
Abstract
Acute lymphoblastic leukemia (ALL) is a common malignancy in children. In this study, we aimed to explore putative mechanisms of microRNA-155-5p (miR-155-5p) involvement in childhood ALL (cALL) via interactions with casitas B-lineage lymphoma (CBL), interferon regulatory factor 4 (IRF4), and cyclin-dependent kinase 6 (CDK6). Bioinformatic analysis was performed initially to identify differentially expressed genes in cALL. The expression levels of miR-155-5p, CBL, IRF4, and CDK6 in peripheral blood lymphocytes from clinical ALL samples were determined using RT-qPCR and Western blot assays. A dual-luciferase reporter gene assay was used to ascertain a possible targeting relationship between miR-155-5p and CBL, CCK-8 assay and flow cytometry were used to measure cell activity and apoptosis of ALL cells. Co-IP was performed to investigate the interaction between CBL and IRF4 and the ubiquitination level of IRF4. Furthermore, in vivo validation was performed inducing xenograft tumor models with ALL cells in nude mice. As indicated by bioinformatic analysis, miR-155-5p and CDK6 were upregulated and CBL was downregulated in ALL. miR-155-5p was found to target CBL to inhibit CBL expression. miR-155-5p promoted the proliferation of ALL cells and inhibited their apoptosis by inhibiting the expression of CBL, which otherwise degraded IRF4 protein through ubiquitination, leading to inhibited CDK6 expression. Collectively, the results show that miR-155-5p can promote the development of cALL via the regulation on CBL-mediated IRF4/CDK6 axis.
Similar content being viewed by others

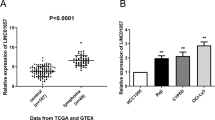

Introduction
Acute lymphoblastic leukemia (ALL) is a frequently occurring childhood malignancy and is a major cause for disease-related death among children [1]. ALL is characterized by uncontrolled immature lymphoid cell proliferation [2]. Genomic features and epigenetic regulation are responsible for the transformation in childhood acute lymphoblastic leukemia (cALL) [3]. The treatment options for cALL include initial high intensity chemotherapy, and subsequent low intensity oral maintenance therapy using oral 6-mercaptopurine and methotrexate [4]. Despite advances in its treatment modalities, the survival of cALL patients with relapse remains unsatisfactory [5].
Of note, the role of microRNAs (miRs) in cALL has been documented [6]. miRs are endogenous 22-nucleotide RNAs with the ability to regulate crucial gene-regulatory events by pairing with the mRNAs of protein-coding genes resulting in gene repression [7]. Previous work has shown differentially upregulated miR-155 in patients with ALL, which can thus serve as diagnostic biomarkers and possibly act as facilitators in leukemogenesis [8]. In particular, overexpressed miR-155-5p has been noted in peripheral blood mononuclear cells of patients with chronic lymphocytic leukemia [9]. In addition, the levels of miR-155 were found as significantly higher in acute myeloid leukemia patients than in controls, and its overexpression has been associated with initial presentation and poor outcome [10]. Mechanistically, miR-155 is capable of targeting casitas B-lineage lymphoma (CBL) in melanoma [11] and is also shown to target CBL to mediate the behaviors of colon cancer cells [12]. CBL is a E3 ubiquitin ligase of multiple tyrosine kinase receptor types [13]. Notably, a lack of CBL E3-ligase activity has been discovered in B-lineage cALL cells [14]. Moreover, it has been previously reported that CBL displays mutations in infants with cALL [15]. In this context, it is noteworthy that CBLs can trigger ubiquitination and degradation of interferon regulatory factor 4 (IRF4) in GC light zone B cells [16]. IRF4, a member of IRF family transcription factors, exerts crucial functions in the development of lymphoid cells as well as in the regulation of immune responses [17] and is highly expressed in cALL [18]. Other research has shown that knockdown of IRF4 downregulated cyclin-dependent kinase 6 (CDK6) in EBV-mediated B cells [19]. CDK6 is a type of D-Cyclin-activated kinase involved in propelling the cell cycle by inactivating pRB in the G1 phase [20]. Inhibition of CDK6 has been reported to contribute to a reduction in IRF4 expression [21]. Interestingly, high expression of CDK6 has been found in childhood patients with B-ALL [22]. This study was designed to test the hypothesis that miR-155-5p mediates regulation of the CBL–IRF4–CDK6 axis and therefore plays a regulatory role in cALL.
Materials and methods
Bioinformatic analysis
The Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/gds) was utilized to obtain the gene expression microarray dataset (GSE141140) and miRNA expression microarray dataset (GSE56489), which pertained to cALL. Differential gene expression analysis was performed using edgeR software package in the R statistical environment. There were 57 samples in the GSE56489 dataset, including 14 normal samples and 43 ALL samples, and there were 17 samples in the GSE141140 dataset, including four normal samples and 13 ALL samples. Significant differentially expressed genes and miRNAs were screened using the criteria logFC and p < 0.05. The starBase (http://starbase.sysu.edu.cn/), TargetScan (http://www.targetscan.org/vert_72/), miRDB (http://mirdb.org/), and mirDIP databases (http://ophid.utoronto.ca/mirDIP/) were used to predict the target genes of the miRNA, and their intersection was selected as the candidate target gene set of miR-155-5p.
Clinical sample collection
Peripheral blood lymphocytes were collected from children with ALL (n = 28), aged 1–15 years old, who were hospitalized between 2017 and 2019 at the Shandong Provincial Hospital Affiliated to Shandong First Medical University. All included patients did not have any prior treatment. Samples were also collected from age-matched normal children (used as normal control material) (n = 28). The specific clinicopathological characteristics of the included patients are shown in Table S1.
Cell culture and treatment
The normal lymphoblastoid cell line AHH-1 and four ALL cell lines (697, KOPN-8, B1, and SUP-B15) were purchased from the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). The cells had been identified by STR DNA analysis (the specific STR data are presented in Table 1), with regular examination for control of mycoplasma contamination. The cells were cultured in Rosewell Park Memorial Institute-1640 (RPMI-1640) medium (72400120, Gibco, Carlsbad, California, USA) containing 10% fetal bovine serum (FBS, 10099141, Gibco) and 1% penicillin/streptomycin (15070063, Gibco). HEK293T cells purchased from American Type Culture Collection (VA, USA) were cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS and 1% penicillin/streptomycin (10569044, Gibco). All cells were cultured in a cell incubator containing 5% CO2 at 37 °C.
The 697 cells were grouped into the following treatment groups: inhibitor-negative control (NC) (cells treated with miR-155-5p inhibitory control), inhibitor-miR-155-5p (cells treated with miR-155-5p inhibitor), short hairpin RNA (sh)-NC (cells treated with lentivirus carrying NC of shRNA vector: 5′-CCTAAGGTTAAGTCGCCCTCG-3′), sh-CBL-1 (cells treated with lentivirus carrying shRNA sequence 1 targeting CBL: 5′-GGGAACATTCTCCAGACAATC-3′), sh-CBL-2 (cells treated with lentivirus carrying shRNA sequence 2 targeting CBL: 5′-GGAGCAATGTGAGGGTGAAGA-3′), inhibitor-NC + sh-NC (cells treated with miR-155-5p inhibitory control and lentivirus carrying NC of shRNA vector), inhibitor-miR-155-5p + sh-NC (cells treated with miR-155-5p inhibitor and lentivirus carrying NC of shRNA vector), inhibitor-miR-155-5p + sh-CBL (cells treated with miR-155-5p inhibitor and lentivirus carrying sh-CBL), overexpression (oe)-NC + dimethylsulfoxide (DMSO) (cells treated with lentivirus carrying NC of overexpression vector and with DMSO), oe-CBL + DMSO (cells treated with lentivirus carrying oe-CBL and with DMSO), oe-NC + MG132 (cells treated with lentivirus carrying NC of overexpression vector and a proteasome inhibitor MG132), oe-CBL + MG132 (cells treated with lentivirus carrying oe-CBL and MG132), oe-NC (cells treated with lentivirus carrying NC of overexpression vector), oe-CBL + oe-NC (cells treated with lentivirus carrying oe-CBL and NC of overexpression vector), oe-CBL + oe-IRF4 (cells treated with lentivirus carrying oe-CBL and oe-IRF4), oe-CBL + oe-CDK6 (cells treated with lentivirus carrying oe-CBL and oe-CDK6), and oe-CDK6 (cells treated with lentivirus carrying oe-CDK6).
KOPN-8 cells were assigned to the following treatment groups: mimic-NC (cells treated with miR-155-5p overexpression control), mimic-miR-155-5p (cells treated with miR-155-5p overexpression), oe-NC (cells treated with lentivirus carrying NC of overexpression vector), oe-CBL-1 (cells treated with lentivirus carrying overexpressed CBL), mimic-NC + oe-NC (cells treated with miR-155-5p overexpression control and lentivirus carrying NC of overexpression vector),e(cells treated with lentivirus carrying NC of shRNA vector), sh-CDK6-1 (cells treated with lentivirus carrying shRNA sequence 1 targeting CDK6: 5′-CTTCTGAAGTGTTTGACATTT-3′), sh-CDK6-2 (cells treated with lentivirus carrying shRNA sequence 2 targeting CDK6: 5′-GAGAAGTTTGTAACAGATATC-3′), sh-CBL + sh-NC (cells treated with lentivirus carrying sh-CBL and NC of shRNA vector), sh-CBL + sh-IRF4 (cells treated with lentivirus carrying sh-CBL and sh-IRF4), and sh-CBL + sh-CDK6 (cells treated with lentivirus carrying sh-CBL and sh-CDK6).
HEK293T cells were divided into the following treatment groups: CBL-wild type (WT) (the normal group of 3′-untranslated region (3′-UTR) binding site between CBL and miR-155-5p), CBL-mutant type (MUT) (the mutation group of 3′-UTR binding site between CBL and miR-155-5p), mimic-NC (cells treated with the control of miR-155-5p overexpression), and mimic-miR-155-5p (cells treated with miR-155-5p overexpression). The core plasmid (PLKO. 1) and the auxiliary plasmid (psPAX2, pMD2. G) were used to package the silencing lentivirus, and, the core plasmid (pHAGE-CMV-MCS-IzsGreen) and auxiliary plasmid (psPAX2, pMD2.G) inserted into the complementary DNA (cDNA) sequence of the target gene were used to package the overexpression lentivirus. All lentiviruses were purchased from Shanghai Sangon Biotechnology Co. Ltd. (Shanghai, China). The primer sequences and plasmid construction were completed by Sangon and the experimental steps were carried out according to the instruction manual. MG132 was purchased from Sigma-Aldrich Chemical Company (St Louis, MO, USA).
Isolation of peripheral blood lymphocytes
A total of 10-mL venous blood was added to Ficoll Hypaque solution (Amersham Pharmacia biotechnology, Uppsala, Sweden) and mixed evenly. The mixture was then centrifuged at 1500 g at room temperature for 25 min. The resulting intermediate layer contained lymphocytes, which were isolated for subsequent experiments.
Cell counting kit-8 (CCK-8) assay
CCK-8 kits (GK10001, GlpBio, Montclair, CA, USA) were used to quantify cell proliferation after different treatments. The experiments were carried out according to the instructions of the kit. At 0, 24, 48, 72, and 96 h after cell treatment, 10 μL of CCK-8 reagent was added to each well and incubated in an incubator for 1 h. The optical density value at 450 nm was then measured.
Flow cytometry
Cell apoptosis was determined using Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) kits (APOAF, Sigma-Aldrich). The cells were subjected to different treatments, cultured in an incubator for 48 h, centrifuged, and resuspended in 200-μL buffer. Next, 10-μL Annexin V-FITC and 5-μL PI were added to the cells, followed by mixing and reaction at room temperature in the dark, for 15 min. The reaction was terminated with addition of 300-μL buffer. A flow cytometer (Becton, Dickinson and Company, Sparks, Md, USA) was used to measure cell apoptosis and the apoptotic rate was determined.
Dual-luciferase reporter gene assay
The normal sequence [CBL-WT] and mutant sequence [CBL-MUT] of miR-155-5p and CBL at the 3′-UTR binding site were synthesized and introduced into the pMIR vector (AM5795, Ambion, Austin, TX, USA). Following restriction endonuclease cleavage, the target fragment was inserted into the vector using T4 DNA ligase (M0204S, New England Biolabs, Ipswich, MA, USA). The fluorescent plasmid of Renilla and the constructed luciferase reporter plasmids were, respectively, co-transfected with mimic-NC and miR-155-5p mimic into HEK293T cells. The cells were cultured in a 37 °C incubator with 5% CO2 and saturated humidity for 48 h. After transfection, the cells were collected and lysed. The luciferase activity was determined using Dual-Luciferase® Reporter Assay System on the GloMax® 20/20 Luminometer detection instrument (E5311, Promega Corporation, Madison, WI, USA). The relative luciferase activity = firefly luciferase activity/Ranilla luciferase activity. All the vectors were constructed by Sangon.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent (16096020, Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA). miRNA (B532451, Sangon) and mRNA (D7168l, Beyotime, Shanghai) were synthesized into cDNA using the first strand cDNA synthesis kits according to the manufacturer’s instructions. RT-qPCR was carried out using RT-qPCR kits (Q511-02, Vazyme Biotechnology, Nanjing, China) according to the instruction manual. PCR amplification was performed on a Bio-Rad real-time qPCR instrument CFX96. U6 was used as internal reference for miR-155-5p and β-actin for CBL, IRF4 and CDK6. All primer sequences were designed and provided by Sangon. The primer sequences are listed in Table S2. The results, using the 2−ΔΔCT method, indicated the expression ratio relationships between the experimental and the control groups.
Western blot assay
Radio-immunoprecipitation assay (RIPA) lysis buffer containing phenylmethylsulfonyl fluoride and the protein inhibitor cocktail (P0013B, Beyotime) was used to lyse tissues and cells to extract total protein. Nuclear and cytoplasmic proteins were extracted using kits (P0028, Beyotime) according to the instruction manual. The supernatant was extracted, and the total protein concentration of each sample was determined with bicinchoninic acid kits (23229, Thermo Fisher Scientific Inc.). The protein concentration was adjusted to 1 μg/μL, and the sample volume of each tube was set at 100 μL. The protein was boiled for 10 min, and the denaturated protein stored at −80 °C for further use. Based on the molecular size of the target protein, 8–12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis was performed, where protein samples were added to the lanes in equal amounts for electrophoresis. The protein was transferred from the gel to the polyvinylidene fluoride membrane (1620177, Bio-Rad, Hercules, CA, USA), which was then blocked with 5% skimmed milk or 5% bovine serum albumin at room temperature for 1 h. The protein samples were then incubated with rabbit antibodies against β-actin (ab8227, 1:5000), CBL (ab32027, 1:1000), IRF4 (4964, 1:1000), and CDK6 (ab124821, 1:1000) at 4 °C overnight. The next day, the protein was further incubated with horseradish peroxidase-labeled goat anti-rabbit against immunoglobulin G (IgG) (ab6721, 1:5000) at room temperature for 1 h. The membrane was immersed in enhanced chemiluminescence reaction solution (1705062, Bio-Rad, USA) at room temperature for 1 min. The excess liquid was removed and the protein was covered with plastic wrap. Band exposure imaging was performed on the Image Quant LAS 4000C gel imager (GE company, USA). Cell total protein was measured using β-actin as internal reference, and the ratio of gray value of target band to that of internal reference band was taken as the relative protein expression. All the aforementioned antibodies were purchased from Abcam Inc. (Cambridge, UK), except the rabbit antibody against IRF4, which was purchased from Cell Signaling Technologies (Beverly, MA, USA).
Co-immunoprecipitation (Co-IP) assay
The interaction between endogenous CBL and IRF4 protein was determined by the Co-IP assay. Pierce IP buffer (1% Triton X-100, 150-mM NaCl, 1-mM ethylenediamine tetraacetic acid, 25-mM Tris HCl, pH 7.5) was used to lyse the cells, followed by addition of protease inhibitors and phosphatase inhibitors. After centrifugation, the supernatant was extracted and incubated with mouse antibody against IRF4 (sc-48338, 1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and IgG (ab200699, 1:100, Abcam) overnight at 4 °C. After that, protein G beads, Dynabeads (Thermo Fisher Scientific Inc.) were added to the above mixture and slowly rotated for 8 h at 4 °C. The protein G beads were then washed with IP buffer three times, eluted with elution buffer and boiled at for 10 min to denature the protein, which was stored at −80 °C for subsequent use. Finally, the Co-IP product was analyzed by the Western blot assay.
Determination of ubiquitination
For determining the endogenous IRF4, the cells were incubated with 10-μM MG132 (HY-13259, MedChemExpress, NJ, USA), a proteosome inhibitor, for 6 h, whereas cells in the control group were added with the same amount of DMSO (D2650, Sigma-Aldrich). Cells were lysed in 1% SDS RIPA buffer and treated with ultrasound. After the IP reaction, the diluted cell lysate (at a final 0.1% SDS concentration) was incubated with IRF4 antibody (sc-48338, 1:100, Santa Cruz) at 4 °C overnight. Protein G beads were added to the cells and incubated at 4 °C for 8 h. Thereafter, the protein G beads were washed in IP buffer three times, eluted with washing buffer, boiled at 100 °C for 10 min to denature the protein, and then stored for subsequent use at −80 °C. The ubiquitination level of IRF4 was determined by Western blot assay using rabbit anti-human ubiquitin (ab7780, 1:1000, Abcam).
Xenograft in nude mice
Male nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (aged 6 weeks) were purchased from Jiangsu ALF Biotechnology Co., LTD. (Nanjing, China) and fed in a specific-pathogen-free animal room with sufficient water and food. After 1 week of adaptive feeding, 697 cells were injected into a tail vein at a dose of 5 × 106 cells/mouse. Ten mice in each group were randomly selected and their conditions were monitored every day. The start and end time-points of the survival of the mice were recorded, and the survival analysis curve was drawn. The surviving mice in each group were euthanized after 30 days; their spleens and livers were collected, and the spleen and liver mass coefficients (ratio of spleen/liver weight to total body weight) were calculated, respectively (Supplementary Fig. 2C). Using human E2A/PBX1 and human CD19 antibodies (ab134114, 1:1000, Abcam), flow cytometry was conducted to analyze bone marrow and peripheral blood cells from mice injected with human 697 cells. The spleens and livers were fixed with formalin, dehydrated, and embedded in paraffin for histopathological analysis by hematoxylin–eosin (HE) staining. Animals were assigned to the blank control group, or treated with antagomir NC + oe-NC (miR-155-5p inhibitory control + lentivirus overexpression control), miR-155-5p antagomir + oe-NC (miR-155-5p inhibition + lentivirus overexpression control), or miR-155-5p antagomir + oe-CDK6 (miR-155-5p inhibition + lentivirus overexpressing CDK6). Engraftments of human 697 cells in NOD/SCID mice are shown in Table 2.
HE staining
The whole sections were dried at room temperature, fixed at room temperature for 30 s, and stained with hematoxylin (60 °C) for 60 s. Next, the sections were treated with 1% hydrochloric acid alcohol differentiation solution for 3 s, stained with eosin for 3 min, dehydrated with 70, 80, 95% ethanol and anhydrous ethanol for 5 min each, and then cleared with xylene three times for 5 min each time. Finally, the sections were sealed with resin and observed under a microscope (BX63, Olympus, Tokyo, Japan).
Statistical analysis
All data were processed using SPSS 21.0 statistical software (IBM Corp., Armonk, NY, USA). The measurement data, obtained from three independent experiments, were expressed as mean ± standard deviation. The comparison between two groups was conducted using unpaired t-test. Data from multiple groups were compared by one-way analysis of variance (ANOVA), with the Tukey’s post hoc test. Data from multiple groups at different time-points were compared by repeated measures ANOVA, followed by the Tukey’s post hoc test. Pearson’s correlation analysis was used to determine the correlations between measures. The Kaplan–Meier method was used to calculate the survival rates and the differences between the survival curves by group were examined using the log-rank test. p < 0.05 indicated statistically significant difference. A schematic diagram illustrating the experimental procedures is depicted in Supplementary Fig. 1.
Results
miR-155-5p and CBL were differentially expressed in cALL
A total of 71 differentially expressed miRNAs were screened using the edge package in R. Among these, 42 were upregulated and 29 were downregulated (Fig. 1A and Table 3). A clustering heatmap of the differentially expressed miRNAs was drawn (Fig. 1B) and miR-155-5p was found to be upregulated in ALL (Fig. 1C). RT-qPCR was used to determine the expression levels of miR-155-5p in peripheral blood lymphocytes from ALL clinical samples. The results showed that the expression of miR-155-5p was significantly increased in peripheral blood lymphocytes from ALL clinical cases as compared with normal controls. To further verify the results, we determined the expression levels of miR-155-5p in ALL cell lines, which showed that, as compared with the AHH-1 cell line, 697, B1, KOPN-8, and SUP-B15 cell lines each displayed notably increased miR-155-5p expression (Fig. 1D, E). The results of the RT-qPCR and Western blot assays showed that CBL expression was significantly decreased in peripheral blood lymphocytes from ALL clinical samples as compared with normal controls (Fig. 1F, G). Similar results were found concerning CBL expression in the above four ALL cell lines (Fig. 1H, I). Pearson’s correlation analysis demonstrated that miR-155-5p was negatively correlated with CBL mRNA (Fig. 1J). These results suggest that miR-155-5p is upregulated and CBL is downregulated in cALL.

A Volcano map of differentially expressed miRNAs in the ALL microarray data. Each dot represents a gene, where red dots indicate upregulation and green dots indicate downregulation. B Clustering heatmap for differentially expressed miRNAs in ALL microarray data. Each row represents a gene, and each column represents a sample. C Expression of miR-155-5p in microarray. Red indicates ALL samples (n = 43) and gray indicates normal bone marrow samples (n = 14). D Scatter plot denoting the expression of miR-155-5p in peripheral blood lymphocytes from ALL clinical samples as determined by RT-qPCR (n = 28). E The expression of miR-155-5p in AHH-1, 697, B1, KOPN-8, and SUP-B15 cell lines as determined by RT-qPCR. F Scatter plot for CBL expression in peripheral blood lymphocytes from ALL clinical samples as determined by RT-qPCR (n = 28). G The protein bands and statistical histogram denoting the expression of CBL in peripheral blood lymphocytes from ALL clinical samples as determined by Western blot assay (n = 28). H The statistical histogram denoting the expression of CBL in AHH-1, 697, B1, KOPN-8, and SUP-B15 cells as determined by RT-qPCR. I The protein bands and statistical histogram denoting the expression of CBL in AHH-1, 697, B1, KOPN-8, and SUP-B15 cell lines as determined by Western blot assay. J The correlation between miR-155-5p and CBL as determined using Pearson’s correlation analysis. *p < 0.05 indicates significant difference. Each cell experiment was conducted three times independently.
miR-155-5p targeted and inhibited CBL
The intersection of four databases predicted that CBL was among the target genes of miR-155-5p (Fig. 2A). We hypothesized that miR-155-5p might target CBL expression in cALL. Using a web-based tool, http://mirdb.org/, a targeted binding site between miR-155-5p and CBL (Fig. 2B) was predicted. mimic-miR-155-5p (overexpression of miR-155-5p) was co-transfected into HEK293T cells with CBL-WT and CBL-MUT, respectively. The expression level of miR-155-5p was determined by RT-qPCR, which showed that the expression of miR-155-5p in CBL-WT and CBL-MUT was significantly increased by mimic-miR-155-5p (Fig. 2C). The results of the dual-luciferase reporter gene assay displayed that mimic-miR-155-5p notably decreased the luciferase activity of CBL-WT, but failed to change that of CBL-MUT (Fig. 2D). Having found that miR-155-5p showed the highest expression in 697 cell line among the four tested cell lines, while the lowest expression was noted in the KOPN-8 cell line, we inhibited the expression of miR-155-5p in 697 cell line and promoted the expression of miR-155-5p in the KOPN-8 cell line. RT-qPCR then confirmed that miR-155-5p expression was significantly lower in 697 cells in response to inhibitor-miR-155-5p treatment than with inhibitor-NC. Furthermore, the expression of miR-155-5p in KOPN-8 cells in response to mimic-miR-155-5p treatment was notably higher than that in the mimic-NC group (Fig. 2E). RT-qPCR and Western blot assay showed that inhibitor-miR-155-5p treatment of 697 cells significantly increased CBL expression, while mimic-miR-155-5p treatment of KOPN-8 cells markedly decreased its expression (Fig. 2F, G). These results suggest that miR-155-5p inhibits CBL expression.

A The intersection of the results obtained from four databases used for prediction of the target genes of miR-155-5p. B The binding site between miR-155-5p and CBL predicted using the web-based tool, http://mirdb.org/. C The statistical histogram denoting miR-155-5p expression in HEK293T cells after treatment with mimic-NC, mimic-miR-155-5p, CBL-WT, or CBL-MUT as determined by RT-qPCR. D The targeted binding between miR-155-5p and CBL after treatment with mimic-NC, mimic-miR-155-5p, CBL-WT, or CBL-MUT in HEK293T cells as examined by dual-luciferase reporter gene assay. E The statistical histogram denoting miR-155-5p expression in 697 and KOPN-8 cells after miR-155-5p was either silenced or overexpressed, as determined by RT-qPCR. F The statistical histogram denoting CBL expression in 697 and KOPN-8 cells after either miR-155-5p silencing or overexpression, as determined by RT-qPCR. G The protein bands and statistical histogram denoting the expression of CBL in 697 and KOPN-8 cells after either miR-155-5p silencing or overexpression, as determined by the Western blot assay. *p < 0.05 indicates significant difference. Each cell experiment was conducted three times independently.
miR-155-5p augmented proliferation and inhibited apoptosis of ALL cells by inhibiting CBL
Next, we focused on elucidating whether miR-155-5p can regulate the cellular functions of ALL cells by inhibiting CBL. RT-qPCR and Western blot assay were used to determine the efficiency of CBL silencing or overexpression in 697 and KOPN-8 cells. The results showed that CBL expression in 697 cells upon sh-CBL-1 and sh-CBL-2 treatment was significantly reduced. Since the silencing efficiency of sh-CBL-1 was higher than that of sh-CBL-2, we used sh-CBL-1 (sh-CBL) for subsequent experiments. CBL expression in KOPN-8 cells in the presence of oe-CBL was markedly increased (Fig. 3A, B). RT-qPCR and Western blot assay showed that miR-155-5p expression was significantly decreased and CBL expression was increased in 697 cells treated with inhibitor-miR-155-5p. Compared with inhibitor-miR-155-5p, inhibitor-miR-155-5p + sh-CBL treatment provoked no significant difference in miR-155-5p expression, but resulted in a marked decline in CBL expression in 697 cells. Moreover, the expression of miR-155-5p in KOPN-8 cells upon mimic-miR-155-5p treatment was significantly increased, along with decreased CBL expression; compared with the effect of mimic-miR-155-5p, whereas mimic-miR-155-5p + oe-CBL treatment of KOPN-8 cells had no significant effects on the expression of miR-155-5p, but increased the expression of CBL (Fig. 3C, D).

A The statistical histogram denoting CBL silencing and overexpression efficiency as determined by RT-qPCR. B The CBL silencing and overexpression efficiency as determined by the Western blot assay. C The statistical histogram denoting miR-155-5p and CBL expression after different treatments as determined by RT-qPCR. D The protein bands and statistical histogram denoting CBL protein expression after different treatments as determined by the Western blot assay. E Line chart depicting cell viability determined by the CCK-8 assay. F The apoptotic rate of cells as examined by flow cytometry. *p < 0.05 indicates significant difference. Each cell experiment was conducted three times independently.
CCK-8 assays showed that inhibitor-miR-155-5p treatment led to markedly decreased cell viability; however, inhibitor-miR-155-5p + sh-CBL reversed this effect. The cell viability was significantly increased by mimic-miR-155-5p, but this effect was neutralized by mimic-miR-155-5p + oe-CBL (Fig. 3E). The results of flow cytometry revealed that the apoptotic rate in the presence of inhibitor-miR-155-5p was significantly increased; however, compared with inhibitor-miR-155-5p treatment, inhibitor-miR-155-5p + sh-CBL provoked a significantly lower apoptotic rate. mimic-miR-155-5p also resulted in a significantly lower apoptotic rate, which was restored in the presence of mimc-miR-155-5p + oe-CBL (Fig. 3F). Overall, miR-155-5p has the potential to promote proliferation and repress apoptosis of ALL cells by inhibiting CBL.
CBL inhibited CDK6 expression through ubiquitination of IRF4
We then investigated the interaction between CBL, CDK6, and IRF4 in ALL. A total of 4396 differentially expressed genes were screened utilizing the edgeR R package, among which 1676 were downregulated and 2720 were upregulated (Fig. 4A and Supplementary Table S3). The clustering heatmap of the differentially expressed genes was delineated (Fig. 4B), among which CDK6 was upregulated in ALL samples (Fig. 4C). Western blot assay was performed to determine the expression levels of IRF4 and CDK6 in clinical peripheral blood lymphocytes. The results demonstrated that the expression levels of IRF4 and CDK6 in ALL were markedly increased compared with those in normal control samples (Fig. 4D). Western blot assay displayed similar results for the expression of IRF4 and CDK6 in AHH-1 and ALL cells (Fig. 4D).

A Volcano map for differentially expressed miRNAs in ALL microarray data. Each dot represents a gene, where red dots indicate upregulation and green dots indicate downregulation. B Clustering heatmap depicting differentially expressed miRNAs in ALL microarray data. Each row represents a gene, and each column represents a sample. C Expression of CDK6 in microarray data. Red indicates ALL samples (n = 13) and gray indicates normal bone marrow samples (n = 4). D The protein expression levels of RF4 and CDK6 in peripheral blood lymphocytes from ALL clinical samples, as determined by the Western blot assay (n = 28). E Protein bands and statistical histograms denoting RF4 and CDK6 expression levels in ALL cells, as determined by the Western blot assay. F Protein bands and statistical histograms denoting RF4 and CDK6 expression in ALL cells after different treatments as determined by the Western blot assay. G The interaction between CBL and IRF4 as determined by the Co-IP assay. H IRF4 ubiquitination level as determined by IP assay. I Protein bands and statistical histograms denoting CBL, IRF4, and CDK6 expression in ALL cells after different treatments as determined by the Western blot assay (n = 28). *p < 0.05 indicates significant difference. Each cell experiment was conducted three times independently.
In addition, our data showed that relative to oe-NC + DMSO, the treatment with oe-CBL + DMSO significantly increased the expression of CBL protein, whereas IRF4 protein expression was notably decreased. When the proteosome inhibitor MG132 was added, CBL protein expression was also significantly increased, but there was no significant difference in the expression of IRF4; however, the expression of IRF4 was higher in the presence of MG132 (Fig. 4E). Compared with treatment with sh-NC + DMSO, the treatment with sh-CBL + DMSO significantly decreased the expression of CBL protein, whereas that of IRF4 protein showed a marked increase. When MG132 was added, CBL protein expression was also significantly decreased, but IRF4 protein expression remained almost unchanged; the protein expression of IRF4 was higher in the presence of MG132 (Fig. 4F).
The results from Co-IP assay showed that CBL interacted with IRF4 (Fig. 4G). IP assays revealed that the ubiquitination level of IRF4 in response to oe-CBL treatment was markedly increased, while sh-CBL significantly decreased the ubiquitination level. Treatment with MG132 also decreased the ubiquitination level of IRF4 (Fig. 4H). Subsequently, we confirmed that CBL inhibited CDK6 expression via ubiquitination of IRF4. Western blot assay results demonstrated that the expression of CBL in the presence of oe-CBL was notably increased, and the expression of IRF4 and CDK6 was significantly decreased. Compared with oe-CBL treatment, oe-CBL + oe-IRF4 did not provoked significantly changed expression of CBL, but did result in a marked increase in the expression of IRF4 and CDK6. CBL expression significantly declined in response to sh-CBL treatment, whereas there was an increase in IRF4 and CDK6 expression. Compared with sh-CBL, sh-CBL + sh-IRF4 treatment resulted in no significant effects on CBL expression, but reduced IRF4 and CDK6 expression (Fig. 4I). These results suggest that CBL downregulates CDK6 by ubiquitination of IRF4.
CBL impaired proliferation and stimulated apoptosis of ALL cells by inhibiting CDK6 expression and promoting the ubiquitination and degradation of IRF4
We then proceeded to examine whether CBL can regulate the cellular functions of ALL cells by regulating the IRF4/CDK6 axis. RT-qPCR showed that expression of CDK6 was increased by oe-CDK6 treatment but decreased by sh-CDK6-1 and sh-CDK6-2 (Fig. 5A). Since the silencing efficiency of sh-CDK6-1 was better than that of sh-CDK6-2, we used sh-CDK6-1 (sh-CDK6) for the follow-up experiments.

A The statistical histogram denoting CDK6 overexpression and silencing efficiency, as determined by RT-qPCR. B The protein bands and statistical histogram denoting CBL, IRF4, and CDK6 protein expression after different treatments, as determined by the Western blot assay. C Line chart showing cell viability determined by the CCK-8 assay. D The apoptotic rate of cells and its statistical histogram, determined by flow cytometry. *p < 0.05 indicates significant difference. Each cell experiment was conducted three times independently.
Western blot assay results revealed that CBL expression was notably increased but IRF4 and CDK6 expression was decreased by oe-CBL. However, compared with oe-CBL treatment, oe-CBL + oe-CDK6 had no significant effect on CBL and IRF4 expression, but provoked a marked increase in CDK6 expression. Treatment with sh-CBL resulted in decreased CBL expression but increased CDK6 and IRF4 expression. Relative to sh-CBL treatment, sh-CBL + sh-CDK6 had no significant effect on CBL and IRF4 expression, but markedly decreased CDK6 expression (Fig. 5B).
CCK-8 assay results demonstrated that oe-CBL decreased cell viability, the effects of which were reversed by oe-CBL + oe-CDK6 combined treatment. sh-CBL notably increased cell viability, while sh-CBL + sh-CDK6 neutralized this effect (Fig. 5C). Flow cytometry showed that the apoptotic rate was increased in the presence of oe-CBL; however, compared with oe-CBL treatment, oe-CBL + oe-CDK6 resulted in a marked decrease in apoptotic rate. Moreover, the apoptotic rate displayed a notable increase in response to sh-CBL. Compared with sh-CBL treatment, sh-CBL + sh-CDK6 resulted in significantly increased apoptotic rate (Fig. 5D). Taken together, CBL enhances the ubiquitination and degradation of IRF4 to reduce CDK6 expression, thus impairing proliferation and promoting apoptosis of ALL cells.
miR-155-5p promoted the development of cALL through the CBL/IRF4/CDK6 axis
Finally, we verified in vivo if miR-155-5p promotes the development of cALL through the CBL/IRF4/CDK6 axis. RT-qPCR was used to determine expression of miR-155-5p in bone marrow and peripheral blood cells, and the Western blot assay was performed to determine the protein expression of CBL, IRF4, and CDK6. The results showed, that compared with blank control, the expression of miR-155-5p, IRF4, and CDK6 in response to antagomir NC + oe-NC treatment was significantly increased, and CBL expression was decreased. miR-155-5p antagomir resulted in a markedly diminished miR-155-5p expression, increased CBL protein expression, but reduced IRF4 and CDK6 protein expression. Relative to miR-155-5p antagomir treatment, miR-155-5p antagomir + oe-CDK6 had no significant effect on miR-155-5p expression or IRF4 and CBL protein expression, but increased CDK6 protein expression (Fig. 6A, B).

A The statistical histogram denoting miR-155-5p expression, as determined by RT-qPCR. B The protein expression levels of CBL, IRF4, and CDK6 as determined by the Western blot assay. C Kaplan–Meier survival curve for the mouse survival rate. D The 697 cell proportion as examined by flow cytometry. E The statistical histogram denoting mouse liver and spleen, and coefficient of liver and spleen. F HE staining for the mouse liver and spleen. *p < 0.05 indicates significant difference between groups (n of mice = 10).
The Kaplan–Meier curve showed that, compared with the blank control, the mice treated with antagomir NC + oe-NC had decreased survival rate; relative to the antagomir NC + oe-NC group, the survival time of mice in the miR-155-5p antagomir + oe-NC group was significantly prolonged, and that of the miR-155-5p antagomir + oe-CDK6 group was decreased (Fig. 6C).
Flow cytometry was used to determine the proportion of human 697 cells in bone marrow and peripheral blood of nude mice. The results demonstrated that the proportion of 697 cells in mice treated with antagomir NC + oe-NC was increased compared with that in the blank control group. miR-155-5p antagomir treatment reduced the proportion of 697 cells, which was reversed in the presence of miR-155-5p antagomir + oe-CDK6 (Fig. 6D and Supplementary Fig. 2A).
Observations of the mouse livers and spleens showed that, compared with blank control, organ swelling was more pronounced in mice treated with antagomir NC + oe-NC, and the coefficient of liver and spleen was increased. The liver and spleen swelling were notably alleviated and the coefficient of liver and spleen was reduced upon treatment with miR-155-5p antagomir, and these effects were neutralized by treatment with miR-155-5p antagomir + oe-CDK6 (Fig. 6E).
HE staining results showed that, as compared with the blank control group, the mice treated with antagomir NC + oe-NC had higher levels of infiltrating cells in the liver and spleen. Treatment with miR-155-5p antagomir led to a marked decrease in infiltrating cells in the livers and spleens of mice. Compared with the miR-155-5p antagomir + oe-NC treatment, miR-155-5p antagomir + oe-CDK6 resulted in notable increase in infiltrating cells in the mouse liver and spleen (Fig. 6F and Supplementary Fig. 2B). These results concurred with other findings indicating miR-155-5p can promote the development of cALL through the CBL/IRF4/CDK6 axis.
Discussion
ALL is a highly prevalent hematological tumor in children, with high mortality despite the best available treatments [23]. In the present study, we showed that miR-155-5p promoted the development of cALL by mediating the CBL–IRF4–CDK6 axis.
In the initial phase of this study, we detected upregulated miR-155-5p and downregulated CBL in cALL samples and confirmed that miR-155-5p decreased the expression of CBL in cALL by targeting CBL. Of note, previous studies have demonstrated the oncogenic function of miR-155 in ALL. One study reported that the lack of miR-155 could lead to suppression of leukemic as well as HCV virus loads in cases of pediatric ALL associated with HCV-4 infection, thus suggesting miR-155 as a potential target for therapy [24]. In addition, downregulation of miR-155 by treatment with IL-27 was reported to aid in suppressing the development of childhood B-ALL, in part, through effects on the proliferation and apoptosis of B-ALL cells [25]. Earlier reports have shown that miR-155-5p is potentially linked to B-cell functions and has an association with the proportion of B-cell population among blood mononuclear cells [26, 27]. In addition, miR-155 is crucial for B-cell responses to thymus-dependent and -independent antigens, whereby B cells without support from miR-155 develop reduced extrafollicular and germinal center responses and cannot produce high-affinity IgG1 antibodies [28]. Previous work has also unveiled the implications of CBL in leukemia. For instance, Martinelli et al. found that CBL E3-ligase activity was absent in B-lineage cALL [14]. In addition, Checquolo et al. revealed that retention of c-CBL due to lack of pTalpha in Notch3 transgenic mice could contribute to leukemogenesis [29]. Of note, the targeting relationship between miR-155 and CBL has been reported and the present findings concur with a previous study demonstrating that miR-155 could target and regulate CBL, thereby regulating the cell biology of melanoma [11]. Furthermore, others have reported that miR-155 could regulate colon cancer cell behaviors by targeting CBL [12]. Taken together, background evidence supports the findings of the current study showing that miR-155-5p targeted and downregulated CBL to promote the development of cALL.
In addition, our study found that CBL suppressed the expression of CDK6 through ubiquitination and degradation of IRF4. In fact, previous studies have highlighted the relation between CBLs and IRFs by showing that high expression of CBLs in GC light zone B cells could enhance the ubiquitination and degradation of IRF4 [16]. Moreover, c-CBL was found to regulate the ubiquitination of another member of the IRFs family, IRF3, to mediate IFN-β production as well as cellular antiviral responses [30]. Similarity to our findings, previous studies have found that IRF4 plays a crucial role in the development of leukemia. As noted by Adamaki et al., IRF4 displayed high expression in cALL, thus functioning as a potential prognostic marker for relapse at diagnosis [18]. In another report, the inhibition of IRF4 was found to partially suppress the development of the pre-B-cell stage in leukemia [31]. Furthermore, the regulation on ALL progression by CDK6 has also been previously demonstrated. Hematopoietic stem/progenitor cells with a lack of CDK6 protein were found to be resistant to ALL induction in a mouse model [31]. In line with our findings, the inhibition of CDK6 was found to reduce the proliferation of ALL cells while promoting their apoptosis [32]. Notably, previous research supports our findings regarding the regulatory relationship between IRF4 and CDK6. It has been previously shown that inhibition of IRF4 could contribute to downregulation of CDK6 in B cells mediated by EBV [19]. In addition, the overexpression of IRF4 was demonstrated to elevate CDK6 expression in KMS11 cells [33]. Therefore, based on the current findings, we conclude that CBL is capable of downregulating IRF4 in cALL through ubiquitination and degradation, thereby repressing the expression of CDK6.
Taken together, the findings of the present study demonstrated that miR-155-5p targets and inhibited the expression of CBL, which suppressed the degradation of IRF4 protein and thus upregulated CDK6, thereby promoting the development of cALL (Fig. 7). These findings provide new mechanistic insights for understanding the role of miR-155-5p in cALL and suggest a basis for potential future therapeutic strategies for cALL. Nevertheless, the specific mechanisms through which the CBL–IRF4–CDK6 axis contributes to the development of cALL warrants further exploration through broader studies in ALL-diagnosed patients and in preclinical models.

miR-155-5p inhibits IRF4 protein degradation through CBL and thus promotes the expression of CDK6, promoting the development of cALL.
Data availability
Data are available upon reasonable request.
References
Gutierrez-Camino A, Martin-Guerrero I, Garcia-Orad A. Genetic susceptibility in childhood acute lymphoblastic leukemia. Med Oncol. 2017;34:179.
Layton Tovar CF, Mendieta, Zeron H. Intracellular signaling pathways involved in childhood acute lymphoblastic leukemia; molecular targets. Indian J Hematol Blood Transfus. 2016;32:141–53.
Bhojwani D, Yang JJ, Pui CH. Biology of childhood acute lymphoblastic leukemia. Pediatr Clin North Am. 2015;62:47–60.
Jost F, Zierk J, Le TTT, Raupach T, Rauh M, Suttorp M, et al. Model-based simulation of maintenance therapy of childhood acute lymphoblastic leukemia. Front Physiol. 2020;11:217.
Paul S, Rausch CR, Nasnas PE, Kantarjian H, Jabbour EJ. Treatment of relapsed/refractory acute lymphoblastic leukemia. Clin Adv Hematol Oncol. 2019;17:166–75.
Schotte D, Akbari Moqadam F, Lange-Turenhout EA, Chen C, van Ijcken WF, Pieters R, et al. Discovery of new microRNAs by small RNAome deep sequencing in childhood acute lymphoblastic leukemia. Leukemia. 2011;25:1389–99.
Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40.
Duyu M, Durmaz B, Gunduz C, Vergin C, Yilmaz Karapinar D, Aksoylar S, et al. Prospective evaluation of whole genome microRNA expression profiling in childhood acute lymphoblastic leukemia. Biomed Res Int. 2014;2014:967585.
Papageorgiou SG, Kontos CK, Diamantopoulos MA, Bouchla A, Glezou E, Bazani E, et al. MicroRNA-155-5p overexpression in peripheral blood mononuclear cells of chronic lymphocytic leukemia patients is a novel, independent molecular biomarker of poor prognosis. Dis Markers. 2017;2017:2046545.
Xu LH, Guo Y, Cen JN, Yan WY, He HL, Niu YN, et al. Overexpressed miR-155 is associated with initial presentation and poor outcome in Chinese pediatric acute myeloid leukemia. Eur Rev Med Pharmacol Sci. 2015;19:4841–50.
Li H, Song JB, Chen HX, Wang QQ, Meng LX, Li Y. MiR-155 inhibits proliferation, invasion and migration of melanoma via targeting CBL. Eur Rev Med Pharmacol Sci. 2019;23:9525–34.
Yu H, Xu W, Gong F, Chi B, Chen J, Zhou L. MicroRNA-155 regulates the proliferation, cell cycle, apoptosis and migration of colon cancer cells and targets CBL. Exp Ther Med. 2017;14:4053–60.
Lo FY, Tan YH, Cheng HC, Salgia R, Wang YC. An E3 ubiquitin ligase: c-Cbl: a new therapeutic target of lung cancer. Cancer. 2011;117:5344–50.
Martinelli S, Checquolo S, Consoli F, Stellacci E, Rossi C, Silvano M, et al. Loss of CBL E3-ligase activity in B-lineage childhood acute lymphoblastic leukaemia. Br J Haematol. 2012;159:115–9.
Shiba N, Park MJ, Taki T, Takita J, Hiwatari M, Kanazawa T, et al. CBL mutations in infant acute lymphoblastic leukaemia. Br J Haematol. 2012;156:672–4.
Li X, Gadzinsky A, Gong L, Tong H, Calderon V, Li Y, et al. Cbl ubiquitin ligases control B cell exit from the germinal-center reaction. Immunity. 2018;48:530–41.e6.
Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature. 2012;490:543–6.
Adamaki M, Lambrou GI, Athanasiadou A, Tzanoudaki M, Vlahopoulos S, Moschovi M. Implication of IRF4 aberrant gene expression in the acute leukemias of childhood. PLoS ONE. 2013;8:e72326.
Banerjee S, Lu J, Cai Q, Saha A, Jha HC, Dzeng RK, et al. The EBV latent antigen 3C inhibits apoptosis through targeted regulation of interferon regulatory factors 4 and 8. PLoS Pathog. 2013;9:e1003314.
Kohrt D, Crary J, Zimmer M, Patrick AN, Ford HL, Hinds PW, et al. CDK6 binds and promotes the degradation of the EYA2 protein. Cell Cycle. 2014;13:62–71.
Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, et al. Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood. 2012;120:1095–106.
Bortolozzi R, Mattiuzzo E, Trentin L, Accordi B, Basso G, Ribociclib VG. a Cdk4/Cdk6 kinase inhibitor, enhances glucocorticoid sensitivity in B-acute lymphoblastic leukemia (B-All). Biochem Pharmacol. 2018;153:230–41.
Suppipat K, Park CS, Shen Y, Zhu X, Lacorazza HD. Sulforaphane induces cell cycle arrest and apoptosis in acute lymphoblastic leukemia cells. PLoS ONE. 2012;7:e51251.
Hassan SS, El-Khazragy N, Elshimy AA, Aboelhussein MM, Saleh SA, Fadel S, et al. In vitro knock-out of miR-155 suppresses leukemic and HCV virus loads in pediatric HCV-4-associated acute lymphoid leukemia: A promising target therapy. J Cell Biochem. 2020;121:2811–7.
Canale S, Cocco C, Frasson C, Seganfreddo E, Di Carlo E, Ognio E, et al. Interleukin-27 inhibits pediatric B-acute lymphoblastic leukemia cell spreading in a preclinical model. Leukemia. 2011;25:1815–24.
Artemaki PI, Letsos PA, Zoupa IC, Katsaraki K, Karousi P, Papageorgiou SG, et al. The multifaceted role and utility of microRNAs in indolent B-cell non-Hodgkin lymphomas. Biomedicines. 2021;9:333.
Katsaraki K, Karousi P, Artemaki PI, Scorilas A, Pappa V, Kontos CK, et al. MicroRNAs: tiny regulators of gene expression with pivotal roles in normal B-cell development and B-cell chronic lymphocytic leukemia. Cancers. 2021;13:593.
Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–59.
Checquolo S, Palermo R, Cialfi S, Ferrara G, Oliviero C, Talora C, et al. Differential subcellular localization regulates c-Cbl E3 ligase activity upon Notch3 protein in T-cell leukemia. Oncogene. 2010;29:1463–74.
Zhao X, Zhu H, Yu J, Li H, Ge J, Chen W. c-Cbl-mediated ubiquitination of IRF3 negatively regulates IFN-beta production and cellular antiviral response. Cell Signal. 2016;28:1683–93.
Jena N, Sheng J, Hu JK, Li W, Zhou W, Lee G, et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia. 2016;30:1033–43.
Rodriguez-Otero P, Roman-Gomez J, Vilas-Zornoza A, Jose-Eneriz ES, Martin-Palanco V, Rifon J, et al. Deregulation of FGFR1 and CDK6 oncogenic pathways in acute lymphoblastic leukaemia harbouring epigenetic modifications of the MIR9 family. Br J Haematol. 2011;155:73–83.
Zhu YX, Shi C-X, Bruins L, Kortuem KM, Stewart K. Cereblon binding IMiD small molecules mediate myeloma cell death via IRF4. Blood. 2012;120:1807.
Acknowledgements
We would also like to thank all participants enrolled in the present study.
Funding
None.
Author information
Authors and Affiliations
Contributions
XS and PZ designed the study. GG was involved in data collection. YD and XL performed the statistical analysis and preparation of figures. LL and QW drafted the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study protocol was approved by the Ethics Committee of Shandong Provincial Hospital Affiliated to Shandong First Medical University and study procedures were performed in strict accordance with the Declaration of Helsinki. Legal guardians of all minor patients provided signed informed consent documentation before study procedures. Animal experiment protocols were approved by the Animal Ethics Committee Shandong Provincial Hospital Affiliated to Shandong First Medical University and conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Extensive efforts were made to ensure minimal suffering of the included animals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sun, X., Guan, G., Dai, Y. et al. microRNA-155-5p initiates childhood acute lymphoblastic leukemia by regulating the IRF4/CDK6/CBL axis. Lab Invest 102, 411–421 (2022). https://doi.org/10.1038/s41374-021-00638-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-021-00638-x
