
Abstract
The pathogenesis of diabetic wounds is closely associated with the dysregulation of macrophage polarization. However, the underlying mechanism remains poorly understood. In this study, we aimed to investigate the potential effects of PAQR3 (progestin and adipoQ receptor 3) silencing in accelerating diabetic wound healing. We showed that PAQR3 silencing promoted skin wound healing and angiogenesis in diabetic mice, which was accompanied by enhanced M2 macrophage polarization and elevated expression of PPARγ (peroxisome proliferator-activated receptor γ). PAQR3 silencing also promoted M2 polarization and increased PPARγ protein level in PMA-treated THP-1 cells. Moreover, knockdown of PAQR3 in macrophages enhanced the migration of HaCaT cells and tube formation of HUVECs. The ubiquitination of PPARγ protein in macrophages was repressed by PAQR3 silencing. STUB1 (STIP1 homology and U-box-containing protein 1) binds with the PPARγ protein to mediate PPARγ ubiquitination and degradation in macrophages, which was impaired by PAQR3 silencing. The PPARγ inhibitor, GW9662, or STUB1 overexpression abrogated the enhanced M2 macrophage polarization induced by PAQR3 silencing. Therefore, these findings demonstrates that PAQR3 silencing accelerates diabetic wound healing by promoting M2 macrophage polarization and angiogenesis, which is mediated by the inhibition of STUB1-mediated PPARγ protein ubiquitination and degradation.
Similar content being viewed by others


Introduction
Diabetic wounds refer to non-healing wounds in diabetes mellitus patients due to the delayed healing process, which is still one of the major diabetic complications worldwide1,2. Recent epidemiological reports have shown that more than 300 million people have been diagnosed with diabetes mellitus worldwide, among which approximately 20% of diabetes mellitus patients suffer from diabetic wounds2,3. Diabetic foot ulcers are the most common form of diabetic wounds and usually lead to other severe pathogenic conditions, such as difficult walking, functional limitation, abscess, cellulitis, osteomyelitis, psychiatric stress, depression, septicaemia and gangrene1,2,4. In addition, more than 50% of global limb amputations have been attributed to non-healing diabetic wounds, superimposed infections and diabetic foot ulcers2,5. Recently, multiple nonsurgical treatments, including wound dressing, negative pressure wound therapy and hyperbaric oxygen treatment as well as debridement, revisional surgery and vascular reconstruction have been widely used for the clinical management of diabetic wounds, which fail to effectively decrease diabetic wound incidence and protect diabetic patients from limb amputation3. The elucidation of pathogenic mechanisms underlying diabetic wound development and progression may allow the development of new therapeutic regimens with improved efficacies.
Macrophages are a major group of white blood cells involved in homoeostasis maintenance and host innate immune defense, and they are well known for their potent capabilities of clearing cellular debris, invading pathogens and cancer cells6,7. Importantly, macrophage dysfunction has been regarded as an important reason for non-healing diabetic wound pathogenesis due to the roles of macrophages in cleansing infectious microorganisms and promoting tissue rebuilding for wound closure8,9. Moreover, the regulation of inflammation progression and resolution by macrophages is also substantially associated with diabetic wound healing and aggravation8,10. Specifically, macrophages under certain conditions further polarize to the classically activated M1 phenotype with pro-inflammatory functions or alternatively to the M2 phenotype acting as inflammation suppressers11. Recent reports have shown that acceleration of M2 macrophage polarization may promote diabetic wound healing12,13,14, while the induction of macrophage polarization towards the M1 phenotype may aggregate diabetic wounds15. Also, M2 macrophage polarization has been reported to activate angiogenesis by promoting EMT (epithelial-mesenchymal transition) during cancer development11,16. However, the roles of M2 macrophages in regulating angiogenesis during diabetic wound healing deserve further investigation.
PPARs (peroxisome proliferator-activated receptors) are a protein family of ligand-activated transcription factors, which belong to the nuclear hormone receptor superfamily and can be divided into three subtypes, PPARα, PPAR β/δ and PPARγ17. PPARγ (peroxisome proliferator-activated receptor γ) is widely implicated in various biological processes and diseases, such as lipid and glucose metabolism, inflammatory activation, insulin resistance, dyslipidaemia, chronic kidney diseases and diabetes17,18. Importantly, the elevated expression of PPARγ in macrophages results in suppressed inflammation and accelerated healing of diabetic wounds by inducing M2 macrophage polarization18. Moreover, recent reports have shown that PPAR proteins, such as PPARγ, are susceptible to ubiquitination-mediated degradation in certain contexts19,20. For instance, PAQR3 (progestin and adipoQ receptor 3) directly interacts with PPARα to promote PPARα polyubiquitination mediated by the E3 ubiquitin ligase HUWE1 (HECT, UBA and WWE domain-containing protein 1), resulting in proteasome-mediated PPARα degradation in hepatic lipid catabolism and response to starvation19. Additionally, PPARγ is ubiquitinated by the E3 ubiquitin ligase SMURF1 (Smad ubiquitin regulatory factor 1), which is associated with lipid accumulation and nonalcoholic fatty liver disease pathogenesis20. However, little is known about the ubiquitination of PPARγ associated with diabetic wound healing or the responsible E3 ubiquitin ligases.
In the present study, we investigate our hypothesis that silencing PAQR3 abrogates PPARγ protein ubiquitination and proteasome-mediated degradation to promote M2 macrophage polarization and angiogenesis, ultimately accelerating the healing of diabetic wounds. Our findings revealed novel molecular mechanisms underlying diabetic wound pathogenesis and served as a basis for developing new therapies for diabetic wound treatment.
Material and Methods
Animals and wound model
Male diabetic C57BL/6J db/db mice aged 7 weeks were purchased from the Model Animal Research Centre of Nanjing University (Nanjing, China), and they were adapted for one week and maintained in a specific pathogen-free atmosphere at the Experimental Animal Centre of the Second Xiangya Hospital, Central South University (Changsha, Hunan, Changsha) at 23 °C under a 12 h/12 h light/dark illumination cycle. All experimental operations on mice were approved in advance by the Experimental Animal Ethics Committee of the Second Xiangya Hospital, Central South University and performed strictly according to the Guide for the Care and Use of Laboratory Animals designated by the National Research Council. 10 mice were included in each group. The diabetic wound model was established using C57BL/6J db/db mice as previously described21. Briefly, the diabetic db/db mice were anaesthetized with pentobarbital and depilated on the back, and 5-mm full-thickness wounds were made on the dorsal side using a biopsy punch.
Adenovirus preparation and injection
The preparation of PAQR3-silencing recombinant adenovirus and its application to mice model were accomplished as previously reported22. Briefly, the PAQR3 shRNA and the NC (negative control) sequences were synthesized by GenePharma and were ligated into the adenovirus serotype 5 vector. The recombinant adenovirus was produced in HEK 293 T cells and tested for the absence of microbiological contamination. Subsequently, 20 μL of viral suspension (109 pfu/mL) was intradermally injected into the dorsal sides of diabetic db/db mice near the wound sites with four injections per wound each day. The healing of the wounds was closely observed once every three days for 12 consecutive days.
Immunohistochemistry (IHC)
The wounded mouse skin tissues were surgically collected on the day 6 after the wound surgery, fixed in 10% formalin, embedded in paraffin and cut into 5 μm sections. Tissue sections were blocked using TBST buffer containing 3% serum for 30 min, stained with haematoxylin and incubated overnight at 4 °C with diluted antibodies targeting CD31 (ab182981, Abcam), iNOS (ab15323, Abcam), or Arg-1 (ab91279, Abcam). Tissue sections were then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After being developed with DAB substrates (ab64238, Abcam), tissue sections were observed under light microscopy (Zeiss, Oberkochen, Germany) to evaluate CD31, iNOS, and Arg-1 protein expression.
Cell culture and transfection
Human monocytic leukaemia cell line THP-1, HUVECs (human umbilical vein endothelial cells), human immortalized keratinocytes HaCaT and HEK 293 T cells were obtained from the Cell Culture Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM (Dulbecco’s Modified Eagle’s Medium; Gibco) supplemented with 10% FBS (foetal bovine serum; Gibco) and 1% penicillin/streptomycin (Sigma-Aldrich) in a humidified atmosphere at 37 °C with 5% CO2. To silence PAQR3 expression, THP-1 cells were transfected with recombinant adenoviral vector containing PAQR3 shRNA sequences. To overexpress STUB1, the CDS (coding sequence) of STUB1 was amplified by RT-PCR, ligated into the pcDNA3.1 plasmid, and transfected into THP-1 cells as described above.
Cell treatments and conditioned medium
The differentiation of THP-1 cells into macrophages was performed by incubation in DMEM containing 60 μg/mL PMA (phorbol-12-myristate-13-acetate; Sigma-Aldrich) for 48 h. Cells were then washed two times with PBS and cultured in normal medium. M1 macrophage polarization of PMA-treated THP-1 cells was induced through treatment with 20 ng/mL IFN-γ (interferon γ; Sigma-Aldrich) and 1 μg/mL LPS (lipopolysaccharide; Sigma-Aldrich). The combination of 20 ng/mL IL-4 and 20 ng/mL IL-13 was used for induction of the M2 macrophage polarization of PMA-treated THP-1 cells. Inhibition of PPARγ activity was realized by treating cells with 10 μM GW9662 (HY-16578, MedChemExpress) for 24 h. For analysis of protein degradation, cells were treated with CHX (Cycloheximide; HY-12320, MedChemExpress) for 0, 4, 8, or 12 h followed by detection of protein level by western blotting. The CM (conditioned medium) from M2 macrophages with PAQR3 silence or not was prepared as previously described23.
Quantitative RT-PCR
For quantitation of mRNA expression, total RNA samples were prepared from cultured cells or mouse skin tissues using TRIzol™ Reagent (#15596026, Thermo Fisher Scientific) following the manufacturer’s instructions. Mouse skin tissues were collected on the day 6 after wound surgery. The concentrations of RNA samples were determined through spectrophotometry, and 3 μg of RNA from each group was used for cDNA synthesis by the reverse transcription method using EasyScript Reverse Transcriptase (AE101-02, TransGen Biotech) according to the manufacturer’s instructions. The relative mRNA levels were measured via real-time quantitative PCR using a SYBR Green PCR Mastermix Kit (SR1110, Solarbio) according to the manufacturer’s instructions. Based on at least three biological replicates, the mRNA expression levels were calculated via the 2-△△Ct method using β-actin as the internal standard.
Western blotting
For comparison of protein abundances, total proteins in cultured cells or mouse skin tissues were isolated using the Total Protein Extraction Kit (AMJ-KT0007, AmyJet Scientific) following the manufacturer’s instructions. Mouse skin tissues were collected at day 6 after wound surgery. After boiling at 100 °C in protein loading buffer for 5 min, approximately 30 μg of protein from each sample was subjected to SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked with 5% lipid-free milk solution for 2 h at room temperature, incubated with diluted primary antibodies overnight at 4 °C, incubated with diluted secondary antibodies for 1–2 h at room temperature, and developed with chemiluminescence substrates (Millipore). Relative protein levels from at least three biological replicates were compared using β-actin as the internal standard. The following antibodies were used in the present study: anti-PAQR3 (ab174327, Abcam), anti-PPARγ (#2443, Cell Signalling Technology), anti-PPARα (ab3484, Abcam), anti-ubiquitin (ab140601, Abcam), anti-MDM2 (ab16895, Abcam), anti-STUB1 (ab134064, Abcam), anti-SMURF1 (ab236081, Abcam) and anti-β-actin (#3700, Cell Signaling Technology).
Macrophage polarization evaluation
The polarization of macrophages was analysed by measuring the percentages of M1 and M2 phenotype cells using flow cytometry. Briefly, after treatments, cells were collected by centrifugation at 800 g for 5 min, washed twice with PBS, and then incubated with diluted primary antibodies (Thermo Fisher Scientific) targeting M1 or M2 macrophages for 25 min at 4 °C. After one wash with PBS, cells were fixed with 4% paraformaldehyde and analysed by flow cytometry (Beckman Coulter). M1 macrophages were defined by measuring the expression of CD11b and CD86 and on the cell surface, while M2 macrophages were defined by the expression of CD11b and CD206.
Cell migration
The migration of HaCaT cells was evaluated using a wound healing assay. Briefly, 2.0 × 105 HaCaT cells were cultured in a 96-well plate at 37 °C for 24–48 h until a complete cell monolayer was formed. Subsequently, a straight line was gently made in the middle of the cell monolayer using a sterile pipette tip to mimic the wound. Cells were then cultured under macrophage-conditioned medium with PAQR3 silence or not at 37 °C for another 24 h. The narrowing of the cell scratch was imaged under light microscopy (Zeiss).
Tube formation
The angiogenic capacities of HUVECs were detected using a tube formation assay. Briefly, HUVECs were cultured at 37 °C for 24 h and then seeded in Transwell plates (Corning) coated with Matrigel (BD Bioscience) containing macrophage-conditioned medium as specified. Cells were cultured at 37 °C for another 24–48 h. Cells forming tubular structures were observed using a phase-contrast microscope, and the tube numbers were counted to evaluate the angiogenic capacity of HUVECs.
Co-immunoprecipitation
After specified transfections or treatments, cultured cells were lysed in RIPA lysis buffer (P0013D, Beyotime Biotechnology) on ice for 30 min. Cell lysates were then centrifuged at 13,000 rpm at 4 °C for 30 min, and the supernatants were mixed with 1 μg of antibodies recognizing IgG (control) or target proteins and incubated overnight at 4 °C. The above mixture was further incubated with 10 μL of Protein A Agarose (P2051–10 mL, Beyotime Biotechnology) at 4 °C for 2–3 h with gentle shaking. After centrifugation at 3000 rpm for 3 min at 4 °C, the agarose beads were washed three times with RIPA lysis buffer and mixed with protein loading buffer for detection through western blotting.
Bioinformatics and statistical analysis
Bioinformatics prediction of E3 ubiquitin ligases involved in the interaction with PPARγ protein was performed using UbiBrowser software (http://ubibrowser.ncpsb.org.cn/ubibrowser/). All experiments were performed in at least three biological replicates, and each biological replicate contained three technical replicates. Data are presented as the mean ± standard deviation and were analysed by SPSS 18.0 software. Differences between two or more groups were evaluated by Student’s t test or one-way ANOVA (analysis of variance) followed by Tukey’s post-hoc test. P < 0.05 was considered a significant difference.
Results
PAQR3 silencing promotes wound healing and angiogenesis in diabetic mice
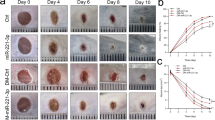
To evaluate the influences of PAQR3 expression on diabetic wound healing, the diabetic wound model was established in C57BL/6 J db/db mice through induction of skin wounds. We found that the wound healing in diabetic mice was significantly less than that in WT (wild type) mice 12 days after the induction of wounds, which validated the delayed wound healing caused by diabetes (Fig. 1A, B). However, silencing of PAQR3 by local injection of PAQR3-silencing recombinant adenovirus around wound sites effectively accelerated the wound healing speed in diabetic db/db mice (Fig. 1A, B), demonstrating the inhibitory effects of PAQR3 on diabetic wound healing. We next measured the expression of the angiogenesis biomarker CD31, which revealed significantly repressed expression of CD31 in wounded skin tissues in db/db mice compared to WT mice (Fig. 1C). Additionally, the injection of PAQR3-silencing recombinant adenovirus significantly enhanced the expression of CD31 in wounded skin tissues of db/db mice (Fig. 1C). These results showed that silencing PAQR3 effectively accelerated wound healing and angiogenesis in diabetic mice.

A The skin wound healing in C57BL/6J db/db mice was promoted by PAQR3 silencing. PAQR3-silencing recombinant adenovirus was injected into skin tissues near the wound site, and wound changes were observed once every three days for 12 consecutive days. B Quantitation of wounded skin areas in C57BL/6J db/db mice injected with PAQR3-silencing recombinant adenovirus. C The expression of CD31 in wounded skin tissues of C57BL/6J db/db mice injected with PAQR3-silencing recombinant adenovirus. CD31 expression in mouse skin tissues was detected on day 6 after the wound surgery by IHC at 100× magnification. Scale bar: 100 μm. WTwild type, Ad adenovirus, PAQR3 progestin and adipoQ receptor 3; NC negative control. 10 mice were included in each group. *P < 0.05 and ***P < 0.001.
PAQR3 silencing activates M2 macrophage polarization and elevates PPARα/γ expression in wounded skin tissues of diabetic mice
To explore the potential mechanisms underlying wound healing and angiogenesis by PAQR3, we subsequently analysed the alteration of macrophage polarization and PPAR protein expression in wounded skin tissues of diabetic mice. IHC analysis demonstrated that the expression of the M1 macrophage marker, iNOS, was significantly increased in wounded skin tissues of db/db mice compared to WT mice (Fig. 2A). In contrast, the expression of the M2 macrophage marker, Arg-1, in wounded skin tissues of db/db mice was remarkably lower than that of WT mice (Fig. 2A). However, PAQR3 silencing effectively suppressed iNOS expression and elevated Arg-1 expression in wounded skin tissues of db/db mice (Fig. 2A). Quantitative RT-PCR analysis showed that PAQR3 silencing significantly repressed the expression of three M1 macrophage markers, namely, iNOS, IL-12 (interleukin 12), and CD86, in wounded skin tissues of db/db mice, while the expression of the M2 macrophage markers, Arg-1, IL-10 (interleukin 10) and Mrc-1 (mannose receptor C type 1), were significantly increased by PAQR3 silencing in wounded skin tissues (Fig. 2B, C). Western blotting analysis demonstrated that the PAQR3 protein level was increased in the wounded skin tissues of db/db mice but significantly downregulated by the PAQR3-silencing recombinant adenovirus (Fig. 2D). Importantly, the abundances of PPARα/γ proteins in the wounded skin tissues of the above mouse models showed opposite alterations to those of the PAQR3 protein in response to wound induction and PAQR3 silencing (Fig. 2D). These results showed that PAQR3 also exerted inhibitory effects on M2 macrophage polarization and PPARα/γ expression in the wound healing of diabetic mice.

A In situ expression of iNOS and Arg-1 proteins in wounded skin tissues of db/db mice with silencing of PAQR3 expression. IHC was used to detect iNOS and Arg-1 protein levels in wounded mouse skin tissues on day 6 after the wound surgery at 200× magnification. Scale bar: 50 μm. Relative mRNA levels of M1 and M2 macrophage markers in wound skin tissues of db/db mice with locally silenced PAQR3 expression. The expression of M1 (B) and M2 (C) macrophage markers was measured on day 6 after the wound surgery using quantitative RT-PCR. (D) Protein levels of PAQR3 and PPARα/γ in wounded skin tissues of db/db mice on day 6 after the wound surgery under PAQR3 silencing. Protein levels in mouse skin tissues were analysed by western blotting using β-actin as the internal standard. WT wild type, Ad adenovirus, NC negative control; PAQR3 progestin and adipoQ receptor 3, iNOS inducible nitric oxide synthase, Arg-1 arginase 1, IL-10/12 interleukin 10/12, Mrc-1 mannose receptor C type 1, PPARα/γ peroxisome proliferator-activated receptor α/γ. 10 mice were included in each group. *P < 0.05, **P < 0.01 and ***P < 0.001.
PAQR3 silencing promotes M2 macrophage polarization and enhances PPARγ expression in PMA-treated THP-1 cells
A macrophage polarization model was established by treating THP-1 cells with PMA followed by induction of M1 or M2 polarization. We first confirmed that transfection of shPAQR3 greatly repressed the expression of PAQR3 protein expression in PMA-induced THP-1 cells (Fig. 3A). Moreover, we observed that silencing PAQR3 in PMA-treated THP-1 cells significantly suppressed their polarization towards the M1 phenotype induced by LPS and IFN-γ treatments (Fig. 3B, C). Consistently, the elevated expression of the M1 macrophage markers, iNOS, IL-12 and IL-1β, in macrophages treated with LPS and IFN-γ were significantly downregulated by shPAQR3 transfection (Fig. 3D). In addition, the expression of PPARγ protein in THP-1 cells was significantly increased by LPS and IFN-γ treatments, which was further enhanced by PAQR3 silencing (Fig. 3E). Correspondingly, the M2 macrophage polarization of PMA-treated THP-1 cells was significantly induced by IL-4 and IL-13 treatments, which was further promoted by PAQR3 silencing (Fig. 3F, G). The expression of the M2 macrophage markers, Arg-1, IL-10, Mrc-1, Ym-1 (chitinase 3-like 3), and Fizz1 (found in inflammatory zone 1), were markedly increased in PMA-treated THP-1 cells by IL-4 and IL-13 treatments, and were further elevated by PAQR3 silencing (Fig. 3H). Furthermore, the PPARγ protein level in PMA-treated THP-1 cells was significantly increased by IL-4 and IL-13 treatments, which was further increased by PAQR3 silencing (Fig. 3I). These results demonstrated that PAQR3 silencing effectively promoted M2 macrophage polarization and enhanced PPARγ expression.

A Relative PAQR3 protein level in PMA-treated PAQR3-deficient THP-1 cells. Protein level was measured by western blotting with β-actin as the internal standard. Suppression of the M1 polarization of PMA-treated THP-1 cells by PAQR3 silencing. M1 macrophage polarization was induced by LPS/IFN-γ treatments, and M1 macrophage polarization was measured by flow cytometry (B) followed by quantitation (C). D Relative mRNA levels of M1 macrophage markers in PMA-treated THP-1 cells regulated by LPS/IFN-γ treatments and PAQR3 silencing. Quantitative RT-PCR was used to detect the mRNA levels of M1 macrophage markers. E PPARγ protein level in PMA-treated THP-1 cells after LPS/IFN-γ treatments combined with PAQR3 silencing. (F, G) Promotion of M2 macrophage polarization of PMA-treated THP-1 cells by IL-4/IL-13 treatments and silencing of PAQR3. H Alterations in the mRNA levels of M2 macrophage markers in PMA-treated THP-1 cells following IL-4/IL-13 treatments and PAQR3 silencing. I PPARγ protein level was increased in PMA-treated THP-1 cells after IL-4/IL-13 treatments and PAQR3 silencing. PAQR3 progestin and adipoQ receptor 3, NC negative control, LPS lipopolysaccharide, IFN-γ interferon γ, iNOS inducible nitric oxide synthase, IL-1β/4/10/12/13 interleukin 1β/4/10/12/13, Arg-1 arginase 1, Mrc-1 mannose receptor C type 1, Ym-1, chitinase 3-like 1, Fizz1 found in inflammatory zone 1. All experiments were performed in at least three biological replicates, and each biological replicate contained three technical replicates. *P < 0.05, **P < 0.01 and ***P < 0.001.
PAQR3 silencing promotes the migration of keratinocytes and enhances the angiogenic ability of HUVECs
For additional cellular insights into the regulation of diabetic wound healing by PAQR3, we cultured keratinocytes HaCaT or HUVECs with CM (conditioned medium) from M2 macrophages with PAQR3 silence or not. The wound-healing assay demonstrated that the migration rate of HaCaT cells was effectively promoted by culture with CM from M2 macrophages, and an even greater migration rate was achieved by culture with CM from M2 macrophages with PAQR3 silencing (Fig. 4A, B). In addition, the tube formation assay indicated that culture with CM from M2 macrophages significantly enhanced the tube formation capacity of HUVECs (Fig. 4C, D), and HUVECs cultured with CM from PAQR3-deficient M2 macrophages showed even greater tube formation (Fig. 4C, D). These results revealed that PAQR3 silencing in macrophages effectively promoted keratinocyte migration and enhanced the angiogenic capacity of vascular endothelial cells.

A The migration of HaCaT cells was accelerated by culturing with conditioned medium from PAQR3-deficient M2 macrophages. The migration rates of cells were evaluated by a wound healing assay at 40× magnification. Scale bar: 500 μm. B Quantitation of HaCaT cell migration shown in Panel A. C The angiogenic capacities of HUVECs were enhanced by culturing with conditioned medium from PAQR3-deficient M2 macrophages. Cell angiogenesis was assessed by a tube formation assay at 100× magnification. Scale bar: 200 μm. D Quantitative analysis of tube formation shown in Panel C. CM conditioned medium, NC negative control; PAQR3 progestin and adipoQ receptor 3, All experiments were performed in at least three biological replicates, and each biological replicate contained three technical replicates. **P < 0.01.
PAQR3 mediates PPARγ protein degradation via the ubiquitination pathway
To investigate the molecular mechanism of PAQR3-induced PPARγ protein suppression, we first evaluated the direct binding between PAQR3 and PPARγ. Co-IP (co-immunoprecipitation) analysis showed that the PAQR3 protein could bind with the PPARγ protein in THP-1 cells (Fig. 5A). However, quantitative RT-PCR analysis showed that the mRNA level of PPARγ in THP-1 cells was not significantly altered by silencing PAQR3 (Fig. 5B). In addition, PAQR3 silencing significantly suppressed the decrease in PPARγ protein levels in THP-1 cells treated with CHX (cycloheximide), which could inhibit protein synthesis (Fig. 5C). To test whether the PAQR3-regulated PPARγ protein degradation is mediated by the ubiquitination pathway, we evaluated the ubiquitination of PPARγ protein using a Co-IP assay. We observed that the ubiquitination of PPARγ protein in THP-1 cells was significantly repressed by PAQR3 silencing (Fig. 5D). These results indicate that PPARγ protein degradation in THP-1 cells is promoted by PAQR3 via the ubiquitination pathway.

A Direct interaction between PAQR3 and PPARγ in THP-1 cells. Protein-protein interactions in cells were verified by Co-IP. B Relative PPARγ mRNA level in THP-1 cells transfected with shPAQR3. Quantitative RT-PCR was performed to detect PPARγ mRNA level. C PAQR3 silencing promoted the stability of the PPARγ protein in THP-1 cells. PPARγ protein levels were analysed by western blotting after cells were treated with CHX for 0, 4, 8 or 12 h. D PAQR3 silencing inhibited the ubiquitination of PPARγ protein in THP-1 cells. The ubiquitination of PPARγ protein was measured by Co-IP. PAQR3 progestin and adipoQ receptor 3, PPARγ peroxisome proliferator-activated receptor γ, IP immunoprecipitation, IB immunoblotting CHX, cycloheximide. All experiments were performed in at least three biological replicates, and each biological replicate contained three technical replicates. ns not significant.
PAQR3 interacts with the E3 ubiquitin ligase STUB1 to promote PPARγ protein degradation in macrophages
For better understanding of the PAQR3-induced PPARγ protein degradation, we analysed the potential E3 ubiquitin ligases responsible for mediating protein ubiquitination and degradation. Bioinformatics prediction using UbiBrowser software showed that top three E3 ubiquitin ligases may bind with the PPARγ protein, including MDM2, STUB1 and SMURF1 (Fig. 6A). Through a Co-IP assay, we found that STUB1, MDM2 and SMURF1 proteins could directly bind with the PAQR3 protein in PMA-treated THP-1 cells. Among these three proteins, STUB1 showed the strongest association with PAQR3, suggesting STUB1 as the E3 ubiquitin ligase mediating PAQR3-induced PPARγ ubiquitination in macrophages (Fig. 6B). For verification, we performed additional Co-IP assay, demonstrating that the STUB1 protein could also associate with the PPARγ protein in PMA-treated THP-1 cells (Fig. 6C). Moreover, silencing STUB1 in THP-1 cells significantly repressed the ubiquitination of PPARγ protein (Fig. 6D). In addition, the binding between STUB1 and PPARγ was impaired by silencing PAQR3 in PMA-treated THP-1 cells (Fig. 6E). These findings demonstrate that PAQR3-induced PPARγ protein ubiquitination and degradation is mediated by the interaction of the E3 ubiquitin ligase STUB1 in macrophages.

A Bioinformatics prediction of the interaction between the PPARγ protein and multiple E3 ubiquitin ligases. Strong association of the PPARγ protein with three E3 ubiquitin ligases, namely, MDM2, STUB1, and SMURF1, was predicted by bioinformatics using UbiBrowser software (http://ubibrowser.ncpsb.org.cn/ubibrowser/). B Direct association of PAQR3 protein with STUB1, MDM2, and SMURF1 proteins in PMA-treated THP-1 cells. Protein-protein interactions in cells were validated by a Co-IP assay using an anti-PAQR3 antibody. An antibody targeting IgG was used as the negative control. C Direct binding of the STUB1 protein with the PPARγ protein in PMA-treated THP-1 cells. Interaction between proteins was confirmed by a Co-IP assay. D STUB1 silencing decreased the ubiquitination of PPARγ protein in PMA-treated THP-1 cells. STUB1 silencing was induced by transfection with shRNA targeting the STUB1, and ubiquitination of the PPARγ protein was detected by western blotting using anti-ubiquitin antibody following immunoprecipitation with an antibody recognizing the PPARγ protein. E PAQR3 silencing impaired the interaction between STUB1 and the PPARγ protein in PMA-treated THP-1 cells. PAQR3 progestin and adipoQ receptor 3 PPARγ peroxisome proliferator-activated receptor γ, MDM2 murine double minute 2, STUB1 STIP1 homology and U-box containing protein 1 SMURF1 Smad ubiquitination regulatory factor-1, IP immunoprecipitation, IB immunoblotting, NC negative control. All experiments were performed in at least three biological replicates, and each biological replicate contained three technical replicates.
PAQR3 silencing-induced M2 macrophage polarization is dependent on STUB1-mediated PPARγ ubiquitination
We further investigated the roles of STUB1-induced PPARγ ubiquitination and degradation in M2 macrophage polarization regulated by PAQR3 silencing. We found that the M1 polarization of PMA-treated THP-1 cells treated with LPS and IFN-γ was significantly inhibited by shPAQR3, but this inhibition was restored by simultaneous treatment with the PPARγ inhibitor GW9662, or overexpression of STUB1 (Fig. 7A). Consistently, we observed that the expression of three M1 macrophage markers, namely, iNOS, IL-12 and IL-1β, in PMA-treated THP-1 cells were significantly decreased by shPAQR3 following LPS and IFN-γ treatments, but GW9662 treatment or STUB1 overexpression significantly increased their expression (Fig. 7B). In contrast, we showed that shPAQR3 promoted the M2 macrophage polarization of PMA-treated THP-1 cells induced by IL-4 and IL-13, which was significantly suppressed by a combination of shPAQR3 with GW9662 treatment or STUB1 overexpression (Fig. 7C). Consistently, the elevations in the M2 macrophage markers, Arg-1, IL-10 and Mrc-1, in PMA-treated THP-1 cells treated with shPAQR3 following IL-4 and IL-13 treatment were also significantly reduced by combined treatment with GW9662 or STUB1 overexpression (Fig. 7D). These results reveal that the promotion of M2 macrophage polarization by PAQR3 silencing is dependent on PPARγ ubiquitination induced by the E3 ubiquitin ligase STUB1.

A Percentages of M1 macrophages following PAQR3 silencing and treatment with LPS/IFN-γ and GW9662 or STUB1 overexpression. M1 macrophage polarization was assessed by flow cytometry. B Relative expression of M1 macrophage markers in PMA-treated THP-1 cells with PAQR3 silencing after treatment with LPS/IFN-γ and GW9662 or STUB1 overexpression. The mRNA levels of iNOS, IL-12, and IL-1β were detected by quantitative RT-PCR. C Inhibition of the M2 macrophage polarization of PMA-treated PAQR3-deficient THP-1 cells by GW9662 treatment or STUB1 overexpression. D Effects of GW9662 treatment or STUB1 overexpression on the expression of M2 macrophage markers in PMA-treated PAQR3-deficient THP-1 cells. Arg-1, IL-10, and Mrc-1 mRNA levels were detected by quantitative RT-PCR. PAQR3 progestin and adipoQ receptor 3 NC negative control, LPS lipopolysaccharide, IFN-γ interferon γ, STUB1 STIP1 homology and U-Box containing protein 1, iNOS inducible nitric oxide synthase, IL-1β/4/10/12/13 interleukin 1β/4/10/12/13, Arg-1 arginase 1, Mrc-1 mannose receptor C type 1. All experiments were performed in at least three biological replicates, and each biological replicate contained three technical replicates. *P < 0.05, **P < 0.01 and ***P < 0.001.
Discussion
Diabetic wounds, such as diabetic foot ulcers, are the severe medical problem associated with diabetes2,3. Previous reports have shown that the pathogenesis of diabetic wounds is linked to dysregulated macrophage differentiation and function, and modulation of macrophage polarization has been suggested as a promising strategy for treating non-healing diabetic wounds8,9,12,13,14. However, the molecular mechanisms driving directed polarization of macrophages in wounded skins of diabetic patients remain poorly elucidated. In the present study, we showed that silencing PAQR3, a membrane receptor protein of the PAQR family24, effectively promoted wound healing and angiogenesis in diabetic mice. Our subsequent investigations revealed that PAQR3 silencing significantly enhanced M2 macrophage polarization and the expression of PPARγ protein using both a mouse diabetic wound model and a cellular model based on the THP-1 monocytic leukaemia cell line. Moreover, silencing PAQR3 in macrophages promoted the migration of HaCaT keratinocytes and the angiogenesis of HUVECs. Finally, we showed that PAQR3 interacted with the STUB1 protein to promote the ubiquitination and degradation of PPARγ protein, which was implicated in PAQR3-mediated M2 macrophage polarization. These results provided novel insights into the molecular mechanisms of macrophage polarization in non-healing diabetic wounds.
PAQR3 is a transmembrane protein mainly distributed in the golgi apparatus, and it is widely implicated in various biological processes and diseases, including diabetes mellitus24,25. Specifically, recent investigations have shown that PAQR3 regulates the deposition of extracellular matrix in glomerular mesangial cells in response to high glucose stimulus24. Moreover, the depletion of PAQR3 results in enhanced resistance to obesity induced by a high-fat diet and activation of leptin signalling25. However, the pathogenic functions of the PAQR3 protein in non-healing diabetic wounds have not been addressed. In the present study, we demonstrated that silencing PAQR3 significantly promoted the healing of diabetic wounds in a mouse model, and the migration of keratinocytes and angiogenesis of HUVECs in an in vitro cellular model, demonstrating for the first time that PAQR3 is an important modulator of diabetic wound healing. Additionally, these observations suggested that PAQR3 may serve as a new target for diabetic wound treatment. As mentioned above, the wound healing process can be accelerated by M2 macrophages at the wound sites12,13,14. In the present study, we revealed that PAQR3 silencing effectively promoted M2 macrophage polarization and repressed M1 macrophage polarization using both animal and cellular models. Together, these results indicated the essential roles of PAQR3 in diabetic wound formation by regulating macrophage polarization.
PPARγ is a ligand-activated transcription factor, and increases in PPARγ expression have been shown to accelerate diabetic wound healing by promoting the polarization of macrophages towards the M2 phenotype18. In the present study, we showed that the expression of PPARγ protein in wounded skin tissues of diabetic mice was significantly elevated by PAQR3 silencing, and the PPARγ inhibitor, GW9662, abrogated the enhanced M2 macrophage polarization induced by PAQR3 silencing. These assays confirmed that the promotion of M2 macrophage polarization and diabetic wound healing by PAQR3 silencing was due to increased PPARγ protein level in macrophages. Moreover, previous reports have shown that the PPARα/γ are degraded through ubiquitination and proteasome-mediated protein degradation pathways, involving two E3 ubiquitin ligases, namely, HUWE1 and SMURF119,20. However, the present study showed that the PPARγ protein directly binds with another E3 ubiquitin ligase STUB1. The STUB1-mediated ubiquitination and degradation of functional proteins have recently been shown to regulate the development of multiple biological and pathogenic processes, such as mitochondrial biogenesis, antiviral innate immune response and advanced prostate cancer treatment26,27,28, but little is known about the roles of STUB1 in diabetes and related complications. Here, we showed that STUB1 associated with the PAQR3 and PPARγ proteins to mediate the ubiquitination and degradation of the PPARγ protein in macrophages. Moreover, PAQR3 silence effectively suppressed PPARγ ubiquitination to promote M2 macrophage polarization, which was mitigated by STUB1 overexpression. Thus, these findings indicated the critical roles of PAQR3 and STUB1-mediated PPARγ protein degradation in diabetic wound development and treatment.
The formation of new blood vessels (angiogenesis) in wounded tissues has been established as an essential biological process substantially contributing to wound healing associated with diabetes and other pathogenic conditions1,29,30. The activation of keratinocytes and endothelial cells as well as their release of angiogenetic factors are critical steps during the reconstruction of wounded microvasculature31. Additionally, activation of the angiogenesis process has been extensively validated as a feasible method to accelerate wound healing in diabetic patients32,33,34,35. For instance, the enhancement of endothelial cell activities by maggot excretions and secretions has been reported to promote the healing of diabetic feet36. In addition, the M2 macrophage polarization also facilitates angiogenesis by regulating the EMT pathway16. In the present study, we showed that PAQR3 silencing promoted angiogenesis in wounded skin tissues of diabetic mice, and we also showed that PAQR3 silencing enhanced the migration of keratinocytes and angiogenic capacities of HUVECs. These observations established the modulation of angiogenesis by PAQR3-regulated M2 polarization of macrophages as a vital cellular process underlying diabetic wound pathogenesis, which may be utilized for accelerating diabetic wound healing.
It should be noted that using PAQR3-flox mice and Mx1-Cre mice with macrophage-specific deletion of PAQR3 expression will further validate the implication of PAQR3-regulated macrophage polarization in diabetic wound progression. Also, the present study is limited by the exclusive inclusion of male mice for model establishment because male mice are relatively stable in physical status, such as hormone levels and bioactive enzyme activities.
In summary, the present study reveals that silencing PAQR3 in macrophages effectively promotes diabetic wound healing by enhancing M2 macrophage polarization and angiogenesis. PAQR3 silencing enhances M2 macrophage polarization by increasing PPARγ expression through inhibiting STUB1-mediated PPARγ protein ubiquitination and degradation. These results provide novel insights into the pathogenic mechanism of diabetic wounds, which could be further explored for diabetic wound treatment.
Data availability
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
References
Lotfy M., Adeghate J., Kalasz H., Singh J., Adeghate E. Chronic complications of Diabetes Mellitus: A mini review. Curr. Diabetes Rev. 13, 3–10 (2017).
Patel S., Srivastava S., Singh M.R., Singh D. Mechanistic insight into diabetic wounds: Pathogenesis, molecular targets and treatment strategies to pace wound healing. Biomed. Pharmacother. 112, 108615 (2019).
Fui L.W., Lok M.P.W., Govindasamy V., Yong T.K., Lek T.K., Das A.K.. Understanding the multifaceted mechanisms of diabetic wound healing and therapeutic application of stem cells conditioned medium in the healing process. J. Tissue Eng. Regen. Med. 13, 2218–2233 (2019).
Uccioli L., Izzo V., Meloni M., Vainieri E., Ruotolo V., Giurato L. Non-healing foot ulcers in diabetic patients: general and local interfering conditions and management options with advanced wound dressings. J. Wound Care 24, 35–42 (2015).
Vijayakumar V., Samal S.K., Mohanty S., Nayak S.K. Recent advancements in biopolymer and metal nanoparticle-based materials in diabetic wound healing management. Int. J. Biol. Macromol. 122, 137–148 (2019).
Beltraminelli T., De Palma M. Biology and therapeutic targeting of tumour-associated macrophages. J. Pathol. https://doi.org/10.1002/path.5403 (2020).
Vorst, E.P.C.V.D., Weber C. Novel features of monocytes and macrophages in cardiovascular biology and disease. Arterioscler. Thromb. Vasc. Biol. 39, e30–e37 (2019).
Ganesh G.V., Ramkumar K.M. Macrophage mediation in normal and diabetic wound healing responses. Inflamm. Res. 69, 347–363 (2020).
Aitcheson S.M., Frentiu F.D., Hurn S.E., Edwards K., Murray R.Z. Skin wound healing: Normal Macrophage function and macrophage dysfunction in diabetic wounds. Molecules 26 (2021).
Kim S.Y., Nair M.G. Macrophages in wound healing: Activation and plasticity. Immunol. Cell Biol. 97, 258–267 (2019).
Lawrence T., Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750-761 (2011).
Lee J., et al. Interleukin-23 regulates interleukin-17 expression in wounds, and its inhibition accelerates diabetic wound healing through the alteration of macrophage polarization. FASEB J. 32, 2086–2094 (2018).
Qing L., et al. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am. J. Transl. Res. 11, 655–668 (2019).
Wilkinson H.N., et al. Tissue iron promotes wound repair via M2 macrophage polarization and the chemokine (C-C Motif) Ligands 17 and 22. Am. J. Pathol. 189, 2196–2208 (2019).
Feng J., et al. Elevated Kallikrein-binding protein in diabetes impairs wound healing through inducing macrophage M1 polarization. Cell Commun. Signal. 17, 60 (2019).
Feng R., et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal 16, 54 (2018).
Ma Y., Shi M., Wang Y., Liu J. PPARγ and its agonists in chronic kidney disease. Int. J. Nephrol. 25 (2020).
Mirza R.E., et al. Macrophage PPARgamma and impaired wound healing in type 2 diabetes. J. Pathol. 236, 433–444 (2015).
Zhao Z., et al. Hepatic PPARα function is controlled by polyubiquitination and proteasome-mediated degradation through the coordinated actions of PAQR3 and HUWE1. Hepatology 68, 289–303 (2018).
Zhu K., X. et al. Non-proteolytic ubiquitin modification of PPARgamma by Smurf1 protects the liver from steatosis. PLoS Biol. 16, e3000091 (2018).
Kolumam G., et al. IL-22R Ligands IL-20, IL-22, and IL-24 promote wound healing in diabetic db/db Mice. PLoS One 12, e0170639 (2017).
Botusan I.R., et al. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc Natl Acad. Sci. USA 105, 19426–19431 (2008).
Shikama Y., et al. Palmitate-stimulated monocytes induce adhesion molecule expression in endothelial cells via IL-1 signaling pathway. J. Cell Physiol. 230, 732–742 (2015).
Li H., Wang Y., Chen B., Shi J. Silencing of PAQR3 suppresses extracellular matrix accumulation in high glucose-stimulated human glomerular mesangial cells via PI3K/AKT signaling pathway. Eur. J. Pharmacol. 832, 50–55 (2018).
Wang L., et al. PAQR3 has modulatory roles in obesity, energy metabolism, and leptin signaling. Endocrinology 154, 4525–4535 (2013).
Rao L., Sha Y., Eissa N.T. The E3 ubiquitin ligase STUB1 regulates autophagy and mitochondrial biogenesis by modulating TFEB activity. Mol Cell Oncol. 4, e1372867 (2017).
Zhou P., et al. MLL5 suppresses antiviral innate immune response by facilitating STUB1-mediated RIG-I degradation. Nat Commun. 9, 1243 (2018).
Liu C., et al. Proteostasis by STUB1/HSP70 complex controls sensitivity to androgen receptor-targeted therapy in advanced prostate cancer. Nat Commun 9, 4700 (2018).
Salazar J.J., Ennis WJ, Koh TJ. Diabetes medications: Impact on inflammation and wound healing. J. Diabetes Complic. 30, 746–752 (2016).
Okonkwo U.A., DiPietro LA. Diabetes and wound angiogenesis. Int. J. Mol. Sci. 18 (2017).
Greaves N.S., Ashcroft KJ, Baguneid M, Bayat A. Current understanding of molecular and cellular mechanisms in fibroplasia and angiogenesis during acute wound healing. J. Dermatol. Sci. 72, 206–217 (2013).
Bazrafshan A., Owji M., Yazdani M., Varedi M. Activation of mitosis and angiogenesis in diabetes-impaired wound healing by processed human amniotic fluid. J. Surg. Res. 188, 545–552 (2014).
Kant V., et al. Curcumin-induced angiogenesis hastens wound healing in diabetic rats. J. Surg. Res. 193, 978–988 (2015).
Gao F., et al. Hyaluronan oligosaccharides promote excisional wound healing through enhanced angiogenesis. Matrix Biol. 29, 107–116 (2010).
Lim Y.C., et al. Proinsulin C-peptide prevents impaired wound healing by activating angiogenesis in diabetes. J. Invest. Dermatol. 135, 269–278 (2015).
Sun X., et al. Maggot debridement therapy promotes diabetic foot wound healing by up-regulating endothelial cell activity. J. Diabetes Complicat. 30, 318–322 (2016).
Author information
Authors and Affiliations
Contributions
J.Q. conceived and designed the work that led to the submission. X.L. acquired data. W.-C.Z. played an important role in interpreting the results. J.Q. drafted, and C.S. revised the manuscript. All authors approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All experimental operations on mice were approved in advance by the Experimental Animal Ethics Committee of the Second Xiangya Hospital, Central South University, and performed strictly according to the Guide for the Care and Use of Laboratory Animals designated by the National Research Council.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Qiu, J., Shu, C., Li, X. et al. PAQR3 depletion accelerates diabetic wound healing by promoting angiogenesis through inhibiting STUB1-mediated PPARγ degradation. Lab Invest 102, 1121–1131 (2022). https://doi.org/10.1038/s41374-022-00786-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-022-00786-8
This article is cited by
-
Systematic pan-cancer analyses of the potential function of the Golgi scaffold protein PAQR3
Scientific Reports (2024)
-
Targeting protein modifications in metabolic diseases: molecular mechanisms and targeted therapies
Signal Transduction and Targeted Therapy (2023)
