
Abstract
Although there have been recent advances in the molecular pathology of ependymomas, little is known about the underlying molecular evolution during its development. Here, we assessed the clinical, pathological and molecular evolutionary process of ependymoma recurrence in a 9-year-old patient who had seven recurrences of supratentorial ependymoma and died from intracranial multiregional recurrences at the age of 19 years old. Whole-genome sequencing (WGS) of 7 tumor samples (1 primary and 6 subsequent recurrent tumors) was performed to elucidate the mutation landscape and identify potential driver mutations for tumor evolution. The genetic profiles of the seven tumor specimens showed significant heterogeneity and suggested a highly branched evolutionary pattern. The mutational signatures and chromothripsis changed with treatments. Strikingly, adhesion G protein-coupled receptor L3 (ADGRL3, also known as Latrophilins 3, LPNH3) was found to be consistently mutated during the entire disease process. However, Sanger sequencing of other 78 ependymoma patients who underwent surgery at our institution showed no genetic alteration of ADGRL3, as found in the present case. The mRNA levels of ADGRL3 were significantly lower in ependymomas (n = 36), as compared with normal brain tissue (n = 3). Grade III ependymomas had the lowest ADGRL3 expression. Moreover, ependymomas with lower mRNA level of ADGRL3 had shorter overall survival. Our findings, therefore, demonstrate a rare evolutionary process of ependymoma involving ADGRL3.
Similar content being viewed by others


Introduction
Ependymoma usually arises from the brain ventricle or ependymal epithelium of the spinal cord and accounts for ~2–3% of all primary tumors of the central nervous system (CNS)1. Its incidences in children and young adults are higher than in older people. Although the prognosis of most ependymomas is comparatively good, about 40% of the cases remain incurable despite radical chemotherapy and irradiation2,3. Furthermore, ependymoma displays remarkable spatial and temporal heterogeneity relating to tumor evolution4. According to the newest (the 5th version) WHO CNS guideline, ependymomas should now be classified based on a combination of histopathological, molecular and anatomic features5; namely as supratentorial, posterior fossa, spinal, myxopapillary and subependymoma2. The first three groups can be further divided into subgroups according to their molecular characteristics, namely, C11orf95 fusion-positive or YAP1 fusion-positive supratentorial, PFA or PFB of the posterior fossa, and MYCN-amplified spinal5. ZFTA is the new designation for C11orf95 and is considered more representative of the tumor type than RELA because it can be fused with partners more than RELA6.
Tumor evolution can appear upon treatment, leading to somatic mutations during the evolutionary process and tumor heterogeneity at different regions of the tumor or recurrence lesions7,8,9. Most of the alterations may not lead to harmful effects but a small set of mutations can lead to driver events for initiating tumor cells’ growth10,11. Delineating the details of tumor evolution could help to identify new treatment approaches, guide precision medicine, and predict patients’ outcomes12,13. Deciphering the mutational processes, the extent of heterogeneity, and its dynamics over time can reveal a tumor’s life history, and therefore, help monitoring subclonal dynamics through treatment14. Whether targeting those driver events leads to improvement of patients’ prognosis is currently being studied in the Deciphering Anti-tumor Response With Intratumor Heterogeneity trial (DARWIN, http://clinicaltrials.gov/show/NCT02183883).
Genetic studies have shown that molecular alterations are very common in ependymomas, which display a broad range of cytogenetic aberrations15,16. Most commonly observed as gains of chromosomes 1q, 5, 7,9,11,18, and 20 and losses of chromosomes 1p, 3,6q, 6, 9q, 13q, 17, and 2215. Here, we report the clinical features and genomic profiles of an ependymoma patient who was initially diagnosed as supratentorial ependymoma, had 7 relapses in 9 years despite intensive treatments but passed away due to intracranial multiple recurrences, as a perfect model for tumor evolution study. Strikingly, mutation of ADGRL3, a member of G protein-coupled receptors (GPCRs), was consistently observed during the entire disease process. Deficient ADGRL3 expression or its mutation has been found to be associated with attention-deficit hyperactivity disorder (ADHD) in human17,18 but its role in tumorigenesis has not yet been described. In this study, we deciphered the role of ADGRL3 mutation in this ependymoma case and other ependymal tumor patients at our Cancer Center.
Materials and methods
Patients and specimens
The peripheral blood monocytes and 7 tumor samples (1 primary and 6 subsequent recurrent tumors) from the patient, anonymously named PYY for this study, were collected. Although the patient had 7 recurrences, no surgery was performed for the last recurrent tumor and only 7 tumor samples were available from the patient. RNA and DNA samples of tumor tissue from other ependymoma patients treated at the Sun Yat-sen University Cancer Center (SYSUCC, Guangzhou, China) were extracted. The patients were all informed and provided signed consent regarding the use of their biological specimens and clinical information for research purposes. This study was approved by the ethics committee and institutional review board of SYSUCC.
Immunohistochemistry (IHC) staining
IHC staining was performed following standard protocol, as previously described19. Briefly, the slides were incubated with mouse monoclonal antibodies for GFAP, EMA, Ki67 and Oligo 2 staining and detection following the manufacturer’s protocol. They were then sequentially incubated with a secondary antibody (Envision; Dako, Glostrup, Denmark) for 1 h at RT, stained with DAB (3, 3-diaminobenzidine), counterstained with Mayer hematoxylin, dehydrated, and mounted. Details of the involved antibodies are listed in Supplementary Table 1.
Detection of RELA fusion gene and 1p19q co-deletion
The DNA probes and the detailed procedure for fluorescence in situ hybridization (FISH) are described in our previous publications19,20. Briefly, all the probes were diluted at a ratio of 1:50 in Den-Hyb buffer (Insitus, Albuquerque, NM, USA) and tested on normal metaphases for their specificity prior to application on the paraffin sections. Hybridization and detection of RELA and deletions on chromosomes 1p and 19q were performed. Hybridization signals for each probe were assessed under an Olympus BX51 TRF microscope (Olympus, Tokyo, Japan) equipped with a triple-pass filter (4',6'-diamidino-2-phenylindole, DAPI/Green/Orange; Abbott Molecular Inc., Des Plaines, IL, USA). The scoring for RELA was conducted in no less than 50 non-overlapping nuclei per core in tumor regions. Tumor specimens were considered to be present for rearrangement if more than 15% of the nuclei demonstrated separate red and green signals.
Sample preparation and whole-genome sequencing (WGS)
The genomic DNAs of the 7 tumor tissue specimens and blood monocytes of PYY were extracted and subjected to WGS. Sequencing was performed using HiSeq X10 (Illumina, San Diego, CA, USA). Standard bioinformatics analyses were done by Gene Denovo (Guangzhou, China). The initial quality control of the data was carried out using the FASTQC tool. To obtain high-quality clean reads, reads that failed quality control were removed from subsequent analysis.
Alignment of sequencing reads and somatic variant detection
150-bp paired-end Fastq files were aligned to the human reference genome (UCSC hg19) using the BWA-MEM (V0.7.10) aligner in “bwa mem” mode. Aligned records were then used for indexing, deduplication, sorted, base recalibration and indel realignment following the GATK best practices (v3.8). MuTect (v1.4) was used to call somatic SNVs by comparing each tumor sample with its matched non-tumor counterpart. Somatic Indels were identified using Strelka (V2.7.1) and MANTA (V1.1.1). Mutations with a filter flag among “PASS” were selected. Imputation step was performed in-house to rescue mutations that were not reported due to inadequate coverage of the target site in a single sample. In order to determine the physical positions of each somatic mutation, ANNOVAR was used to align and annotate SNPs or Indels to the following database: 1000 Genomes Project, HAMAP, ESP6500, Clinvar, dbSNP. Both silent and non-silent somatic mutations were classified as either truncal or branched, and the mutational signatures of these variants were generated separately.
Copy-number and structural variant analysis
Control-FREEC software (V0.3.2) was used to identify somatic copy number variations (CNVs) from the tumor and germline control bam files with all default parameters. Somatic structural variations were classified using breakDancer (Max1.1.2).
Mutational signature analysis
We applied deconstructSigs to extract mutational signatures and to statistically quantify the contribution of each signature for each tumor. We confirmed the mutation site G1382A bidirectionally by Sanger sequencing (Tsingke Co., Guangzhou, P.R.China). Briefly, the genomic DNA was amplified using the following primers: 5'-TGGCATCCTGATACCTT-3' (forward) and 5'-TGATTTCTCGGGCTTC-3'(reverse). The products were then sequenced following standard procedures21.
Construction of phylogenetic tree
To further explore the clonal architecture of the disease, phylogenetic trees were constructed using the ‘discrete-characters Wagner parsimony’ method in PHYLOGENY INFERENCE PACKAGE (PHYLIP) version 3.695 to generate phylogenetic trees with the matched nonmalignant tissue as the out-group (input to PHYLIP). The trees were drawn using the DrawTree tool from the PHYLIP package. Selected driver genes were marked in the trunk and branch of evolutionary trees by the neighbor-joining (NJ) method using all somatic mutations identified in each tumor region in consideration. The trunk, ‘shared’ branches, and ‘private’ branches of the tree represented mutations in all tumor regions, in some but not all tumor regions, and in only one tumor region, respectively. The lengths of trunks and branches were proportional to the number of mutations acquired.
Quantitative real-time PCR
Total RNAs were extracted from 36 ependymoma and 3 normal brain samples using the RNA Quick Purification kit (ESscience, Shanghai, China) and reverse-transcribed into cDNA (Tiangen, Beijing, China) following the manufacturer’s protocol. Real-time quantitative PCR (RT-qPCR) was performed using the Bio-Rad CFX96 Real-Time PCR System (Bio-Rad Laboratories, Inc., Hercules, CA). All samples were analyzed in triplicates, the mRNA expression level was normalized to that of the housekeeping gene GAPDH. The primer sequences used were: ADGRL3, Forward: 5'-GATGGGGAGGCAAATCTGACA-3', Reverse: 5'- CCGTAGGGTGTAAGGGTTCAAT-3'; GAPDH, Forward: 5'- ACCACAGTCCATGCCATCAC -3', Reverse: 5'- CACCACCCTGTTGCTGTAGCC -3'. The expression level of ADGRL3 was divided into a high and low group based on the median cycle number.
GEPIA-based prognoses analysis
Gene Expression Profiling Interactive Analysis (GEPIA) was performed, as reported in our previous publication, to demonstrate the role of ADGRL3 in the prognosis of low grade glioma (LGG)22. GEPIA is an interactive web application for gene expression analysis based on 9736 tumors and 8587 normal samples from the TCGA and the GTEx databases, using the output of a standard processing pipeline for RNA sequencing data23. GEPIA is a time-saving and intuitive tool for making full use of big genomic data in TCGA and GTEx (http://gepia.cancer-pku.cn/index.html)24. OS (overall survival) and DFS (disease-free survival) were assessed based on the expression level of ADGRL3 in LGG.
Statistical analysis
The relationship between the expression level of ADGRL3 and prognosis (OS and DFS) was determined using the Kaplan–Meier method and Cox regression model. Data are shown as the mean ± standard error. P < 0.05 was considered statistically significant. Statistical analysis was performed using Graph Pad Prism 5.0.
Results
Disease progression of the patient
PYY was 9 years old when she was admitted and diagnosed with anaplastic supratentorial ependymoma in September 2007. Only surgery was performed on the initial primary tumor (T1). Surgery followed by radiotherapy was given for the first recurrence (T2). Surgery followed by adjuvant chemotherapy together with radiotherapy was given as from the second relapse (T3-T7). For the last intracranial multiregional recurrence, only chemotherapy was administered because neither surgery nor radiotherapy was applicable. Nine years after the initial diagnosis, the patient died from the 7th recurrence in August 2016. The tumor progression of the patient is summarized in Fig. 1A–C and Table 1.

A The disease progression timeline and therapeutic strategies, from diagnosis of the primary tumor (T1) to the patient’s death. The interval between the two tumors ranged from 5 months to 18 months, especially shorter in the later stages (5 and 9 months). The red box represents surgery, green box represents radiation and blue box represents chemotherapy. B Graphical representation of the tumor lesions’ locations. Colored numbers indicate the order of tumor recurrence and region, from the right or left frontal lobe (T1, T2, and T3) to left parietal lobe (T4), right frontal/occipital lobe (T5), right frontal/temporal/parietal lobe (T6), right frontal/temporal lobe (T7) and the last tumor recurrence occurred multi-regionally on April 2015 (T8). C MRI images of the tumor during the progression process from T1 to T8. The images shown are of the axial, sagittal and transverse plane (tumors were indicated by arrows).
Histologically, ependymoma is a circumscribed glioma composed of uniform small cells with round nuclei in a fibrillary matrix and characterized by perivascular anucleate zones (pseudorosettes) and ependymal rosettes. H&E was reviewed to confirm the diagnosis, and IHC staining for Ki67 was performed. The Ki67 index of the 7 tumor samples (T1 to T7) was 5%, 7%, 30%, 15%, 30%, 25% and 20% respectively25. Hypercellularity, cellular atypia, vascular proliferation, necrosis, mitosis and high Ki67 index indicated the diagnosis for grade III or anaplastic ependymoma. The tumor cells showed dot-like positive staining for EMA as well as for GFAP and Olig2, supporting the diagnosis of anaplastic ependymoma (Fig. 2A). In the recurrence cases, increasing cellular atypia, necrosis and higher Ki67 index were observed. It is well known that fusion of C11orf95 to RELA occurred in more than 70% of pediatric supratentorial ependymomas1,26,27,28, and thus, we performed FISH to detect RELA fusion. We found that RELA fusion was present in the primary lesion and all recurrent tumors (Fig. 2B). The status of 1p19q co-deletion and IDH1 mutation by FISH or WGS were also assessed but both were negative in the 7 specimens (Fig. 2B, Supplementary Fig. S1). Taken together, all tumors of the patient were classified as grade III supratentorial ependymoma with C11orf95 fusion gene based on the updated WHO guideline for CNS tumors (2016 version)1,5.

A Staining of the key parameters for pathological diagnosis, including H&E, Ki67, EMA, GFAP, and Oligo2. B Molecular features of 1p/19q and RELA by FISH, respectively. The arrow indicates the fusion of RELA.
Mutation signature alterations during the tumor progression
To investigate the landscape of genomic profile, we systematically analyzed the genomic alterations of the patient with WGS data. A total of 1149 somatic mutations were identified by removal of germline mutations from the peripheral blood monocytes control sample, including nonsynonymous (n = 746), stopgain (n = 30), stoploss (n = 1), nonframeshift substitution (n = 7)/deletion (n = 136)/insertion (n = 18), frameshift substitution (n = 6)/deletion (n = 171)/insertion (n = 34). The mutation load of T3 was the highest while that of T5 was the lowest, which were 102, 277, 295, 147, 94, 131, and 103 from T1 to T7, respectively. The mutation fraction patterns were dominant by C > T in early stages (T1 to T3), while majored by T > C in late stages (T4 to T7, Fig. 3A). Signatures of mutagenic alteration were analyzed based on the mutational spectra which identify the linear combination of predefined signatures from COSMIC that most accurately reconstruct the mutational profile of a single tumor sample29,30. Ten signatures were identified with remarkable differences in the 7 tumor tissues (Signatures 1, 2, 5, 8, 11, 16, 19, 21, 23, and 26), and their contribution to each tumor was calculated (Fig. 3B and Supplementary Fig. S2). Similar to the tendency of mutation fraction pattern, T1 to T3 shared a signature combination different to those shared by T4 to T7. Signature 1, 2, 5, 11, and 19 were more present in early-stage tumors (T1 to T3), signature 16, 21, 26 and 23 in the later stage tumors (T4 to T7), while signature 8 was consistently present from T1 to T7 except for T4 (Fig. 3B and Supplementary Fig. S2). These data suggest that signatures could be related to specific tumor stages.

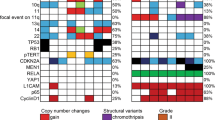
A Mutational fractions of the 7 tumor tissues. The mutational patterns of T1 to T3 (C > T, red) were different compared with T4 to T7 (T > C, yellow). B Contributions of the mutation signatures found in the seven tumor samples and their contributions to each tumor. C Phylogenetic tree constructed with all somatic variants. In the left panel, the gray, orange and red lines indicate the trunk, shared branch, and private branch, respectively and the length of trunk and branch was proportional to their number of mutations. Putative driver mutations were mapped along the trees as indicated. Heat maps show the presence (blue) or absence (gray) of a mutation in each tumor or normal (PBMC). D The chromothripsis of 7 tumor tissues. E CNVs of the seven tumor tissues. Blue and orange represent the chromosomes. The dots above or below the baseline indicate the amplification or deletion on each chromosome.
Heterogeneity and architecture of tumor evolution
To illustrate the tumor evolution process of this patient, a phylogenetic tree was constructed with identified mutations across all tumor specimens. Those mutations harbored by all 7 tumor specimens were considered as shared mutations that built the trunk of the phylogenetic tree (black color, Fig. 3C). The mutations shared by more than 2 samples or held by only one sample were considered as partial mutation and private mutation, respectively. Each tumor contained a large number of private mutations while only a few mutations were shared in those samples. The driver gene list was identified by annotating the private mutations with COSMIC database (Supplementary Table. 2). Although only one cancer-related gene (NCOR2) was found in the initial tumor (T1), it accumulated huge numbers of mutations (the branch was very long, Fig. 3C). Additionally, two private mutated genes were found in T2 (MLLT1 and MUC4), five in T3 (AR, EP300, GATA3, SETBP1, ELF4) and T4 (CARD11, KTN1, PTPN6, ZNF429, SGK1), and six in T5 (ARID2, PDE4DIP, SMARCA4, DDX6, TOP1, SUFU) and T7 (CREB3L1, DCC, NUMA1, PER1, ZFHX3, STRN). Ten mutated genes, the most among 7 tumors, were observed in T6 (ABL1, JAZF1, APC, ETV5, CHD2, ARID1B, PCM1, ATM, HNRNPA2B1, and KDM5A). These data indicated that the genetic alternation burden increased gradually upon disease progression and treatment selection.
Profiles of chromothripsis and CNV
Chromothripsis is a one-step catastrophic event reported in a variety of cancers, in which chromosomal shattering and rearrangement occurs in a few or one chromosome, and further lead to gene amplification or deletion30,31. In this case, chromothripsis was detected in all of 7 tumor tissues, and their incidence increased alone with disease progression from T1 to T7, particularly significant in T5 and T6, except in T2 (Fig. 3D). CNV is a phenomenon in which structural variations of the genome (including insertion, deletion, and duplication) are repeated and the numbers of repeats vary between individuals in the human population32. In process of tumor evolution, the whole genome encountered a large number of CNVs and rearrangements, at both the onset (T1) and later stage (T7) of the tumor (Fig. 3E). These data support the notion that the evolution of the tumor could be a product of many genetic alterations, including chromothripsis and CNV.
Identification of driver mutation on ADGRL3
To further investigate driver mutations involved in the disease evolution, we determined the mutational profiles of the 7 tumors samples. The number of mutation intersections per million base pairs was almost similar from T1 to T4 (57, 60, 62, and 65, respectively). Tumors in the later stages (T5 to T7) acquired more mutations (239, 256, and 132, respectively) (Fig. 4A). There were six genes with the same mutation found in 5 of the 7 specimens (PCDHA4, PCDHA8, SEC14L6, SETD2, RIOK2, and SLCO2A1) and three genes in 6 of 7 the samples (RYR1 in T2 to T7, SNX25 in T1 to T7 except T3, DSC2 in T1 to T7 except for T2). Strikingly, ADGRL3 was consistently mutated in the entire disease progression process (Fig. 4A, indicated by arrow).

A The number of mutation intersections in each tumor and the mutations shared by the tumor specimens. The mutation number is shown on the x-axis. ADGRL3 mutation occurred in all seven specimens. B Confirmation of the mutation site on ADGRL3 of the seven tumor tissues by Sanger sequencing.
The nonsynonymous exonic mutation of ADGRL3 was located in the CDS region (c.1382 G > A, p.G461E) and identified in all seven tumor specimens but not in the monocytes of the patient. Sanger sequencing confirmed the mutation to be heterozygous in T1 and T3 to T7, and homogenous in T2, which might be affected by the purity of sampled tissues (Fig. 4B). This mutation led to an amino acid switch from glycine (G), the smallest (molecular weight 75.067), nonpolar aliphatic (neutral) amino acid, to glutamic (E), a much bigger (molecular weight 146.146) polar acidic (negative) amino acid. The score by pholyphen2 prediction based on HDIV was 0.728, indicating that this was a potential damaging mutation (Supplementary Table 3). The mutation rates, the ratio of mutated reads to the sum of mutated reads and the reference reads in the tumor (R = Reads-Mu/[Reads-Mu + Reads-Ref]) ranged from 27.27% to 51.35% in the 7 tumor specimens (Supplementary Table 4). The mutation of ADGRL3 on the same site was reported to be pathogenic in malignant melanoma with a high FATHMM prediction score (0.98) based on the Catalogue Of Somatic Mutations In Cancers (COSMIC) database (mutation ID: COSM3917981)33. These data indicated that ADGRL3 mutation might have important roles in the progression of ependymoma.
Next, we collected the genomic DNAs of 78 ependymomas who underwent surgery at SYSUCC from the year 2014 to 2021 (Supplementary Table 5) and were sent for Sanger sequencing to assess the mutation status of ADGRL3. However, no mutation was found on the exact site of ADGRL3 existing in the case. Next, we checked the online dataset regarding the mutation status of ADGRL3. Since there is no data based on ependymal tumors in TCGA, the ICGC (The Cancer Genome Collaboratory data portal) database was assessed. The Pediatric brain tumor with multiple subtypes project (PBCA-US, https://dcc.icgc.org/projects/PBCA-US) which included 25 ependymal tumor cases upon 290 donors with molecular data was investigated. Most of the variants including single base substitution and insertion of ADGRL3 were located on intron, while missense variants were found only in three donors, and the sites were different from our reported site (p.G461E).
Prognostic role of ADGRL3 in ependymoma and low-grade glioma (LGG) patient
We detected the mRNA level of ADGRL3 in normal brain and ependymal tumor tissues. The simplified information of 36 patients collected from 2014 to 2019 is summarized in Supplementary Table 6. The mRNA level of ADGRL3 was higher in normal brain tissue (n = 3) and grade II (n = 15) than in grade III (n = 21), p = 0.0028 and 0.0192, respectively (Fig. 5A), while no significance was found between normal brain and grade II (p = 0.0525). Although there was no significant difference in the overall survival (OS) between the patients with a higher level of ADGRL3 (n = 19) and those with lower levels (n = 17) (p = 0.0643), the patients’ median survival time was better in the higher-level group (40 months vs. 31 months, Fig. 5B). We then checked the TCGA database but there was no data focusing on ependymal tumors. We then analyzed the prognostic role of ADGRL3 in LGG patients using the GEPIA database which is based on the data from TCGA and GTEx. The expression level of ADGRL3 was observed to be significantly higher in normal brain (n = 207) than LGG (n = 518), p < 0.001(Fig. 5C). Further, both the OS and DFS were better in patients with higher ADGRL3 level (n = 256) than those with lower ADGRL3 level (n = 256), p = 0.006 and 0.0023 respectively (Fig. 5D, E). Data of glioma from TCGA showed that the OS and DFS were better in patients with intact ADGRL3 compared to those with alterations (Supplementary Fig. S3). Our data was limited by both cohort size and available database from concluding the role of ADGRL3 in ependymoma. However, lower mRNA level or the gene alteration on ADGRL3 might be related to poor prognosis of ependymoma and LGG patients, supporting the hypothesis that ADGRL3 might serve as a tumor suppressor gene in the evolution of tumor.

A The expression of ADGRL3 level in normal brain (n = 3) tissues and ependymoma tissues (n = 36; grade II, n = 15; grade III, n = 21) (*p < 0.05, **p < 0.001). B OS of the ependymoma patients with high or low mRNA level of ADGRL3. C The expression level of ADGRL3 was significantly higher in LGG (n = 518) than normal brain tissues (n = 207) from GEPIA. (***p < 0.0001). D, E The OS and DFS of LGG patients with high expression level of ADGRL3 were superior to those with lower expression level of ADGRL3. p = 0.006 and p = 0.0023 respectively.
Discussion
Tumor recurrence is a major cause of cancer treatment failure. The genetic landscape of the tumor evolves together with tumor development upon treatment selection34. Johnson et al. found that the recurrent tumor was derived from the initial tumor at a very early stage of their evolution35. At the same time, irradiation and chemotherapy (TMZ) could induce an alternative evolution, driver mutations and selective advantages characterized by hypermutation status36. In this reported case, PYY underwent surgery for T1, surgery followed by radiotherapy for the first recurrence (T2) and chemotherapy was applied after the removal of T3. With the administration of chemotherapy, the mutational pattern shared by T1 to T3 (majored by C > T) evolved into a pattern dominated by T > C in T4 to T7, and the mutated gene numbers were more in T4 to T7 than in T1 to T3 (Figs. 1A, 3A, B and 4A). The hypermutation status was probably induced by chemotherapy. Our findings on the mutational pattern, signatures and the phylogenetic tree support that chemotherapy (TMZ) could push tumors to an evolutionary process.
Although the mutation profile of ependymoma has not been widely reported, Liu et al investigated radiographically distinct regions of an ependymoma tumor using transcriptomic, genetic and epigenomic profiling, and discovered axes of gene expression programs that could recapitulate normal brain development in addition to phylogenies37. Their work reflected neuronal development gene expression programs through spatially distinct clusters, while our work aimed at clarifying the genetic events occurring during the timeline of the developmental process of an ependymoma patient with 7 recurrences. Here, we found that PYY had mutation of histone methyltransferase SETD2 in 5 of her 7 tumor specimens while Liu et al.37 found that mutant SETD2 enhanced the proliferation of ependymoma cells. Pediatric high-grade glioma patients with mutant SETD2 were shown to have significantly lower OS compared to patients with SETD2 wild-type cancers38,39. In contrast, in the TCGA database, we observed that both the OS and DFS of glioma patients were better in those with intact ADGRL3 compared to those with alterations. Therefore, these findings are consistent in regard to the driver genetic alterations during the development of ependymoma.
Somatic mutational signatures are the consequences of multiple mutational processes during the evolutionary process, and might be used as readouts of the biological history and can reveal novel mechanisms or new targets for anticancer therapy40,41. There are ten mutational signatures identified in the multi-recurrences in this patient. Individually, signatures 1 and 5 are clock-like age-associated signatures and are almost universally active in human cancer and normal cells. Signature 11 was detected in glioblastoma and melanoma and was found to be related to TMZ-induced O6-methylguanine aberrations42,43. Signature 19 was found in pilocytic astrocytoma44. Signature 2 has been reported in various cancer types including breast cancer, leukemia, melanoma, and stomach cancer42. Signature 16 has only been reported in liver cancer, signature 21 was found in stomach cancer and related to microsatellite unstable tumors, and signature 26 was associated with defective DNA mismatch repair and found in breast cancer, cervical cancer, stomach cancer, and uterine carcinoma. Signature 8 was reported in medulloblastoma and breast cancer42. We, herein, provided evidence that mutational signatures might reflect the evolutionary process of ependymoma.
The current case was a female young patient with multiple recurrences of ependymoma and with this unique model, we provided evidence that although heterogeneity was found in her tumor, there were still common mutations during the development of the disease. GPCRs have been shown to modulate a variety of physiological processes through cross-talk between signaling pathways and have been widely studied as druggable sites due to their cell surface localization45. Mutation of ADGRL3, a member of GPCRs, was found in all 7 tumor tissues along the disease progression. It is well known that alterations on coding and non-coding regions of ADGRL3 could lead to an increased risk of attention-deficit/hyperactivity disorder (ADHD) in both animal and cellular models46,47,48. However, the exact role of ADGRL3 in human cancer was rarely reported although it was predicted as a tumor suppressor in multiple types of human cancer49,50. Kan et al. reported 13 significant genes mutation among the 156 GPCRs in lung adenocarcinoma51. ADGRL3 mutation was just next to that of TP53 and KRAS on the list and was predicted as a putative tumor suppressor according to the functional effects of the mutation. The haplotype (rs2271338G > A) on the evolutionarily conserved region reduced 40% of enhancer activity in neuroblastoma and astrocytoma cell line, thereby, decreasing the expression of ADGRL3 in the thalamus in post-mortem materials46. Crystal structure studies showed that the olfactomedin domain (OLF) on ADGRL3 was sufficient for binding with fibronectin leucine-rich repeat transmembrane proteins (FLRT3) and further led to intercellular adhesion. There is a conserved calcium-binding site located in the central pore of OLF so that the mutations in the binding pocket might disrupt their interaction11,52. The mutation on ADGRL3 in our case was located at the very beginning of the OLF domain, the structure of which has not yet been reported, so further investigations are necessary to clarify whether the mutation affects the binding ability of ADGRL3 and FLRT3.
As a summary, based on this study findings, we suggest that ADGRL3 has critical roles in the initiation and recurrence of ependymoma, and further investigation should be performed to confirm its role in tumor development and evolution.
Data availability
The datasets used and/or analyzed in the current study are available from the corresponding author on reasonable request or through Research Data Deposit (http://www.researchdata.org.cn), the approval number is RDDB2020000810.
References
Louis, D. N. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta. Neuropathol. 131, 803–820 (2016).
Pajtler, K. W. et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27, 728–743 (2015).
Godfraind, C. et al. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta. Neuropathol. 124, 247–257 (2012).
Mack, S. C. et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450 (2014).
Ellison, D. W. et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 30, 863–866 (2020).
Louis, D. N. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro. Oncol. 23, 1231–1251 (2021).
Nowell, P. C. The clonal evolution of tumor cell populations. Science 194, 23–28 (1976).
Ding, S., Chen, X. & Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 40, 329–344 (2020).
Burrell, R. A., McGranahan, N., Bartek, J. & Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501, 338–345 (2013).
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2018).
Bradley, C. A. Tracing the steps of cancer evolution. Nat. Rev. Urol. 15, 395 (2018).
Huang, H. M. & Li, H. X. Tumor heterogeneity and the potential role of liquid biopsy in bladder cancer. Cancer Commun. 41, 91–108 (2021).
McGranahan, N. & Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 27, 15–26 (2010).
Kilday, J. P. et al. Pediatric ependymoma: biological perspectives. Mol. Cancer Res. 7, 765–786 (2009).
Korshunov, A. et al. Molecular staging of intracranial ependymoma in children and adults. J. Clin. Oncol. 28, 3182–3190 (2010).
Arcos-Burgos, M. et al. A common variant of the latrophilin 3 gene, LPHN3, confers susceptibility to ADHD and predicts effectiveness of stimulant medication. Mol. Psychiatry 15, 1053–1066 (2010).
Ozaslan, A., Guney, E., Ergun, M. A., Okur, I. & Yapar, D. CDH13 and LPHN3 Gene Polymorphisms in Attention-Deficit/Hyperactivity Disorder: Their Relation to Clinical Characteristics. J. Mol. Neurosci. 71, 394–408 (2021).
Xi, S. et al. Clinical significance of the histological and molecular characteristics of ependymal tumors: a single institution case series from China. BMC Cancer 19, 717 (2019).
Hu, W. M. et al. Practice of the New Integrated Molecular Diagnostics in Gliomas: Experiences and New Findings in a Single Chinese Center. J. Cancer 11, 1371–1382 (2020).
Catteau, A. et al. A new sensitive PCR assay for one-step detection of 12 IDH1/2 mutations in glioma. Acta. Neuropathol. Commun. 2, 58 (2014).
Wu, J. et al. Skp2 modulates proliferation, senescence and tumorigenesis of glioma. Cancer Cell Int. 20, 71 (2020).
Tang, Z., Li, C., Kang, B., Gao, G., & Zhang, Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102.
Tang, Z., Kang, B., Li, C., Chen, T., & Zhang, Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560.
Preusser, M. et al. Ki67 index in intracranial ependymoma: a promising histopathologicalcandidate biomarker. Histopathology 53, 39–47 (2008).
Nowak, J., et al. MRI phenotype of RELA-fused pediatric supratentorial ependymoma. Clin. Neuroradiol 29, 595–604 (2019).
Gamboa, N. T., et al. Preoperative and intraoperative perfusion magnetic resonance imaging in a RELA fusion-positive anaplastic ependymoma: a case report. Surg. Neurol. Int. 9, 144 (2018).
Parker, M., et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 506, 451–455 (2014).
Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S. & Swanton, C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 17, 31 (2016).
Maher, C. A. & Wilson, R. K. Chromothripsis and human disease: piecing together the shattering process. Cell 148, 29–32 (2012).
Forment, J. V., Kaidi, A. & Jackson, S. P. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer 12, 663–670 (2012).
Hu, L., et al. Clinical significance of germline copy number variation in susceptibility of human diseases. J. Genet. Genomics 45, 3–12 (2018).
Study: ICGC(SKCM-US): Skin Cutaneous melanoma—TCGA, US; Study ID: COSU540; Gene Analysed: 24136; Mutated Samples: 399; Total No. of Samples: 477. (https://cancer.sanger.ac.uk/cosmic/mutation/overview?id=149185751).
Bozic, I., et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife 2, e00747 (2013).
Johnson, B. E. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014).
Kim, C., et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879–893 e813 (2018).
Liu, S. J. et al. Multiplatform molecular profiling reveals epigenomic intratumor heterogeneity in ependymoma. Cell Rep. 30, 1300–1309 e1305 (2020).
Fontebasso, A. M. et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta. Neuropathol. 125, 659–669 (2013).
Mackay, A. et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537 e525 (2017).
Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl Acad. Sci. USA 107, 961–968 (2010).
Xing, R. et al. Whole-genome sequencing reveals novel tandem-duplication hotspots and a prognostic mutational signature in gastric cancer. Nat. Commun. 10, 2037 (2019).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Helleday, T., Eshtad, S. & Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 15, 585–598 (2014).
Reitman, Z. J. et al. Mitogenic and progenitor gene programmes in single pilocytic astrocytoma cells. Nat. Commun. 10, 3731 (2019).
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017).
Martinez, A. F. et al. An Ultraconserved Brain-Specific Enhancer Within ADGRL3 (LPHN3) Underpins Attention-Deficit/Hyperactivity Disorder Susceptibility. Biol. Psychiatry 80, 943–954 (2016).
Bruxel, E. M., et al. LPHN3 and attention-deficit/hyperactivity disorder: a susceptibility and pharmacogenetic study. Genes Brain Behav. 14, 419–427 (2015).
Fallgatter, A. J., et al. Influence of a latrophilin 3 (LPHN3) risk haplotype on event-related potential measures of cognitive response control in attention-deficit hyperactivity disorder (ADHD). Eur. Neuropsychopharmacol. 23, 458–468 (2012).
O’Hayre, M. et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 13, 412–424 (2013).
Maru, Y. et al. Identification of novel mutations in Japanese ovarian clear cell carcinoma patients using optimized targeted NGS for clinical diagnosis. Gynecol. Oncol. 144, 377–383 (2017).
Kan, Z. et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873 (2010).
Ranaivoson, F. M. et al. Structural and Mechanistic Insights into the Latrophilin3-FLRT3 Complex that Mediates Glutamatergic Synapse Development. Structure 23, 1665–1677 (2015).
Funding
This study was supported by grants from the National Natural Science Foundation of China (82073109, 81872059, 81772677, 81772675, 81572479, 81372685, and 81101521), the Guangzhou Science Technology and Innovation Project (201508020125), the Science and Technology Planning Project of Guangzhou (2016A020213004), the Natural Science Foundation of Guangdong Province (2016A030313309). All these funding had no influential role in the study design, data collection, analysis, data interpretation or writing of the report.
Author information
Authors and Affiliations
Contributions
Z.P.C., J.W., S.-Y.X., Q.Z., and K.S. conceived and designed the project. H.-P.C., F.W., H.-J.H., Y.-Y.Z., Z.-H.Y., F-.R.C., and H.-K.S. developed and performed the functional experiments. J.W., J.Z., C.K., X.-H.Z., F.-H.L., C.-C.G., Y.-C.L.., C.L., H.-T.X., F.H., Z.-J.L., K.S., and Z.P.C. provided medical care and managed patients. J.W., S.-Y.X., Q.Z., H.-P.C., F.W., H.-J.H., Y.-Y.Z., K.S., and Z.P.C. analyzed and interpreted data. Q.C., H.-M.W., Y.-H.L., Z.L., J.Z. provided materials and technical support. J.W., Z.P.C., S.-Y.X., Q.Z., and K.S. wrote, reviewed and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This work has been approved by the ethics committee and institutional review board of Sun Yat-sen University Cancer Center.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, J., Xi, Sy., Zhao, Q. et al. Driver mutations in ADGRL3 are involved in the evolution of ependymoma. Lab Invest 102, 702–710 (2022). https://doi.org/10.1038/s41374-021-00721-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-021-00721-3
