
Abstract
GPM6A is a glycoprotein in endothelial cells, and its biological function in the development of hepatocellular carcinoma (HCC) is unknown. Through Affymetrix gene expression microarray and bioinformatic analysis, very low GPM6A expression was found in HCC tissue. The present study aims to explore the function and regulatory mechanism of GPM6A in HCC development and progression. Levels of GPM6A expression in HCC specimens from different disorders and various hepatoma cell lines were determined, and its role on cell proliferation was evaluated in hepatoma cells stably overexpressing GPM6A. Modulation of a specific microRNA (miRNA) on its expression and function was evaluated with miRNA mimetic transfection. Herein, it is reported that much lower GPM6A levels were found in HCC tissues than pericancerous liver tissues and correlated to a poor prognosis. GPM6A overexpression inhibited cell proliferation, suppressed colony formation, migration and invasion in two hepatoma cell types. Available evidence does not support that genetic and epigenetic dysregulation contributes significantly to GPM6A inactivation in HCC. Additional findings demonstrated that miR-96-5p acted directly on the 3′-UTR of the GPM6A gene and significantly decreased its mRNA and protein levels. MiR-96-5p transfection promoted proliferation, migration and invasion of SMMC-7721 and MHCC-97H hepatoma cells; whereas the function of oncogenic microRNA-96 was significantly inhibited in GPM6A-overexpressed hepatoma cells. In conclusion, GPM6A expression in HCC is commonly suppressed regardless its base disease types, and its low expression in HCC tissues is most likely attributed to upregulated miR-96-5p. GPM6A may function as a valuable biomarker for HCC progression and prognosis.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and the third leading cause of cancer-related death worldwide. Hepatitis B and C viral infection, aflatoxin intake, alcohol abuse and nonalcoholic steatohepatitis (NASH) are common base diseases for liver cancer1. With increased epidemics of obesity and diabetes, the incidence of nonalcoholic fatty liver disease (NAFLD) displays in an increasing trend in latest two decades2. With the progression of the disease, nonalcoholic fatty liver (NAFL) progresses to NASH, and the latter may further advance to liver fibrosis, cirrhosis and even liver cancer3. Studies have shown that NASH is the main cause of chronic liver disease and an important pre-carcinogenic factor for HCC4. NASH-associated HCC (NAH-HCC) accounts for the major etiology for increased incidence in HCC in the US and many other regions5. Despite studies on NASH-HCC have increased, available data on its pathogenic pathway, molecular interaction and prophylactic measures are limited. We have previously demonstrated that junctional protein-associated with coronary artery disease (JCAD) exerts a promoting role in NASH-HCC development through the Hippo signaling pathway6. The FDA has approved several medications, including check-point inhibitors and molecular targeting agents, for the treatment of advanced HCC since sorafenib, a multi-kinase inhibitor, was approved a decade ago. However, these medications have not significantly improved the disappointing long-term survival for advanced HCC7,8. Therefore, more studies on molecular interplays and carcinogenic pathways are needed to elucidate how HCC develops in a steatotic, inflamed and fibrotic liver, and to identify potential therapeutic targets or valuable biomarkers for NASH-HCC.
GPM6A, a member of the glycoprotein family, is located in the pre33-34 region of chromosome 4, in the same region (q34-35) with another type 2 diabetes-associated gene, NEIL39. Previous studies have shown that GPM6A is closely associated to neuronal differentiation, synaptic formation and stress response10. Only one study suggested that GPM6A and GPM6B might be oncogenes for malignant lesions in B-lymphocyte cell lines11. To our knowledge, no studies are available so far regarding its possible function in the liver under a pathophysiologic condition. Through Affymetrix gene expression microarray analysis, GPM6A expression was found to be extremely suppressed in human NASH-HCC specimens, which prompted to further investigate its biological function and possible role in NASH-HCC development and progression. The aim of the present study was to understand the function and regulation of GPM6A in normal liver and NASH-HCC. In the present study, both clinical HCC specimens and GPM6A-overexpressed hepatoma cells were employed to study molecular modulation of GPM6A expression in normal and malignant tissue, as well as its effects on cell proliferation, migration and colonization. The findings have demonstrated that GPM6A expression was significantly lower in NASH-HCC and HCC from other base diseases than pericancerous liver tissue, and there was no significant difference in GPM6A expression between NASH-HCC and HCC derived from other base diseases. Moreover, existing evidence does not support that gene mutation and methylation regulation are the main molecular basis for the suppressed expression of GPM6A in the progression of liver cancer. On the other hand, its low expression in HCC tissues is most likely attributed to upregulated microRNA-96, for which GPM6A is its target gene. With additional evidence demonstrating its suppressive effects on proliferation and migration of hepatoma cells, it is conceivable that GPM6A expression is suppressed in chronic information, fibrosis and oncogenic transformation and may function as a valuable biomarker for HCC progression and prognosis.
Materials and methods
Tissue sample collection
Fourteen pairs of paraffin-embedded tissues with pathologically confirmed diagnosis of NASH-HCC were identified after excluding any other base liver diseases, such as viral infection, alcoholic liver diseases or genetic deficiencies, and four pairs of specimens were used for Affymetrix microarray assay of gene expression with sufficient RNA extraction from the paraffin-embedded sections. A portion of Affymetrix analysis data was reported previously6, and is accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99807.
For further GPM6A detection, paired human HCC specimens and pericancerous liver tissues (n = 21) were collected from the Biorepository of Fudan University Institute of Liver Cancer. For microRNA (miRNA) detection, additional 16 pairs of HCC and pericancerous liver tissues were collected from Huashan Hospital. All fresh samples were immediately frozen in liquid nitrogen and then stored at −80 °C for subsequent experiments. The use of human tissue was approved by the Ethic Committee of Fudan University School of Basic Medical Sciences, and followed guidelines of the Helsinki Declaration and the national, municipal, and university regulations. mRNA and DNA data from the cancer genome atlas (TCGA) was retrieved for analysis of GPM6A expression and mutation in HCC (https://portal.gdc.cancer.gov).
Methylation analysis of the GPM6A promoter in genomic DNA from HCC specimens
Human genomic DNA was extracted from resected liver cancer specimens and paired pericancerous liver tissues (n = 5), and treated with sodium bisulfite using EZ DNA Methylation-Gold kit (Zymo Research, CA, USA) according to manufacturer’s instructions. Five hundred ng of extracted DNA was used for this step. This process converted all unmethylated cytosine to uracil, which could be recognized as thymidine by Taq polymerase; and the treatment did not affect methylated cytosine. Bisulfate-treated DNA was amplified using Qiagen PyroMark PCR Kit (Qiagen, Hilden, Germany). The PCR products were amplified using the following primers: BSP-forward: AGTTGTTTGTGGTGATTTTAAAATT and BSP-reverse: TCAAAAATACATCACCCTTCTAAA. The amplification was carried out as follows: denaturation at 94 °C for 5 min followed by 40 cycles at 94 °C for 30 s, at 58 °C for 30 s, at 72 °C for 30 s for each cycle; and a final extension at 72 °C for 10 min. Amplified PCR products were gel-purified and cloned into T vector for sequencing. At least ten colonies were randomly chosen for sequence comparison by BiQ Analyzer with technical assistance from Oebiotech, Shanghai, China.
Cell culture
Huh-7 cell line was provided by Professor Mark Feitelson, PhD from Temple University (Philadelphia, PA). Huh-7-Trans cell line was previously derived from Huh-7 cell line as we reported previously12. HLE and HLF cells were obtained from the Health Science Research Resources Bank, Japan in 2003. HepG2 and Hep3B cells were obtained from the American Type Culture Collection. SMCC-7721, MHCC-97H, and MHCC-97L cell lines were obtained from the Institute of Liver Cancer, Zhongshan Hospital of Fudan University (Shanghai, China). The above mentioned cell lines were all authenticated using short tandem repeat (STR) analysis (GENEWIZ, Inc., Suzhou, China) and tested periodically for mycoplasma by polymerase chain reaction (PCR)7. Hep3B and HepG2 cells were cultured in MEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Remaining cell lines were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were incubated in humidified atmosphere with 5% CO2 at 37 °C.
Overexpression of GPM6A
The full-length open reading frame sequence of GPM6A was obtained by PCR amplification of immortalized hepatic cell cDNA. The PCR product was ligated into the pLVX-IRES-puro (Addgene, Cambridge, MA, USA) expression vector, and then verified by DNA sequencing. The pLVX-IRES-puro-GPM6A plasmid and packaging plasmids (pMD2.G and psPAX2) (Addgene) were co-transfected into 293T cells using Lipofectamine 2000 (Invitrogen) to generate recombinant lentiviruses for GPM6A overexpression. SMMC-7721 and MHCC-97H cell lines were stably transfected with GPM6A overexpression vector using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. Stably transfected cells were selected for 2 weeks with puromycin antibiotics. Synthetic miR-96 mimetics and mimetic controls were synthesized by RiboBio (Guangzhou, China). Transfection of miR-96 mimetics and mimetic controls were performed with riboFECT CP Transfection Kit (RiboBio) according to the manufacturer’s instruction. The final concentration of miR-96 mimetics was 100 nM in culture medium.
Dual-luciferase reporter assay
In order to identify whether GPM6A is a direct target of miR-96, wild-type, and mutated GPM6A 3′-UTR was cloned to downstream of the firefly luciferase gene in a pmir-GLO vector (Promega, Madison, WI). For luciferase assay, HEK293T cells were seeded in 12-well plates and then co-transfected with pmir-GLO-GPM6A 3′UTR-wt or pmir-GLO-GPM6A 3′-UTR-mut vectors and miR-96 mimetics for 48 h using riboFECT CP Transfection Kit (RiboBio). Dual-luciferase reporter gene assay kit (Yeasen, Shanghai, China) was employed to detect the luciferase activity, and firefly luciferase activity was measured and normalized to the control Renilla luciferase levels7.
Immunofluorescent staining
Human liver biopsy specimens with NASH were obtained from the First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang Province, China13, embedded in paraffin, and sliced into 4 μm sections for the subsequent immunohistochemical staining with an approval of the Ethic Committee of Fudan University School of Basic Medical Sciences. Tissue sections were first incubated at 65 °C for 1 h, and then subjected to deparaffinization in xylene and rehydrated in a graded series of alcohol. Next, antigen retrieve was conducted in boiled citrate buffer for 15 min, and endogenous peroxidase activity was blocked with 0.1% H2O2 for 15 min. The sections were permeabilized with 0.5% Triton X-100 in PBS for 15 min, then blocked with 5% goat serum for 2 h followed by overnight incubation with anti-GPM6A antibody at 4 °C, and then incubated with Alexa Fluor 594-conjugated secondary antibody at room temperature for 1 h. Lastly, tissue sections were dehydrated and mounted for fluorescent microscopy examination14.
Protein extraction and Western blotting analysis
For HCC tissue, cell fractionation kit (Ivent) was used to isolate plasma membrane protein, and GPM6A level was normalized by probing the same blots with anti-Na-K ATPase antibody purchased from Abcam (Cambridge, MA, USA). Cells were washed three times with ice-cold phosphate-buffered sodium (PBS) solution and then lysed with RIPA buffer (Beyotime, Shanghai, China). Whole cell protein extracts were quantified using the Bradford method. Protein samples were separated using 10% SDS-PAGE (sodium dodecyl sulphate–polyacrylamide gel electrophoresis), and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA, USA). The membrane was incubated with primary antibodies overnight at 4 °C, and then incubated with a secondary antibody for 2 h at room temperature. The blots were finally visualized by an enhanced chemiluminescence detection kit. Primary GPM6A antibody was purchased from Proteintech (Chicago, IL, USA), and GPM6A level was normalized by probing the same blots with anti-β-actin antibody purchased from Santa Cruz Biotechnology (Santa Cruz, CA)6.
RNA extraction and quantitative RT-PCR
Total RNA extraction from frozen tissues and cell lines was performed using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. qRT-PCR was performed to detect mRNA levels of GPM6A, β-actin and miR-96 of frozen tissues and cultured cells. cDNA was synthesized using PrimeScript™ RT reagent kit (Takara, Kusatsu, Japan). qRT-PCR was performed using SYBR green (Invitrogen) with the specific primers for GPM6A (Forward: 5ʹ-CTGCTTGGACCTTCGTCAGT-3ʹ and Reverse: 5ʹ-GCTCAGTAGATTCGCACATCC-3ʹ), β-actin (Forward: 5ʹ-GGCATCCTCACCCTGAAGTA-3ʹ and Reverse: 5ʹ-AGGTGTGGTGCCAGATTTTC −3ʹ). For miRNA analysis, reverse transcription was performed using miRNA 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China). qRT-PCR was performed using miRNA Universal SYBR qPCR Master Mix (Vazyme) with the specific primers for miR-96-5p15. Gene expression levels (log 2(−ΔΔCT)) were calculated using β-actin as a house-keeping gene as previously described14.
Cell proliferation assay
Cell proliferation was examined by CellTiter-Lumi™ luminescent cell viability assay kit (Beyotime, Shanghai, China) according to the manufacturer’s instruction. In brief, cells were seeded on 96-well plates at a concentration of 5 × 103/well. After incubation for 24 h, detection buffer was added to culture medium, and luminescent intensity was detected with a microplate reader. Each assay was performed in triplicate and repeated at least three times.
Colony formation assay
Stably transfected cells were cultured in 6-well plates with agar gel at a density of 1000/well and incubated for 14 days. When colonies were clearly visible, they were fixed with 4% paraformaldehyde for 15 min, stained by 0.1% crystal violet for 15 min followed by washing in running water, and dried. Fixed colonies were counted using Image-J software as reported previously6.
Wound healing assay
Wound healing assay was used to assess cell migration ability. Cells were seeded in 6-well plates at a density of 1 × 106 cells per well. After reaching 90% confluence, cells were wounded by scratching with a 200-μl pipette tip and then washed three times with PBS buffer to remove impaired cells. The culture medium was then replaced with serum-free medium, and cells were allowed to grow and close the wound for 24 h. Micrographical images of a specific position on the scratched areas at two different time points (0 and 24 h) were taken by an inverted microscope (Leica, Germany). The percentage of the area with migrated cells compared to the initial wound region was defined as a wound closure rate. Image-J software was used to measure the wound region area.
Cell invasion assay
For the detection of cell invasion ability, transwell invasion assays were performed using the 24-well transwell with 8-µm pore size polycarbonate membrane. Cells in the logarithmic phase were digested and resuspended to a density of 105/ml and seeded onto a Matrigel-coated membrane matrix present in the insert of a 24-well culture plate, containing serum-free DMEM (200 μl). The lower chambers were filled with 500 μl DMEM supplemented with 20% FBS. After being incubated for 48 h, the medium remaining in the upper chambers was discarded and the chambers were washed in PBS for 2 times. The invaded cells were fixed with 4% paraformaldehyde at room temperature for 30 min, and stained with 0.1% crystal violet at room temperature for 15 min. The membrane was then washed again with 500 mL of PBS, and cells remained on the upper side of the membrane were wiped by a cotton swab. The invaded cells were stained, photographed for at least 5 microscope fields, and counted using Image-J software12.
Statistical analysis
All experiments were independently repeated for at least three times and the results of multiple experiments are presented as mean ± SD. Statistical analyses were performed using SPSS 22.0 statistical software. Student’s t test was performed for data that had a normal distribution and homogeneity of variance. The data that are not in a normal distribution were analyzed by nonparametric Mann–Whitney U test. Survival data were computed using the Kaplan–Meier method, and survival difference between two groups was conducted by the log-rank test. Correlation between miRNA and GPM6A expression levels was determined by Pearson correlation analysis. A p value < 0.05 was considered statistically significant.
Results
GPM6A was downregulated in HCC tissues and hepatoma cells
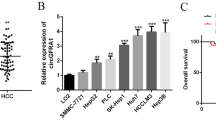
In our previous study, aberrant expression of more than 50 genes was found from four sets of NASH-HCC and adjacent liver tissues (Fig. 1A). Among them 12 genes were strikingly different in expression (3 downregulated and 9 upregulated) (Fig. 1B). One of them, JCAD was found to favor the transformation of NASH to hepatic carcinogenesis by inhibiting large tumor suppressor kinase-2 (LATS2) kinase activity in the Hippo signaling pathway6; whereas, GPM6A was significantly downregulated by 5-fold in HCC specimens compared to pericancerous tissue. To verify reduced GPM6A expression levels in HCC from other base diseases, GPM6A expression data were retrieved from a large cohort of HCC patients (n = 50) available from TCGA database. As shown in Fig. 2A, GPM6A was remarkably downregulated in HCC most derived from HCV infection compared to controls (n = 324). In 21 pairs of HCC tissue and matched pericancerous liver tissue specimens (most of them were arisen from HBV infection), quantitative RT-PCR and Western blot analyses revealed that both mRNA and protein GPM6A levels were consistently decreased in almost all cancer tissues compared to pericancerous liver tissues (p < 0.001) (Fig. 2B, C). There was no significant difference in GPM6A expression between NASH-HCC and HCC derived from other base diseases. Similar results were obtained in 5 out of 9 hepatoma cell lines compared to control cells (Fig. 2D). Immunofluorescent staining of a liver biopsy specimen from a NASH patient confirmed GPM6A expression in the cytoplasmic membrane (Fig. 2E). The survival data from the Cancer Genome Atlas (TCGA) cohort (n = 185) showed that patients with low expression of GPM6A had a shorter overall survival duration (p = 0.018) and disease-free survival duration (p = 0.004) than those with relatively high GPM6A expression (Fig. 2F).

A Heatmap of differential expression of significant genes from four pairs of NASH-HCC specimens (T) and pericancerous tissue (P). Green and red colors indicate relatively low and high expression levels, respectively. B Fold changes of GPM6A expression levels compared with their corresponding nontumorous livers. It was noted that GPM6A expression levels were very low compared to pericancerous tissue. Other overexpressed genes, such as CD36 and KIAA1462 (JCAD) were also noted.

A GPM6A expression in 50 paired HCC tissues and 324 non-tumor tissues from TCGA-LIHC cohort. B GPM6A was significantly downregulated in 21 paired non-NASH-related HCC tissues compared with their corresponding pericancerous livers. C Western blot analysis of GPM6A protein level in 5 pairs of non-NASH-related HCC tissues compared with their corresponding pericancerous livers. Na-K ATPase was used as a loading control. D GPM6A mRNA level in human hepatoma cell lines was analyzed using qRT-PCR. Immortalized hepatic (LO2) cells served as a control. E Immunostaining of GPM6A. GPM6A was mainly expressed in the cell membrane of liver biopsy of an NASH patient (630x, left panel) with a local amplification on the right. F Decreased expression of GPM6A was correlated with a low overall survival rate and disease-free survival rate compared with high expression group in TCGA-LIHC cohort (n = 185). TGCA the Cancer Genome Atlas (TCGA). ***p < 0.005 compared with the control.
Effects of genetic and epigenetic changes on suppressed GMP6A in HCC specimens
In order to delineate the molecular basis for suppressed GMP6A expression in HCC, the mutation at the genome level was initially approached. To determine whether genetic aberration leads to the loss of GPM6A in HCC, the sequencing data of 364 cases of liver cancer from TCGA cohort was retrieved to identify the mutation of the GPM6A gene. Surprisingly, only one case of HCC samples had a missense mutation at the 762 nucleotide of the GPM6A gene, resulting in the conversion of tryptophan to leucine. In addition, further analysis revealed that the copy number of this missense mutation in none of these samples was changed more than twofold, indicating the significance of this missense in leading to suppressed GPM6A could be minimal.
To further determine whether epigenetic deregulation affects GPM6A expression in HCC, CpG islands in the GPM6A gene was screened using bioinformatic tools. To our surprising, no CpG islands were found but there were only several CpG sites in the promoter region of the GPM6A gene (Fig. S2A). Three CpG sites were selected for further mythylation analysis at the promoter region. For this analysis, five pairs of HCC specimens and adjacent liver tissue were selected to extract genomic DNA, and methylation status in the promoter region was analyzed by bisulfite sequencing PCR (BSP). The results showed that three methylation sites in the GPM6A promoter region were hypermethylated in cancer specimens compared to pericancerous liver tissue (Fig. S2B, C). In order to further determine whether the methylation at the promoter region accounts for the low GPM6A expression, SMMC-7721 and MHCC-97H hepatoma cells were treated with a methyltransferase inhibitor, 5-aza-2-deoxycitidine (5-Aza). As shown in Fig. S3A the 5-Aza treatment did significantly inhibit methyltransferase activity in SMMC-7721 and MHCC-97H hepatoma cells; whereas, GPM6A expression levels were not significantly affected by the 5-Aza treatment (Fig. S3B), indicating that inhibition of methyltransferase activity by 5-Aza did not restore GPM6A expression in these cells (Fig. S3A, B). Therefore, it is our speculation that neither genetic aberration nor methylation at the promoter region plays a major role in affecting GPM6A expression in HCC specimens and hepatoma cells investigated.
Effects of GPM6A expression on cell proliferation and migration activity
The remarkable reduction of GPM6A expression in liver cancer tissues prompted us to investigate the possible biological function in tumorigenesis. To explore the role of GPM6A in cell proliferation, the full sequence of GPM6A cDNA was cloned to form pLVX-IRES-puro-GPM6A lentiviral vector; hepatoma SMMC-7721 and MHCC-97H cells were transduced with the recombinant lentivirus. Overexpression of GPM6A (GPM6A-OE) in these cell lines was confirmed by qRT-PCR and Western blot analysis at the mRNA and protein levels (Fig. 3A, B), respectively. Cell viability assay was conducted to examine the effect of GPM6A overexpression on proliferation. As shown in Fig. 3C, proliferation of hepatoma cells with GPM6A-OE was significantly suppressed in comparison to those transduced with control lentiviral vector (p < 0.05).

A qRT-PCR was conducted to detect GPM6A mRNA levels in stable GPM6A-OE (SMMC-7721, MHCC-97H) and control cells. B Western blot was conducted to detect GPM6A protein levels in stable GPM6A-OE and control cells. β-Actin was used as an endogenous control. C Proliferation of stable GPM6A-OE and control cells was detected using CellTiter-Lumi™ assay. D Representative images of colony formation assays in stable GPM6A-OE and control cells. E Representative images of a wound healing assay in stable GPM6A-OE and control cells. F Representative images (left) of a transwell assay and quantitative analysis (right) in stable GPM6A-OE and control cells (400×). *p < 0.05; **p < 0.01; ***p < 0.005 compared with the control.
To further confirm the role of GPM6A in tumorigenesis in vitro, colony formation assay of GPM6A-OE cells and corresponding control cells was performed. Colony number of GPM6A-OE cells was much lower than that of the control cells (down by 30–40% (Figs. 3D, S1A, p < 0.005). Consistent with an inhibitory role of GPM6A on hepatoma cell proliferation, wound healing assay exhibited that GPM6A overexpression was significantly associated with a slower wound closure than control cells (Figs. 3E, S1B, p < 0.05–0.01). Transwell Matrigel invasive assays demonstrated that GPM6A-OE cells had lower invasive capability than control cells (Fig. 3F; p < 0.01). In summary, these results suggest that GPM6A overexpression suppressed proliferation, migration and invasive capability of hepatoma cells.
miR-96 counteracted on suppressive effects of GPM6A in HCC
With significant role determined with GPM6A overexpression, the lack of genetic or epigenetic deregulation of GPM6A inactivation in HCC prompted us to seek additional answers, and speculate that miRNAs may be involved in regulation of GPM6A expression in HCC tissue. To support this assumption, potential miRNAs which might bind to the GPM6A sequence were searched using different databases. Among microRNAs that potentially bind to the 3′-untranslated region (3′-UTR) of the GPM6A sequence, miR-96 is of interest to be investigated, because it’s frequently overexpressed in tumor tissue and is well-known for its importance in both tumor angiogenesis and metastasis. In order to prove its involvement in suppressed GPM6A expression, additional 10 pairs of primary HCC specimens and their corresponding pericancerous liver tissue were analyzed by qRT-PCR and Western blotting analyses. Consistent with the data shown in Fig. 2B, C, GPM6A expression was significantly reduced in HCC specimens compared to pericancerous liver tissues (Fig. 4A; p < 0.005). In contrast, miR-96 expression level was significantly upregulated in these HCC specimens compared to pericancerous liver tissue (Fig. 4B; p < 0.005). Interestingly, the HCC specimens with higher miR-96 levels tended to possess lower GPM6A mRNA levels, and there was a negative correlation between miRNA 96 and GPM6A expression in the same HCC specimens (r = 0.64, p < 0.05, Fig. 4C). These results implied that suppressed GPM6A expression may be associated with upregulated miR-96 expression in these specimens.

A, B Quantitative RT-PCR analysis of the relative expression levels of GPM6A (A), miR-96 (B) in 10 pairs of liver cancer tissues and matched adjacent noncancerous tissues. C Pearson’s correlation scatter plot of the fold-change of miR-96 and GPM6A mRNA in human liver cancer tissues. D Luciferase binding assay for miR-96 and GPM6A. miR-96 putative binding sites and corresponding mutant sites of the 3′-UTR of GPM6A are presented. Relative luciferase activity was analyzed after the above reporter plasmids or mock reporter plasmid were co-transfected into 293T cells infected with miR-96 mimics. E Effect of miR-96 overexpression on GPM6A protein level of immortalized hepatic (LO2) cells. ***p < 0.005 compared to the control.
miR-96 directly bound to the GPM6A 3′-UTR and suppressed GPM6A expression
The up-regulation of miR-96, down-regulation of GPM6A and the negative correlation between them prompted us to speculate that miR-96 might be a direct regulator of GPM6A at a translational level. To test this hypothesis, the wild-type (WT) 3′-untranslated region (UTR) or the mutant 3′-UTR target sequences of the GPM6A gene was cloned into pmir-GLO luciferase reporter vector to generate a new reporter vector, pmir-GLO-GPM6A-3′-UTR. Dual-luciferase reporter analysis documented that transfection of miR-96 mimetics in immortalized hepatic cells significantly inhibited the activity of firefly luciferase that carried the wild-type, but the mutated 3′-UTR of GPM6A did not, indicating that miR-96 may suppress gene expression through its binding sequence at the 3′-UTR of GPM6A (Fig. 4D; p < 0.005). These results were supported by the data of Western blotting analysis, showing that transfection of miR-96 mimetics suppressed GPM6A expression at protein levels in immortalized hepatic cells (Fig. 4E). Taking together, these data indicate that miR-96 negatively regulates the GPM6A expression by directly targeting its 3′-UTR at a translational level.
MiR-96 promoted proliferation and migration of HCC cells and overexpression of GPM6A counteracted the oncogenic role of miR-96
After having verified that GPM6A is a direct target of miR-96, the biological role of miR-96 was further investigated in hepatoma cell lines. To approach to this question, miR-96 was ectopically expressed in SMMC-7721 and MHCC-97H cells by transfecting miR-96 mimetics, and increased miR-96 levels in transfected cells were verified by qRT-PCR (Fig. 5A, p < 0.005). Along with increased levels of miR-96 mRNA, GPM6A level was significantly suppressed in SMMC-7721 and MHCC-97H cells (Fig. 5B, p < 0.05). At the same time, proliferation rate was markedly enhanced in SMMC-7721 and MHCC-97H cells transfected with miR-96 mimetics compared to control cells (Fig. 5C, p < 0.05). Moreover, it is evident that miR-96 overexpression significantly accelerated cell migration, and enhanced invasive capability (Fig. 5D, E, p < 0.05). However, such effects were not seen in GPM6A-OE cells (Fig. 6A–D). Presumably, this may be attributed to high GPM6A expression levels in these cells (Fig. S4). These findings suggest that miR-96 may function as an oncolytic regulator through suppressing GMP6A that may suppress proliferation, cloning formation, migration, and invasion of hepatoma cells.

A miR-96 was overexpressed in SMMC-7721 and MHCC-97H by transfecting with miR-96 mimics. B qRT-PCR was conducted to detect the effects of miR-96 on GPM6A mRNA levels. C Effects of miR-96 overexpression on proliferation of SMMC-7721 and MHCC-97H was conducted using CellTiter-Lumi™ assay. D Wound healing assay was conducted to detect the effects of miR-96 on migration of SMMC-7721 and MHCC-97H. Quantification (up) of percentage of wound healing area and representative images (down). E Transwell assay was conducted to detect the effects of miR-96 on migration of SMMC-7721 and MHCC-97H (400×). *p < 0.05; **p < 0.01; ***p < 0.005 compared with the control.

A, B Effects of miR-96 overexpression on proliferation of GPM6A-OE (7721-OE, 97H-OE) and control cells (7721-PLVX, 97H-PLVX) was conducted using CellTiter-Lumi™ assay. C, D Wound healing assay was conducted to detect the effects of miR-96 on migration of GPM6A-OE (7721-OE, 97H-OE) and control cells (7721-PLVX, 97H-PLVX). Representative images (up) and quantification (down) of percentage of the wound healing area are shown in (C, D). *p < 0.05; **p < 0.01 compared with the control.
Discussion
Many tumor suppressor genes, such as P53, Pten, are mutated or deleted in an oncogenic process, resulting in enhanced proliferation and/or reduced apoptosis, and a net consequence of malignant progression. Mutation and deletion of the p53 gene in HCC is very common, and lead to inactivation of p53 and/or activation oncogenic genes16,17, such as C-myc, wnt-β-catenin, Hippo or hedgehog signaling in the tumor tissue6,14,18, and the imbalance between tumor suppressing and oncogenic or morphogenic factors, contributing to its malignant behaviors. Oncogenic process and progressive behaviors are well studied in HCC post hepatitis viral infection, such as HBV and HCV, as well as co-infection with HDV, alcoholic liver diseases or arisen from other inherited genetic deficiencies, such as α1-antitrypsin deficiency, Wilson’s disease, etc. However, no previous study has been seen regarding whether a tumor suppressor gene may take a part in oncogenesis, progression and metastasis of HCC in the base of metabolism-associated NASH. When GPM6A was found to be suppressed in NASH-HCC specimens and hepatoma cell lines in our Affymetrix gene expression microarray analysis, it was intriguing to elucidate its biologic effects and involvement in an oncogenic process, given that this gene is located at almost the same locus as type 2 diabetes-associated gene, NEIL39. The following critical questions have been addressed in the present study.
At the first, whether the suppressed GPM6A expression is unique for NASH-HCC or universal for HCC arisen from various base diseases? In order to answer this question, the data of GPM6A expression levels were retrieved from the TCGA database, and most subjects had HCC arisen from HCV infection. Another domestic cohort of HCC subjects were most HBV-positive. For these two cohorts of the patients, mRNA levels of GPM6A were all suppressed compared to pericancerous liver tissue, and the suppressed expression was verified at protein levels by Western blot analysis and immunohistochemical staining in the domestic cohort of the subjects. It seems that there was no significant difference in GPM6A expression (although all reduced) regardless the etiology for the base liver diseases (HBV, HCV infection or NASH) from this observation. Moreover, strikingly reduced GPM6A expression was found in 5 out of 9 hepatoma cell lines, and none was increased when compared to immortalized hepatic cells. Therefore, it appears that suppressed GPM6A expression at both mRNA and protein levels is universal in HCC from different base liver diseases.
To approach the 2nd question regarding the molecular basis for the reduced expression of GPM6A in HCC, genetic and epigenetic analyses were undertaken with the sequencing data retrieved from the TCGA database, and methylation status in the promoter region of the GPM6A gene. It was surprising that no significant mutation was identified to support the speculation that strikingly reduced GPM6A expression in HCC was due to marked mutation of the gene. Moreover, enhanced methylation of 3 CpG sites was confirmed in 5 HCC specimens compared to pericancerous liver tissue. Nevertheless, due to the fact that no CpG islands were found in the promoter region of the gene, and that inhibition of methyltransferase activity by 5-Aza in hepatoma cells did not restore GPM6A expression, it is attempting to speculate that both genetic and epigenetic modulation at the transcription level on GPM6A expression has less impact on the reduced GPM6A expression in HCC, and further studies are needed to reveal the underlying mechanisms.
The 3rd question focuses on functional determination of GPM6A with stably transduced hepatoma SMMC-7721 and MHCC-97H cells, and the findings demonstrate that GPM6A overexpression suppressed the proliferation, colony formation, migration and metastatic capability of these hepatoma cells in vitro when compared to those with control vector transduction. It is evident that GPM6A may exhibit inhibitory effects on these progressive behaviors although the level of overexpression may exceed a physiologic level of this membrane glycoprotein, and the molecular basis in exerting these critical roles remains to be investigated. At the same time, it is valuable to find that patients with low expression of GPM6A had a significantly lower overall survival rate and disease-free survival rate in comparison to patients with high expression of GPM6A, indicating that GPM6A may serve as a valuable biomarker for prognosis prediction and that this glycoprotein may have an important function in cancer biology.
The additional efforts were made to answer the 4th question on what is the regulator for inhibited GPM6A expression in HCC tissue. miRNAs are the pivotal members of a noncoding RNA family in the human genome. A total of more than 2000 miRNAs have been discovered, acting as important gene regulators in almost every aspect of biologic events. In the liver, miRNAs are critical for proliferation, inflammation, fibrosis and carcinogenesis through translational repression15,19,20. miRNAs elicit mRNA degradation by Dicer after its specific binding to the 3′-untranslated region (3′-UTR) of target mRNAs21. Many reports have shown that miRNA dysregulation was associated with the pathogenesis for various types of cancers17,22,23,24. In HCC, dysregulation of numerous miRNAs such as miR-21, miR-25, miR-122, miR-221, as well as, miR-96 and miR-96-5p was documented and may be responsible for the progressive behaviors, such as reduced apoptosis, enhanced growth, migration and invasion17,25,26,27,28. In the present study, the inhibition of GPM6A expression was negatively correlated with increased levels of miR-96 in HCC specimens; whereas an increase in intracellular miR-96 mimetics by transfection resulted in enhanced proliferation and invasive capability in hepatoma cells. Therefore, with these pieces of evidence, it is reasonable to conclude that low GPM6A expression in HCC tissues is most likely attributed to upregulated microRNA-96, for which GPM6A is its target gene. There was further supporting evidence with the reporter assay for which the mutation at the 3′-UTR blunted the inhibition of miR-96 on luciferase activity. This finding is consistent with a recent report which disclosed that GPM6A was significantly suppressed in HCC probably via a circulating RNA circCCNB1/miR-106b-5p/GPM6A network29. With additional evidence demonstrating promoting effects after transfection of miR-96 on proliferation and migration of hepatoma SMMC-7721 and MHCC-97H cells, it is conceivable that GPM6A expression is suppressed in chronic information, fibrosis and oncogenic transformation. It is not surprising that no significant promoting effects were observed in GPM6A overexpression 7721-OE and 97H-OE cells since transfected miR-96 may not have sufficient inhibition on persistent production of GPM6A from integrated lentiviral transduction.
In conclusion, GPM6A expression in HCC is commonly suppressed in HCC, and correlated to its poor prognosis. Its low expression in HCC tissues is most likely attributed to upregulated miR-96-5p, which may function as an oncogenic regulator in HCC progression. The findings of this investigation underscore the molecular function of novel GPM6A glycoprotein, and point to its biomarker potential for HCC progression and prognosis; whereas, miR-96-5p acts as an oncogenic facilitator in a carcinogenic process.
Data availability
Affymetrix analysis data is accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99807.
References
Zhu H, Wu J, Shen X. Genome-wide association study: new genetic insights into HBV/HCV-related hepatocellular carcinoma genomes. Scand J Gastroenterol 52, 209-215 (2017).
Fan JG, Kim SU, Wong VW. New trends on obesity and NAFLD in Asia. J Hepatol 67, 862-873 (2017).
Diehl AM, Day C. Cause, Pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med 377, 2063-2072 (2017).
Yang JD, Ahmed F, Mara KC, Addissie BD, Allen AM, Gores GJ et al. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology 71, 907-916 (2020).
Starley BQ, Calcagno CJ, Harrison S A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 51, 1820-1832 (2010).
Ye J, Li TS, Xu G, Zhao YM, Zhang NP, Fan J et al. JCAD promotes progression of nonalcoholic steatohepatitis to liver cancer by inhibiting lats2 kinase activity. Cancer Res 77, 5287-5300 (2017).
Zhou XT, Ding J, Li HY, Zuo JL, Ge SY, Jia HL et al. Hedgehog signalling mediates drug resistance through targeting TAP1 in hepatocellular carcinoma. J Cell Mol Med 24, 4298-4311 (2020).
Zhang L, Ding J, Li HY, Wang ZH, Wu J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim Biophys Acta Rev Cancer 1874, 188441 (2020).
Park MH, Kwak S H, Kim KJ, Go MJ, Lee H J, Kim KS et al. Identification of a genetic locus on chromosome 4q34-35 for type 2 diabetes with overweight. Exp Mol Med 45, e7 (2013).
Formoso K, Billi S C, Frasch AC, Scorticati C. Tyrosine 251 at the C-terminus of neuronal glycoprotein M6a is critical for neurite outgrowth. J Neurosci Res 93, 215-229 (2015).
Charfi C, Edouard E, Rassart E. Identification of GPM6A and GPM6B as potential new human lymphoid leukemia-associated oncogenes. Cell Oncol (Dordr) 37, 179-191 (2014).
Fan YH, Ding J, Nguyen S, Liu X J, Xu G, Zhou HY et al. Aberrant hedgehog signaling is responsible for the highly invasive behavior of a subpopulation of hepatoma cells. Oncogene 35, 116-124 (2016).
Wang ZH, Zheng KI, Wang XD, Qiao J, Li YY, Zhang L et al. LC-MS-based lipidomic analysis in distinguishing patients with nonalcoholic steatohepatitis from nonalcoholic fatty liver. Hepatobiliary Pancreat Dis Int 20, 452-459 (2021).
Xu G, Ye J, Liu XJ, Zhang NP, Zhao YM, Fan J et al. Activation of pluripotent genes in hepatic progenitor cells in the transition of nonalcoholic steatohepatitis to pre-malignant lesions. Lab Invest 97, 1201-1217 (2017).
Chen X, Murad M, Cui YY, Yao LJ, Venugopal SK, Dawson K et al. miRNA regulation of liver growth after 50% partial hepatectomy and small size grafts in rats. Transplantation 91, 293-299 (2011).
Long J, Wang A, Bai Y, Lin J, Yang X, Wang D et al. Development and validation of a TP53-associated immune prognostic model for hepatocellular carcinoma. EBioMedicine 42, 363-374 (2019).
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133, 647-658 (2007).
Cancer Genome Atlas Research Network. Electronic address WBE, Cancer Genome Atlas Research N. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327-1341. e1323 (2017).
Bartel D P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281-297 (2004).
Venugopal SK, Jiang J, Kim T H, Li Y, Wang SS, Torok NJ et al. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol 298, G101-106 (2010).
Shukla GC, Singh J, Barik S. MicroRNAs: Processing, maturation, target recognition and regulatory functions. Mol Cell Pharmacol 3, 83-92 (2011).
Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64, 3753-3756 (2004).
Yang X, Liang L, Zhang XF, Jia H L, Qin Y, Zhu XC et al. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology 58, 158-170 (2013).
Michael M Z, SM O C, van Holst Pellekaan N G, Young G P, James R J. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 1, 882-891 (2003).
Feng X, Jiang J, Shi S, Xie H, Zhou L, Zheng S. Knockdown of miR-25 increases the sensitivity of liver cancer stem cells to TRAIL-induced apoptosis via PTEN/PI3K/Akt/Bad signaling pathway. Int J Oncol 49, 2600-2610 (2016).
Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A 107, 264-269 (2010).
Iwai N, Yasui K, Tomie A, Gen Y, Terasaki K, Kitaichi T et al. Oncogenic miR-96-5p inhibits apoptosis by targeting the caspase-9 gene in hepatocellular carcinoma. Int J Oncol 53, 237-245 (2018).
Wang TH, Yeh CT, Ho J Y, Ng KF, Chen TC. OncomiR miR-96 and miR-182 promote cell proliferation and invasion through targeting ephrinA5 in hepatocellular carcinoma. Mol Carcinog 55, 366-375 (2016).
Liu YM, Cao Y, Zhao PS, Wu L Y, Lu YM, Wang YL et al. CircCCNB1 silencing acting as a miR-106b-5p sponge inhibited GPM6A expression to promote HCC progression by enhancing DYNC1I1 expression and activating the AKT/ERK signaling pathway. Int J Biol Sci 18, 637-651 (2022).
Acknowledgements
Part of this work was presented in the American Association of Cancer Research (AACR) Annual Virtual Meeting II, Session title: Mechanisms of Tumor Suppressor Genes, AACR Abstract # 5970, June 24-26, 2020, Philadelphia, PA, USA; and published as an abstract in the Proceedings of AACR Annual Meeting 2020 (DOI: 10.1158/1538-7445). The authors are grateful to senior technologist Ke Qiao in the Technology Platform of Fudan University School of Basic Medical Sciences and Dr. Juan Ye in the Dept. of Medical Microbiology and Parasitology, School of Basic Medical Sciences, Fudan University, Xiang-Nan Yu in the Dept. of Gastroenterology & Hepatology, Zhongshan Hospital of Fudan University, and Dr. Shengyang Ge in the Department of Abdominal Surgery, Huashan Hospital of Fudan University for their technical assistance.
Funding
This work is supported by the National Natural Science Foundation of China (NSFC #81572356, 81871997 and 82170624), the National Key R&D Program of China (#2016YFE0107400) to J.W.
Author information
Authors and Affiliations
Contributions
Z.R.L.: Design and conduct of most experiments, data collection and analysis, paper preparation; G.X., L.Y.Z. and H.C.: Conduct of some experiments, data collection, and analysis. J.M.Z.: providing clinical specimens and patient information. J.W.: Concept development, experimental design and data analysis, overall supervision and funding support, paper preparation, and finalization.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The use of human tissue was approved by the Ethic Committee of Fudan University School of Basic Medical Sciences, and followed guidelines of the Helsinki Declaration and the national, municipal, and university regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, ZR., Xu, G., Zhu, LY. et al. GPM6A expression is suppressed in hepatocellular carcinoma through miRNA-96 production. Lab Invest 102, 1280–1291 (2022). https://doi.org/10.1038/s41374-022-00818-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-022-00818-3
