state conditions.
On the basis of the fact that heme oxygenase-1 (HO-1) is a negative regulator of HSPC mobilization [24], we evaluated the effect of ATP, which, as we report here, stimulates mobilization, and the effect of adenosine, which inhibits this process and the expression of HO-1. As shown in Fig. 5c, BM-MNCs exposed to ATP downregulated the expression of HO-1 mRNA, and exposure to adenosine upregulated mRNA encoding this anti-inflammatory enzyme.
Discussion
The seminal observation of this paper is that EXNs, particularly their most significant members, ATP and its degradation metabolite adenosine [41], are important factors that combine purinergic signaling with innate immunity in triggering egress of HSPCs from BM into PB. Although ATP released in the pannexin-1-dependent pathway from Gr-1+ cells as DAMP molecules induces sterile inflammation in BM and activates the MBL-pathway of ComC activation and involves activation of P2X7 receptor, adenosine has the opposite, anti-inflammatory, and inhibitory effect (Fig. 5d).
The egress of HSPCs from BM into PB is still not well understood, despite the importance of this process in understanding the response of the organism to inflammation, tissue and organ injury, and pharmacological mobilization to obtain HSPCs for hematopoietic transplantation [1,2,3,4,5,6,7]. Several pathways and cells have been proposed to have a pivotal role in this phenomenon [1, 9, 24, 27, 28, 30, 33, 42, 43]. From an historical point of view, mobilization has been connected to induction of proteolytic activity in the hematopoietic microenvironment, and this effect seems to be mediated by several redundant proteolytic enzymes that attenuate retention axes of HSPCs in BM niches (e.g., SDF-1–CXCR4 and VLA-4–VCAM-1) and are involved in permeabilization of the BM–PB endothelial barrier [5, 26]. Recently, it has been demonstrated that the lipolytic enzyme PLC-β2, which disrupts the membrane lipid rafts required for CXCR4 and VLA-4 receptor-mediated retention of HSPCs in BM niches, also has a crucial role [28].
Important cellular players facilitating the mobilization process have been identified. It has been demonstrated in a few elegant papers that both Gr-1+ granulocytes and monocytes are required for egress of HSPCs into PB [44, 45]. Granulocytes have been reported to be a rich source of proteolytic [46] and lipolytic [28, 47] enzymes, being the first cells that transmigrate through the BM–PB endothelial barrier and pave the way for egress of HSPCs, which follow in their footsteps [25, 26, 48]. By contrast, monocytes secrete certain chemokines that participate in mobilization [49]. The involvement of another cell type that lines trabecular bone, monocyte-derived osteoclasts [50], is still disputed at present. Nevertheless, the egress of cells into blood involves the coordinated effort of many cell types residing in BM as well as β-adrenergic neural fibers [51].
Purinergic signaling was proposed almost 100 years ago by Albert Szent-Gyorgi, who reported the effect of intravenously injected adenine compounds on heart rate in experimental animals [52]. In 1972, Geoffrey Burnstock ignited decades of controversy by proposing the existence of a non-adrenergic, non-cholinergic neurotransmitter, which he identified as ATP, and after years of prolonged skepticism, the concept of EXNs as effectors of purinergic signaling is currently widely accepted as a general intercellular communication system [53]. EXNs, including nucleotides and nucleosides, bind to several receptors from the P1, P2X, and P2Y families. It is well established that stimulation of mammalian cells, including BM cells, leads to release of purine and pyrimidine nucleotides and nucleosides and activation of autocrine/paracrine feedback loops [54]. The most important players, however, are ATP and its metabolite adenosine [41].
As mentioned in the introduction, EXNs, in particular ATP and adenosine, have been reported to stimulate not only proliferation but also to regulate development of normal HSPCs [12,13,14,15], which opens a new chapter in the study of non-peptide-based factors regulating hematopoiesis. By contrast, UTP has been reported to be an inhibitor of malignant hematopoietic cell proliferation and migration [15]. Purinergic receptors, belonging to several families, are among the most abundant receptors in living organisms, and appeared early in evolution, over a billion years ago. They have been demonstrated to be expressed on the surface of HSPCs, all types of differentiated hematopoietic cells, BM SCs, and endothelium [11]. As ATP is an important mediator of signaling in neural fiber synapses [55], one has to consider their potential involvement in regulating, besides catecholamines [56], in release of HSPCs from BM niches.
In our work, we demonstrated that mobilizing agents induce secretion of ATP from BM cells in a pannexin-1 (PANX1)-dependent manner. Mice exposed to PANX1 inhibitors release fewer cells from BM into PB in response to G-CSF and AMD3100. Interestingly, infusion of additional ATP into WT mice augmented their responsiveness to G-CSF. By contrast, inhibition of PANX1 by probenecid impairs both ATP release and the mobilization process. In addition, both probenecid and a PANX1-blocking peptide decrease migration of clonogenic HSPCs, but both at the same time increase HSPC adhesion. Inhibition of ATP secretion also decreases activation of the ComC, which clearly shows that ATP acts as a DAMP molecule in the MBL-pathway of ComC activation.
As pannexin channels are not the only channels that release ATP into the extracellular space, future work is needed to see whether impaired mobilization in connexin-43-deficient mice [57] could be explained by impaired secretion of ATP as well. Another potential source of ATP is extracellular microvesicles that are released from activated cells [58]. Nevertheless, pannexin channels, and in particular PANX1, seem to be a key contributor to ATP release.
ATP secreted from the cells is processed to adenosine by several exonucleases in the extracellular space [39, 40]. As we have demonstrated, a lack of CD73, which metabolizes AMP to adenosine, enhances mobilization in CD73-KO mice, indicating that adenosine is an important inhibitor of mobilization. Corroborating this finding, mice exposed to adenosine along with G-CSF also turned out to be poor mobilizers. On the basis of these findings, adenosine, which is a well-known anti-inflammatory and immunosupressive nucleoside [37, 38], provides a negative feedback in mobilization triggered by ATP (Fig. 5d). Therefore, small molecule inhibitors of CD73 could be novel mobilization-potentiating agents, and this idea is currently being tested in our laboratory. As shown in Fig. 5d, mobilization is also inhibited by HO-1, and, as we observed, expression of this anti-inflammatory, ComC-inhibiting enzyme [24, 59] is downregulated by ATP and upregulated by adenosine.
The level of adenosine in the extracellular space is also regulated by nucleoside transporters, which are a group of membrane transport proteins that transport nucleoside substrates across the cell membrane [60]. Thus, in future it would be interesting to see how knockout of these transporters (the SLC28 concentrative nucleoside transporters and the SLC29 equilibrative nucleoside transporters) affect mobilization.
In conclusion, we have demonstrated for the first time that purinergic signaling involving ATP and its metabolite adenosine regulates mobilization of HSPCs. Thus, administration of ATP or inhibition of CD73 by small molecule antagonists may provide new strategies for developing more efficient mobilization. At the same time, we demonstrated that the most abundant EXN, ATP, provides an important link between purinergic signaling and activation of the ComC in BM and better explains the role of sterile inflammation in the egress of cells from the BM microenvironment into PB. Further studies are also needed if similar mechanism is involved in egress of other types of BM stem cells [61,62,63,64].
Change history
08 March 2019
Following the publication of this article, the authors noted that the following should be included in the Acknowledgements section: “MA is the recipient of a START scholarship (0785) from FNP”. The authors wish to apologise for any inconvenience caused.
References
Ratajczak MZ, Adamiak M, Plonka M, Abdel-Latif A, Ratajczak J. Mobilization of hematopoietic stem cells as a result of innate immunity-mediated sterile inflammation in the bone marrow microenvironment—the involvement of extracellular nucleotides and purinergic signaling. Leukemia. 2018. https://doi.org/10.1038/s41375-018-0087-z.
Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008.
Wojakowski W, Tendera M, Kucia M, Zuba-Surma E, Paczkowska E, Ciosek J, et al. Mobilization of bone marrow-derived Oct-4+SSEA-4+ very small embryonic-like stem cells in patients with acute myocardial infarction. J Am Coll Cardiol. 2009;53:1–9.
Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 1995;201:1307–18.
Link DC. Mechanisms of granulocyte colony-stimulating factor-induced hematopoietic progenitor-cell mobilization. Semin Hematol. 2000;37:25–32.
Matsunaga T, Sakamaki S, Kohgo Y, Ohi S, Hirayama Y, Niitsu Y. Recombinant human granulocyte colony-stimulating factor can mobilize sufficient amounts of peripheral blood stem cells in healthy volunteers for allogeneic transplantation. Bone Marrow Transplant. 1993;11:103–8.
Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002;527:255–62.
Adamiak M, Abdelbaset-Ismail A, Suszynska M, Abdel-Latif A, Ratajczak J, Ratajczak MZ. Novel evidence that the mannan-binding lectin pathway of complement activation plays a pivotal role in triggering mobilization of hematopoietic stem/progenitor cells by activation of both the complement and coagulation cascades. Leukemia. 2017;31:262–5.
Adamiak M, Ratajczak MZ. Innate immunity and mobilization of hematopoietic stem cells. Curr Stem Cell Rep. 2017;3:172–80.
Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37.
Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–92.
Jing L, Tamplin OJ, Chen MJ, Deng Q, Patterson S, Kim PG, et al. Adenosine signaling promotes hematopoietic stem and progenitor cell emergence. J Exp Med. 2015;212:649–63.
Shah D, Romero F, Stafstrom W, Duong M, Summer R. Extracellular ATP mediates the late phase of neutrophil recruitment to the lung in murine models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306:L152–L161.
Junger WG. Purinergic regulation of neutrophil chemotaxis. CMLS. 2008;65:2528–40.
Salvestrini V, Zini R, Rossi L, Gulinelli S, Manfredini R, Bianchi E, et al. Purinergic signaling inhibits human acute myeloblastic leukemia cell proliferation, migration, and engraftment in immunodeficient mice. Blood. 2012;119:217–26.
Kook S, Cho J, Lee SB, Lee BC. The nucleotide sugar UDP-glucose mobilizes long-term repopulating primitive hematopoietic cells. J Clin Invest. 2013;123:3420–35.
Feng W, Wang L, Zheng G. Expression and function of P2 receptors in hematopoietic stem and progenitor cells. Stem Cell Investig. 2015;2:14.
Kaebisch C, Schipper D, Babczyk P, Tobiasch E. The role of purinergic receptors in stem cell differentiation. Comput Struct Biotechnol J. 2015;13:75–84.
Boeynaems JM, Communi D, Gonzalez NS, Robaye B. Overview of the P2 receptors. Semin Thromb Hemost. 2005;31:139–49.
Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev. 2011;63:1–34.
Zhang D, Jiang X, Fang P, Yan Y, Song J, Gupta S, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation. 2009;120:1893–902.
Sunderkötter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–7.
Borkowska S, Suszynska M, Mierzejewska K, Ismail A, Budkowska M, Salata D, et al. Novel evidence that crosstalk between the complement, coagulation, and fibrinolysis proteolytic cascades is involved in mobilization of hematopoietic stem/progenitor cells (HSPCs). Leukemia. 2014;28:2148–54.
Wysoczynski M, Ratajczak J, Pedziwiatr D, Rokosh G, Bolli R, Ratajczak MZ. Identification of heme oxygenase 1 (HO-1) as a novel negative regulator of mobilization of hematopoietic stem/progenitor cells. Stem Cell Rev. 2015;11:110–8.
Lee HM, Wysoczynski M, Liu R, Shin DM, Kucia M, Botto M, et al. Mobilization studies in complement-deficient mice reveal that optimal AMD3100 mobilization of hematopoietic stem cells depends on complement cascade activation by AMD3100-stimulated granulocytes. Leukemia. 2010;24:573–82.
Lee HM, Wu W, Wysoczynski M, Liu R, Zuba-Surma EK, Kucia M, et al. Impaired mobilization of hematopoietic stem/progenitor cells in C5-deficient mice supports the pivotal involvement of innate immunity in this process and reveals novel promobilization effects of granulocytes. Leukemia. 2009;23:2052–62.
Ratajczak MZ, Lee H, Wysoczynski M, Wan W, Marlicz W, Laughlin MJ, et al. Novel insight into stem cell mobilization plasma sphingosine-1-phosphate is a major chemoattractant that directs the egress of hematopoietic stem progenitor cells from the bone marrow and its level in peripheral blood increases during mobilization due to activation of complement cascade/membrane attack complex. Leukemia. 2010;24:976–85.
Adamiak M, Poniewierska-Baran A, Borkowska S, Schneider G, Abdelbaset-Ismail A, Suszynska M, et al. Evidence that a lipolytic enzyme-hematopoietic-specific phospholipase C-b2-promotes mobilization of hematopoietic stem cells by decreasing their lipid raft-mediated bone marrow retention and increasing the promobilizing effects of granulocytes. Leukemia. 2015;30:919–28.
Golan K, Vagima Y, Ludin A, Itkin T, Cohen-Gur S, Kalinkovich A, et al. S1P promotes murine progenitor cell egress and mobilization via S1P1-mediated ROS signaling and SDF-1 release. Blood. 2012;119:2478–88.
Adamiak M, Abdelbaset-Ismail A, Moore JB, Zhao J, Abdel-Latif A, Wysoczynski M, et al. Inducible nitric oxide synthase (iNOS) is a novel negative regulator of hematopoietic stem/progenitor cell trafficking. Stem Cell Rev. 2017;13:92–103.
Sáez PJ, Vargas P, Shoji KF, Harcha PA, Lennon-Duménil AM, et al. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X7 receptors. Sci Signal. 2017;10:eaah7107.
Silverman W, Locovei S, Dahl G. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am J Physiol Cell Physiol. 2008;295:C761–C767.
Ratajczak MZ, Suszynska M, Borkowska S, Ratajczak J, Schneider G. The role of sphingosine-1-phosphate (S1P) and ceramide-1-phosphate (C1P) in the trafficking of normal and malignant cells. Expert Opin Ther Targets. 2014;18:95–107.
Ratajczak MZ, Suszynska M. Emerging strategies to enhance homing and engraftment of hematopoietic stem cells. Stem Cell Rev. 2016;12:121–8.
Volonté C, Apolloni S, Skaper SD, Burnstock G. P2X7 receptors: channels, pores and more. CNS Neurol Disord Drug Targets. 2012;11:705–21.
Abbracchio MP, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, et al. Characterization of the UDP-glucose receptor (re-named here the P2Y14 receptor) adds diversity to the P2Y receptor family. Trends Pharmacol Sci. 2003;24:52–55.
Da Rocha Lapa F, da Silva MD, de Almeida Cabrini D, Santos ARS. Anti-inflammatory effects of purine nucleosides, adenosine and inosine, in a mouse model of pleurisy: evidence for the role of adenosine A2 receptors. Purinergic Signal. 2012;8:693–704.
Haskó G, Cronstein B. Regulation of inflammation by adenosine. Front Immunol. 2013;4:85.
Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65.
Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–405.
Brake AJ, Julius D. Signaling by extracellular nucleotides. Annu Rev Cell Dev Biol. 1996;12:519–41.
Bonig H, Papayannopoulou T. Mobilization of hematopoietic stem/progenitor cells: general principles and molecular mechanisms. Methods Mol Biol. 2012;904:1–14.
Ratajczak MZ, Borkowska S, Ratajczak J. An emerging link in stem cell mobilization between activation of the complement cascade and the chemotactic gradient of sphingosine-1-phosphate. Prostaglandins Other Lipid Mediat. 2013;104:122–9.
Lévesque JP, Hendy J, Takamatsu Y, Williams B, Winkler IG, Simmons PJ. Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp Hematol. 2002;30:440–9.
Wysoczynski M, Adamiak M, Suszynska M, Abdel-Latif A, Ratajczak J, Ratajczak MZ. Poor mobilization in T-cell-deficient nude mice is explained by defective activation of granulocytes and monocytes. Cell Transplant. 2017;26:83–93.
Rijken F, Bruijnzeel PL. The pathogenesis of photoaging: the role of neutrophils and neutrophil-derived enzymes. J Investig Dermatol Symp Proc. 2009;14:67–72.
Jiang H, Kuang Y, Wu Y, Xie W, Simon MI, Wu D. Roles of phospholipase C β2 in chemoattractant-elicited responses. Proc Natl Acad Sci USA. 1997;94:7971–5.
Ratajczak MZ. A novel view of the adult bone marrow stem cell hierarchy and stem cell trafficking. Leukemia. 2015;29:776–82.
Drechsler M, Duchene J, Soehnlein O. Chemokines control mobilization, recruitment, and fate of monocytes in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:1050–5.
Feng X, McDonald JM. Disorders of bone remodeling. Annu Rev Pathol. 2011;6:121–45.
Hanoun M, Maryanovich M, Arnal-Estapé A, Frenette PS. Neural regulation of hematopoiesis, inflammation and cancer. Neuron. 2015;86:360–73.
Drury AN, Szent-Gyorgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol. 1929;68:213–37.
Burnstock G. Purinergic nerves. Pharmacol Rev. 1972;24:509–81.
Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11:201–12.
Ren J, Bertrand PP. Purinergic receptors and synaptic transmission in enteric neurons. Purinergic Signal. 2008;4:255–66.
Burnstock G. An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology. 2016;104:4–17.
Dahl G. ATP release through pannexon channels. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140191.
Iraci N, Leonardi T, Gessler F, Vega B, Pluchino S. Focus on extracellular vesicles: physiological role and signalling properties of extracellular membrane vesicles. Int J Mol Sci. 2016;17:171.
Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895–903.
Nguyen MD, Ross AE, Ryals M, Lee ST, Venton BJ. Clearance of rapid adenosine release is regulated by nucleoside transporters and metabolism. Pharmacol Res Perspect. 2015;3:e00189.
Ratajczak MZ, Bartke A, Darzynkiewicz Z. Prolonged growth hormone/insulin/insulin-like growth factor nutrient response signaling pathway as a silent killer of stem cells and a culprit in aging. Stem Cell Rev. 2017;13:443–53.
Bhartiya D. Pluripotent stem cells in adult tissues: struggling to be acknowledged over two decades. Stem Cell Rev. 2017;13:713–24.
Smadja DM. Bone marrow very small embryonic-like stem cells: new generation of autologous cell therapy soon ready for prime time? Stem Cell Rev. 2017;13:198–201.
Williams AR, Hare JM. Mesenchymal stem cells: Biology, patho-physiology, translational findings, and therapeutic implications for cardiac disease. Circ Res. 2011;109:923–40.
Acknowledgements
This work was supported by NIH grants 2R01 DK074720 and R01HL112788, the Stella and Henry Endowment, and the OPUS grant DEC-2016/23/B/NZ3/03157 to MZR. Dr. Abdel-Latif is supported by the University of Kentucky COBRE Early Career Program (P20 GM103527) and the NIH Grant R01 HL124266. MP was supported by NIH T32 HL134644 to MZR. HU’s is supported by grants from the São Paulo Research Foundation FAPESP (Project No. 2012/50880-4) and the National Council for Scientific and Technological Development (CNPq), Brazil.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Adamiak, M., Bujko, K., Cymer, M. et al. Novel evidence that extracellular nucleotides and purinergic signaling induce innate immunity-mediated mobilization of hematopoietic stem/progenitor cells. Leukemia 32, 1920–1931 (2018). https://doi.org/10.1038/s41375-018-0122-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0122-0
This article is cited by
-
Hematopoiesis Revolves Around the Primordial Evolutional Rhythm of Purinergic Signaling and Innate Immunity – A Journey to the Developmental Roots
Stem Cell Reviews and Reports (2024)
-
Murine and Human-Purified very Small Embryonic-like Stem Cells (VSELs) Express Purinergic Receptors and Migrate to Extracellular ATP Gradient
Stem Cell Reviews and Reports (2024)
-
Pineal Gland Hormone Melatonin Inhibits Migration of Hematopoietic Stem/Progenitor Cells (HSPCs) by Downregulating Nlrp3 Inflammasome and Upregulating Heme Oxygenase-1 (HO-1) Activity
Stem Cell Reviews and Reports (2024)
-
Lactate—a new player in G-CSF-induced mobilization of hematopoietic stem/progenitor cells
Leukemia (2023)
-
Hematopoietic stem cells on the crossroad between purinergic signaling and innate immunity
Purinergic Signalling (2023)
