
Abstract
Numerous studies have revealed that hyperglycemia is a pivotal driver of diabetic vascular complications. However, the mechanisms of hyperglycemia-induced endothelial dysfunction in diabetes remain incompletely understood. This study aims to expound on the underlying mechanism of the endothelial dysfunction induced by hyperglycemia from the perspective of long non-coding RNAs (lncRNA). In this study, a downregulation of SNHG15 was observed in the ischemic hind limb of diabetic mice and high glucose (HG)-treated HUVECs. Functionally, the overexpression of SNHG15 promoted cell proliferation, migration, and tube formation, and suppressed cell apoptosis in HG-treated HUVECs. Mechanistically, SNHG15 reduced thioredoxin-interacting protein (TXNIP) expression by enhancing ITCH-mediated ubiquitination of TXNIP. TXNIP overexpression abrogated the protective effect of lncRNA SNHG15 overexpression on HG-induced endothelial dysfunction. The following experiment further confirmed that SNHG15 overexpression promoted angiogenesis of the ischemic hind limb in diabetic mice. In conclusion, SNHG15 is a novel protector for hyperglycemia-induced endothelial dysfunction via decreasing TXNIP expression.
Similar content being viewed by others
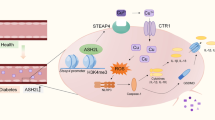


Introduction
Diabetes is a common metabolic disorder characterized by hyperglycemia; it affects more than 400 million people worldwide [1]. Diabetic vascular complications, predominantly characterized by damaged vascular endothelial function, are the leading cause of mortality in diabetic patients [2]. Increasing evidence shows that hyperglycemia is a vital factor that triggers vascular endothelial dysfunction [3, 4]. During the progression of diabetes, a hyperglycemic state leads to aberrant angiogenesis and enhances endothelial permeability, ultimately resulting in the onset of diabetic vascular complications [5]. However, to date, the mechanisms underlying hyperglycemia-induced endothelial dysfunction remain unclear.
Small nucleolar RNA host genes (SNHGs) are a group of long non-coding RNAs (lncRNAs) that have been reported to participate in the regulation of vascular endothelial cell function. For instance, SNHG1 relieves hypoxia-reoxygenation-induced vascular endothelial cell impairment [6]. SNHG6 expression is obviously elevated in the serum of patients with atherosclerosis; its overexpression aggravates human umbilical vein endothelial cell (HUVEC) injury induced by low-density lipoproteins [7]. Moreover, SNHG12 facilitates angiogenesis during ischemic stroke [8], while SNHG15 and SNHG16 are involved in the growth of endothelial cells in glioma and hemangioma tissues, respectively [9, 10]. However, whether these lncRNAs are associated with hyperglycemia-induced endothelial dysfunction remains unclear. In our preliminary experiment, the expression levels of the aforementioned lncRNAs were assessed in high glucose (HG)-treated HUVECs. qRT-PCR results revealed a significant decrease in only the SNHG15 expression level after HG treatment (Supplementary Fig. 1A–E), indicating that SNHG15 may be a potential regulator of hyperglycemia-induced endothelial dysfunction.
Therefore, in the present study, we investigated the role of SNHG15 in hyperglycemia-induced endothelial dysfunction and the underlying mechanism. Moreover, we assessed whether SNHG15 could function as a potential target for the treatment of diabetic endothelial dysfunction.
Materials and methods
Diabetic model of murine hind limb ischemia
Male 6-week-old C57BL/6J mice were commercially obtained from Shanghai SLAC Laboratory Animal Co., Ltd. (China). All animal experiments were approved by the Ethics Committee of The First Affiliated Hospital, School of Medicine, Zhejiang University. Diabetes was induced by intraperitoneally injecting streptomycin (STZ, 50 mg/kg) for five consecutive days. The blood glucose levels of the mice were monitored. Two weeks after the injection, the mice whose blood glucose levels were more than 16 mmol/L were considered to be diabetic (n = 25) [11,12,13]. The mice injected with the same volume of citrate buffer (pH 4.5, the solvent of STZ) were considered to be nondiabetic (n = 25). Following this, 8-week-old diabetic and nondiabetic mice were subjected to left limb ischemia, as described previously [11]. A sham procedure was performed on the right hind limb. Laser Doppler perfusion imaging (LDPI; Moor Instruments, UK) was utilized to monitor the blood flow in the hind limb immediately after surgery as well as 7, 14, 21, and 28 days after surgery. The mice were sacrificed at five time-points (immediately after surgery and 3, 7, 14, and 28 days after surgery) and the gastrocnemius tissues of their hind limbs were collected.
To assess the effect of SNHG15 on hyperglycemia-induced endothelial dysfunction in vivo, 1 × 108 TU SNHG15-overexpressing lentiviral vector (Lenti-SNHG15) or its negative control (Lenti-NC) was injected into the diabetic mice through the tail vein. Left hind limb ischemia was induced and the blood flow in the hind limbs was then measured as described above. All the diabetic mice survived during the surgery. The limb function was measured on the basis of the foot movement score (0 = normal, 1 = plantar but not toe flexion, 2 = no flexion, and 3 = dragging of the foot) before surgery and 3, 7, 14, 21, and 28 days after surgery. Twenty-eight days after surgery, five Lenti-SNHG15-treated mice and five Lenti-NC-treated mice were sacrificed; the gastrocnemius tissues of their hind limbs were then collected.
Isolation of primary endothelial cells
Primary endothelial cells were isolated from the gastrocnemius tissues of the ischemic hind limbs of the mice, as described previously [14]. In Brief, tissues were cut into small sections and immersed in 0.2% type IV collagenase (Shanghai YESEN Biotechnology Co., Ltd. China) for 30 min. Next, the tissues were filtered and centrifuged. The pellet formed was resuspended in PBS, and endothelial cells were isolated using DSB-X biotinylated mouse CD31 antibody (eBioscience, CA) and FlowComp Dynabeads (Invitrogen, USA).
Immunohistochemical staining
The gastrocnemius tissues of the hind limbs were processed into 4-μm-thick paraffin-embedded sections. Immunohistochemical staining was performed using CD31 antibody to examine the capillary density, as described previously [15].
Cell culture and transfection
HUVECs were commercially obtained from Wuhan Procell Life Technology Co., Ltd (China) and cultured in Endothelial Cell Medium (Sciencell, USA) containing 5% fetal bovine serum and 1% growth supplement. The cells were then treated with a normal concentration of glucose (5 mM d-glucose, NG group) or high concentration of glucose (25 mM d-glucose, HG group) for 24 h.
For constructing the lentiviral vector, SNHG15/thioredoxin-interacting protein (TXNIP) sequences were synthesized and subcloned into the pWPXL vector (Hunan Fenghui Biological Technology Co., Ltd. China). The pWPXL/pWPXL-SNHG15/pWPXL-TXNIP, psPAX2, and pMD2G plasmid were co-transfected into HEK293T cells using lipofectamine 3000. Virus particles were harvested 72 h after transfection, filtered through a 0.45-µm filter, and centrifuged (20,000 rpm, 4 °C). Following this, Lenti-SNHG15 or Lenti-TXNIP was collected and used to infect HUVECs with 8 µg/ml polybrene (Biosharp Life Science, China).
qRT-PCR
The RNAprep Pure Cell Kit and RNAprep Pure Tissue Kit (Tiangen, China) were used to extracting the total RNA from HUVECs, gastrocnemius tissues, and primary endothelial cells, respectively. Reverse transcription was performed to synthesize cDNA using the PrimeScript RT Reagent Kit (Takara, Japan). PCR was performed using the SYBR Premix Ex Taq II Kit (Takara). The relative SNHG15 and TXNIP expression levels were calculated using the 2−ΔΔCT method. GAPDH was used as the endogenous control.
Cell counting kit-8 assay
Cell proliferation was assessed using the cell counting kit-8 (CCK-8) assay. After Lenti-NC, Lenti-SNHG15, or Lenti-SNHG15 + Lenti-TXNIP infection, HUVECs were seeded in 96-well plates. Following this, HUVECs were treated with NG or HG for 0, 24, 48, and 72 h and then incubated with CCK-8 (10 μl, CoWin Biosciences, China) for 2 h. The absorbance of each well was detected at 450 nm utilizing a microplate reader (Bio-Rad, USA).
Transwell assay
Cell migration was assessed using the transwell assay. HUVECs were incubated with HG or NG for 24 h. Next, the serum-free medium (100 µl) containing 3 × 104 HUVECs was transferred to the upper Transwell chamber (BD Bioscience, USA). Medium (500 µl) containing 15% FBS and 5 mM or 25 mM d-glucose was added to the lower chamber. Twenty-four hours later, the cells that passed through the filter were fixed with paraformaldehyde and then stained with 0.1% crystal violet. Finally, the stained cells were counted under a microscope.
Tube formation assay
Matrigel (BD Bioscience) was added into 24-well plates and solidified by incubating at 37 °C for 30 min HUVECs were incubated with HG or NG for 42 h. The cells were then seeded on Matrigel at a density of 1 × 105 cells/well, followed by incubation with HG or NG for another 6 h. Tube formation was evaluated using an inverted microscope.
Cell apoptosis assay
After incubation with NG or HG for 48 h, HUVECs were seeded in six-well plates. Cell apoptosis was measured using the Annexin V-FITC Apoptosis Detection Kit (Univ-bio, China). In brief, HUVECs were resuspended in 195 μl annexin V-FITC binding buffer and then incubated with 5 μl annexin V-FITC for 15 min at room temperature without light. Next, HUVECs were stained with 10 μl propidium iodide staining solution. After 5 min, cell apoptosis was measured using a flow cytometer (BD Biosciences).
RNA pull-down assay
The biotinylated SNHG15 (sense) probe and negative control [biotinylated antisense of SNHG15 (antisense)] probe were commercially obtained from RiboBio (China). The lysates of HUVECs were incubated with the sense probe or antisense probe overnight at 4 °C, followed by incubation with streptavidin magnetic beads (Invitrogen) for 1 h at 4 °C. Subsequently, TXNIP expression in the complex bound by the sense or antisense probe was detected using western blot analysis.
RNA-binding protein immunoprecipitation (RIP) assay
The EZ-Magna RIP Kit (Millipore Corporation, USA) was used to perform the RIP assay. HUVECs were lysed and reacted with RIP buffer containing magnetic beads that were conjugated with anti-TXNIP or anti-IgG. After isolating purified RNA samples from the protein-RNA complex, the SNHG15 expression level was determined by qRT-PCR. IgG served as the normalization control.
Western blot
Western blot was conducted using a previously described method [16]. The primary antibodies used in the present study were as follows: anti-TXNIP (1:1000, Abcam, UK), anti-beta actin (1:1000, Abcam), anti-FLAG (0.5 µg/ml, Abcam), anti-HA (1:4000, Abcam), and anti-ITCH (1:1000, Abcam). The secondary antibody was Goat Anti-Rabbit IgG H&L (HRP) (1:5000, Abcam).
Cycloheximide (CHX)-chase assay
HUVECs were transfected with Lenti-SNHG15 or Lenti-NC. Forty-eight hours after transfection, the original medium was replaced with the fresh medium containing CHX (10 μg/ml). Then, the expression of TXNIP was determined using the western bolt at 0, 2, 4, 6 h after CHX treatment.
Co-immunoprecipitation (Co-IP) assay
To evaluate the effect of SNHG15 on the ubiquitination of TXNIP, HUVECs were co-transfected with HA-ubiquitin (Ub), FLAG-TXNIP, and Lenti-SNHG15 or Lenti-NC, followed by treatment with MG132 (10 μM) for 4 h. The Classic IP kit (ThermoFisher, USA) was used for the IP assay. HUVECs were lysed using lyse buffer and incubated with anti-FLAG antibody (Abcam) overnight at 4 °C. Protein complexes were precipitated using protein A/G agarose beads. Subsequently, the precipitates were eluted and analyzed using western blot analysis.
To evaluate the effect of SNHG15 on the interaction between ITCH and TXNIP, HUVECs were transfected with Lenti-SNHG15 or Lenti-NC, followed by treatment with MG132 (10 μM) for 4 h. Next, HUVECs were subjected to co-IP using an anti-ITCH antibody (Abcam) and then assessed using western blot analysis.
Statistical analysis
Data are expressed as the mean ± SD. Statistical analysis was performed using GraphPad Prism 6.0. Comparisons between two experimental groups were made using Student’s t-test. For multiple experimental groups, data were analyzed by one-way ANOVA, followed by the Tukey post hoc test for multiple comparisons. Results were considered statistically significant when P < 0.05.
Results
SNHG15 was downregulated in HG-treated HUVECs and ischemic hind limb of diabetic mice
Compared with NG-treated HUVECs, SNHG15 expression was downregulated in HG-treated HUVECs (Supplementary Fig. 1D). Hence, SNHG15 expression level was further assessed under different glucose concentrations and different treatment times. As shown in Fig. 1A, B, the expression level of SNHG15 was decreased in HG-treated HUVECs in a dose- and time-dependent manner. Next, unilateral femoral artery ligation was performed in diabetic and nondiabetic mice. Assessment of the blood flow by LDPI revealed that compared with nondiabetic mice, perfusion recovery was attenuated in diabetic mice at 7, 14, 21, and 28 days after surgery (Fig. 1C), indicating that the diabetic model of murine hind limb ischemia was successfully established. Meanwhile, compared with nondiabetic mice, a significant decrease in the SNHG15 expression level was observed in the gastrocnemius tissues of the ischemic hind limbs of diabetic mice (Fig. 1D). However, the SNHG15 expression level was not significantly different in the gastrocnemius tissues of the ischemic and nonischemic hind limbs of diabetic mice (Supplementary Fig. 1F). In addition, as shown in Fig. 1E, endothelial cells isolated from the gastrocnemius tissues of the ischemic hind limbs of diabetic mice had significantly lower SNHG15 expression levels than those isolated from the gastrocnemius tissues of the ischemic hind limbs of nondiabetic mice.

A SNHG15 expression was measured by qRT-PCR in HUVECs treated with 5, 15, or 25 mM d-glucose or 5 mM d-glucose + 20 mM mannitol. B SNHG15 expression was measured by qRT-PCR at 0, 12, 24, and 48 h after normal glucose (NG; 5 mM d-glucose) or high glucose (HG; 25 mM d-glucose) treatment of HUVECs. Unilateral femoral artery ligation was performed on diabetic (n = 25) and nondiabetic mice (n = 25). C Laser doppler perfusion imaging (LDPI) monitored the foot blood flow of ischemic (left) and nonischemic (right) hind limbs in diabetic and nondiabetic mice at five time-points (immediately after, and 7, 14, 21, 28 days after surgery). Mice were sacrificed at five time-points (immediately after, and 3, 7, 14, 28 days after surgery) and the gastrocnemius tissues of ischemic hind limbs in diabetic and nondiabetic mice were collected. n = 5 in each group. D SNHG15 expression was measured by qRT-PCR. E The primary endothelial cells (ECs) were isolated from gastrocnemius tissues of diabetic/nondiabetic mice’s ischemic hind limbs 28 days after surgery and the SNHG15 expression was measured. *P < 0.05, **P < 0.01, ***P < 0.001 vs NG-treated HUVECs or nondiabetic mice.
SNHG15 overexpression promoted proliferation, migration, and tube formation, and suppressed apoptosis in HG-treated HUVECs
To assess the biological function of SNHG15 in hyperglycemia-induced endothelial dysfunction, SNHG15 was overexpressed in HG-treated HUVECs through Lenti-SNHG15 infection (Fig. 2A). Compared with NG-treated HUVECs, cell proliferation and cell migration were decreased (Fig. 2B, C) and cell apoptosis was increased (Fig. 2E) in HG-treated HUVECs. Meanwhile, in HG-treated HUVECs, tube formation was suppressed, as evidenced by the shorter tube length and fewer branch points (Fig. 2D). In contrast, Lenti-SNHG15 increased cell proliferation, promoted cell migration and tube formation, and decreased cell apoptosis in HG-treated HUVECs (Fig. 2B–E).

HUVECs were divided into four groups: NG, HG, HG+ lentivirus (Lenti)-SNHG15, and HG+ the negative control of Lenti-SNHG15 (Lenti-NC). A SNHG15 expression was measured by qRT-PCR. B Cell proliferation was detected by cell counting kit-8 (CCK-8) assay. C Cell migration was measured by the transwell assay. D Representative images of tube formation assay performed on HUVECs were shown (scale bar = 400 µm). The tube length and branch points were measured utilizing Image-Pro Plus software. E Cell apoptosis was measured using flow cytometry. *P < 0.05, **P < 0.01, ***P < 0.001 vs NG; #P < 0.05, ##P < 0.01, ###P < 0.001 vs HG + Lenti-NC.
SNHG15 interacted with TXNIP in HUVECs
A bioinformatics database (RNA-Protein Interaction Prediction) forecasted an interaction between SNHG15 and TXNIP. As TXNIP has been identified to participate in hyperglycemia-induced endothelial dysfunction [17], we assessed whether SNHG15 interacted with TXNIP. As shown in Fig. 3A, the sense of SNHG15, rather than its antisense, specifically pulled down endogenous TXNIP protein. Moreover, the RIP assay revealed a higher SNHG15 level in the anti-TXNIP antibody precipitation complex than in the anti-IgG antibody precipitation complex (Fig. 3B). Furthermore, SNHG15 overexpression lessened the TXNIP protein level without changing the TXNIP mRNA level (Fig. 3C, D) in HG-treated HUVECs.

A Detection of TXNIP expression level using western blot in the sample pulled down by the biotinylated SNHG15 (sense) probe. The biotinylated antisense of SNHG15 was served as a negative control (antisense) probe. B RNA-binding protein immunoprecipitation (RIP) of HUVECs, followed by qRT-PCR analysis to detect the SNHG15 expression level (**P < 0.01). HUVECs were treated with Lenti-SNHG15 + HG or Lenti + HG, followed by the measurement of C mRNA and D protein levels of TXNIP utilizing qRT-PCR and western blot, respectively. β-actin was used as an internal control. *P < 0.05 vs HG + Lenti-NC; ns = no significance.
SNHG15 decreased TXNIP expression by reinforcing its ubiquitination
To further investigate the mechanism by which SNHG15 modulates TXNIP expression, HUVECs were transfected with Lenti-SNHG15 or Lenti-NC, following which protein synthesis was blocked using CHX (10 μg/ml). As shown in Fig. 4A, the TXNIP protein level was continuously reduced in SNHG15-overexpressing HUVECs in the presence of CHX, indicating that SNHG15 facilitated the degradation of TXNIP. We then verified whether SNHG15 affected TXNIP stability through proteasome-mediated degradation. As shown in Fig. 4B, the supplementation of MG132 significantly increased the TXNIP expression level in SNHG15-overexpressing HUVECs. Besides, abundant HA-Ub was pulled down by FLAG-TXNIP in SNHG15-overexpressing HUVECs in the presence of MG132 (Fig. 4C). A previous study revealed that the ubiquitin proteasomal degradation of TXNIP protein was mediated by ITCH, an E3 biquitin ligase, in cardiomyocytes [18]. To confirm the interaction between TXNIP and ITCH in HUVECs, a co-IP assay was performed. The results in Fig. 4D depicted that the overexpression of SNHG15 promoted the interaction between TXNIP and ITCH. These data indicated that SNHG15 decreased TXNIP expression by enhancing the ITCH-mediated ubiquitination of TXNIP. In addition, the protein level of TXNIP was increased, while the protein level of ITCH was decreased in HG-treated HUVECs (Supplementary Fig. 2). This suggested the potential roles of ITCH and TXNIP in HG-induced endothelial dysfunction.

A After transfected with Lenti-SNHG15 or Lenti-NC, HUVECs were incubated with cyclohexane (10 μg/mL) for 0, 2, 4, and 6 h. TXNIP protein levels were measured by western blot. β-actin was used as an internal control. ***P < 0.001 vs Lenti-NC. B After transfected with Lenti-SNHG15 or Lenti-NC, HUVECs were treated with or without MG132 (10 μM) for 4 h. TXNIP protein level was measured by western blot. ***P < 0.001. C HUVECs were co-transfected with HA-Ubiquitin (Ub), FLAG-TXNIP, and Lenti-SNHG15 or Lenti-NC, followed by MG132 (10 μM) treatment for 4 h. Then, cells were subjected to co-immunoprecipitation (co-IP) using an anti-FLAG antibody, followed by western blot using anti-HA or anti-FLAG antibody. WCL = whole-cell lysate. D HUVECs were transfected with Lenti-SNHG15 or Lenti-NC, followed by MG132 (10 μM) treatment for 4 h. Then, cells were subjected to co-IP using an anti-ITCH antibody, followed by western blot using anti-TXNIP or anti-ITCH antibody. β-actin was used as an internal control.
TXNIP mediated the regulatory effect of SNHG15 on proliferation, migration, tube formation, and apoptosis in HG-treated HUVECs
To determine whether SNHG15 could modulate HG-induced endothelial dysfunction by regulating TXNIP expression, HUVECs were transfected with Lenti-SNHG15 or Lenti-SNHG15 + Lenti-TXNIP. As shown in Fig. 5A, Lenti-TXNIP infection removed the inhibitory effect of Lenti-SNHG15 on TXNIP expression. In addition, TXNIP overexpression reduced cell proliferation, suppressed cell migration, damaged tube formation, and promoted cell apoptosis in HG-treated HUVECs in the presence of Lenti-SNHG15 (Fig. 5B–E). These data suggested that overexpression of SNHG15 relieved HG-induced endothelial dysfunction by suppressing TXNIP expression.

HUVECs were transfected with Lenti-NC, Lenti-SNHG15, Lenti-SNHG15 + Lenti-TXNIP, followed by HG treatment. A TXNIP protein level was determined using western blot. β-actin was used as an internal control. B Cell proliferation was detected by CCK-8 assay. C Cell migration was measured by the transwell assay. D Representative images of tube formation assay performed on HUVECs were shown (scale bar = 400 µm). The tube length and branch points were measured utilizing Image-Pro Plus software. E Cell apoptosis was measured using flow cytometry.*P < 0.05, **P < 0.01, ***P < 0.001 vs HG + Lenti-NC; #P < 0.05, ##P < 0.01, ###P < 0.001 vs HG + Lenti-SNHG15.
SNHG15 overexpression promoted angiogenesis in ischemic hind limbs of diabetic mice
To determine whether SNHG15 overexpression could alleviate hyperglycemia-induced endothelial dysfunction in vivo, Lenti-SNHG15 or Lenti-NC was injected into diabetic mice through the tail vein, followed which unilateral femoral artery ligation was performed. As shown in Fig. 6D, following Lenti-SNHG15 injection, SNHG15 was successfully overexpressed in the gastrocnemius tissues of the ischemic hind limbs of diabetic mice. The results of LDPI showed that the overexpression of SNHG15 significantly facilitated blood flow recovery in the ischemic hind limb of diabetic mice at 7, 14, 21, and 28 days after surgery (Fig. 6A). In line with LDPI data, the foot movement score demonstrated that the ischemic hind limb function also improved following SNHG15 overexpression (Fig. 6B). Immunohistochemical staining showed that the capillary density was upregulated in the ischemic hind limbs of diabetic mice treated with Lenti-SNHG15 (Fig. 6C). In addition, Lenti-SNHG15 suppressed TXNIP expression in the gastrocnemius tissues of the ischemic hind limbs of diabetic mice (Fig. 6E), suggesting that TXNIP also participated in the angiogenesis-promoting effect of SNHG15 overexpression in vivo.

Diabetic mice were injected into Lenti-SNHG15 (1 × 108 TU, n = 5) or Lenti-NC (1 × 108 TU, n = 5) through the tail vein, followed by unilateral femoral artery ligation. A LDPI monitored the foot blood flow of ischemic (left) and nonischemic (right) hind limbs in diabetic mice at five time-points. B The function of the ischemic hind limb was measured by foot movement score at six time-points (pre-surgery, and 3, 7, 14, 21, 28 days after surgery). ***P < 0.001. Then, mice were sacrificed 28 days after surgery and the gastrocnemius tissues of diabetic mice’s ischemic hind limbs were collected. C The capillary density was examined by immunohistochemical staining as the CD31+ capillary to the muscle fiber ratio (scale bar = 100 µm). D LncRNA SNHG15 expression was measured by qRT-PCR. E TXNIP protein level was measured by western blot. β-actin was used as an internal control. *P < 0.05, ***P < 0.001 vs Lenti-NC.
Discussion
Numerous studies have revealed that hyperglycemia is a pivotal driver of diabetic vascular complications [3, 4]. However, the mechanisms underlying hyperglycemia-induced endothelial dysfunction in diabetic patients remain unclear. The main findings of the present study are as follows: (1) SNHG15 expression was downregulated in HG-treated HUVECs and ischemic hind limbs of diabetic mice; (2) overexpression of SNHG15 mitigated HG-induced endothelial dysfunction by reducing TXNIP expression; and (3) overexpression of SNHG15 promoted angiogenesis in vivo. These findings provide evidence that SNHG15 is a novel regulator of hyperglycemia-induced endothelial dysfunction during the progression of diabetes.
SNHG15 is a strongly conserved lncRNA located on chromosome 7p13 [19]. Previous studies on SNHG15 have focused on its role in cancer. For instance, SNHG15 has been defined as an oncogene involved in several types of cancer, such as hepatocellular carcinoma, non-small cell lung cancer, and osteosarcoma [20,21,22]. However, the function of SNHG15 in diabetic endothelial dysfunction has rarely been studied. In the present study, we assessed SNHG15 expression level in the hind limbs of diabetic mice and HG-treated HUVECs. The downregulation of SNHG15 expression in these samples suggested that SNHG15 was associated with hyperglycemia-induced endothelial dysfunction. The impairment of angiogenesis is the major characteristic of diabetic vascular complications [23]. Several studies have reported that HG treatment suppressed tube formation in HUVECs [24, 25]. Consistent with previous findings, decreased tube formation was observed in HG-treated HUVECs in the present study; it was relieved by SNHG15 overexpression. In addition, SNHG15 overexpression increased cell proliferation, promoted cell migration, facilitated tube formation, and decreased cell apoptosis in HG-treated HUVECs. Moreover, the high expression of SNHG15 promoted angiogenesis and ischemic hind limb function in vivo. Based on these findings, we identified the involvement of SNHG15 in diabetic endothelial dysfunction for the first time.
LncRNAs modulate the expression level of the target protein by affecting its stability [26]. For example, SNHG1, a member of lncRNA SNHGs, disrupts the stability of p-p38 protein by enhancing its ubiquitination [27]. In the present study, TXNIP was found to be the downstream protein of SNHG15 using RNA pull-down and RIP assays. Furthermore, the CHX-chase assay and co-IP assay revealed that SNHG15 promoted TXNIP degradation by enhancing the ubiquitination of TXNIP. ITCH is an E3 ubiquitin ligase and its four WW domains interact with the PPxY motif of TXNIP, thereby resulting in the ubiquitination of TXNIP [28]. ITCH-mediated ubiquitination of TXNIP has been reported in the lung cancer cell line h1299, rat neonatal cardiomyocytes, and macrophages [18, 29, 30]. In line with these findings, we found that SNHG15 enhanced the ubiquitination of TXNIP by increasing the interaction between TXNIP and ITCH in HUVECs.
TXNIP is a multifunctional protein that plays an important role in glucose uptake and metabolism [31]. Upregulated TXNIP levels have been detected in the plasma samples of diabetic patients [32]. A previous study revealed that STZ-induced diabetes could be prevented in TXNIP-deficient HcB-19 mice [33]. In addition, Dunn et al. [17] reported that TXNIP expression was induced in HG-treated HUVECs and that the interference of TXNIP efficiently reversed the inhibitory effect of HG on cell migration, cell proliferation, and tube formation in HUVECs. Consistent with these findings, we observed that TXNIP overexpression reversed the protective effect of SNHG15 on endothelial dysfunction under HG conditions. These findings clearly indicate that TXNIP is a promising target for the treatment of hyperglycemia-induced endothelial dysfunction in diabetes. Dunn et al. [24] reported that TXNIP expression was upregulated in endothelial cells under HG condition and that TXNIP overexpression triggered endothelial dysfunction by suppressing the production of vascular endothelial growth factor (VEGF). Therefore, we speculated that VEGF could be the target gene of SNHG15-TXNIP under hyperglycemic conditions in endothelial cells; this hypothesis will be verified in future research.
To sum up, we found that SNHG15 expression was downregulated under hyperglycemic conditions and that SNHG15 overexpression improved hyperglycemia-impaired endothelial dysfunction by reducing TXNIP expression. Our findings indicate that SNHG15 is a novel regulator of endothelial function and could provide new insights into the mechanisms underlying the onset of endothelial dysfunction during the progression of diabetes.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract. 2017;128:40–50.
Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. Jama. 1979;241:2035–8.
Meza, CA, La Favor, JD, Kim, DH & Hickner, RC Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int J Mol Sci. 2019;20:3775.
Aronson D, Rayfield EJ. How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovasc Diabetol. 2002;1:1–1.
Roberts AC, Porter KE. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diab Vasc Dis Res. 2013;10:472–82.
Liang S, Ren K, Li B, Li F, Liang Z, Hu J, et al. LncRNA SNHG1 alleviates hypoxia-reoxygenation-induced vascular endothelial cell injury as a competing endogenous RNA through the HIF-1α/VEGF signal pathway. Mol Cell Biochem. 2020;465:1–11.
Shan H, Guo D, Zhang S, Qi H, Liu S, Du Y, et al. SNHG6 modulates oxidized low-density lipoprotein-induced endothelial cells injury through miR-135a-5p/ROCK in atherosclerosis. Cell Biosci. 2020;10:4–4.
Zhao M, Wang J, Xi X, Tan N, Zhang L. SNHG12 promotes angiogenesis following ischemic stroke via regulating miR-150/VEGF pathway. Neuroscience. 2018;390:231–40.
Ma Y, Xue Y, Liu X, Qu C, Cai H, Wang P, et al. SNHG15 affects the growth of glioma microvascular endothelial cells by negatively regulating miR-153. Oncol Rep. 2017;38:3265–77.
Zhao W, Fu H, Zhang S, Sun S, Liu Y. LncRNA SNHG16 drives proliferation, migration, and invasion of hemangioma endothelial cell through modulation of miR-520d-3p/STAT3 axis. Cancer Med. 2018;7:3311–20.
Biscetti F, Straface G, De Cristofaro R, Lancellotti S, Rizzo P, Arena V, et al. High-mobility group box-1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a VEGF-dependent mechanism. Diabetes. 2010;59:1496–505.
Shi Y, Huang C, Zhao Y, Cao Q, Yi H, Chen X, et al. RIPK3 blockade attenuates tubulointerstitial fibrosis in a mouse model of diabetic nephropathy. Sci Rep. 2020;10:10458–10458.
Himeno T, Kamiya H, Naruse K, Cheng Z, Ito S, Shibata T, et al. Angioblast derived from ES cells construct blood vessels and ameliorate diabetic polyneuropathy in mice. J Diabetes Res. 2015;2015:257230–257230.
Imoukhuede PI, Dokun AO, Annex BH, Popel AS. Endothelial cell-by-cell profiling reveals the temporal dynamics of VEGFR1 and VEGFR2 membrane localization after murine hindlimb ischemia. Am J Physiol Heart Circ Physiol. 2013;304:H1085–1093.
Xu X, Wei T, Zhong W, Zhu Z, Liu F, Li Q. IL-17 regulates the expression of major histocompatibility complex II and VEGF in DLBCL mice on tumor growth. Aging Pathobiol Therap. 2020;2:96–100.
Niu Y, Zhou B, Wan C, wu R, Sun H, Lu D. Down-regulation of miR-181a promotes microglial M1 polarization through increasing expression of NDRG2. Aging Pathobiol Therap. 2020;2:52–57.
Dunn LL, Simpson PJL, Prosser HC, Lecce L, Yuen GSC, Buckle A, et al. A critical role for thioredoxin-interacting protein in diabetes-related impairment of angiogenesis. Diabetes. 2014;63:675–87.
Otaki, Y, Takahashi, H, Watanabe, T, Funayama, A, Netsu, S, Honda, Y et al. HECT-type ubiquitin E3 ligase ITCH interacts with thioredoxin-interacting protein and ameliorates reactive oxygen species-induced cardiotoxicity. J Am Heart Assoc. 2016;5:e002485.
Shuai Y, Ma Z, Lu J, Feng J. LncRNA SNHG15: A new budding star in human cancers. Cell Prolif. 2020;53:e12716–e12716.
Dai W, Dai JL, Tang MH, Ye MS, Fang S. lncRNA-SNHG15 accelerates the development of hepatocellular carcinoma by targeting miR-490-3p/ histone deacetylase 2 axis. World J Gastroenterol. 2019;25:5789–99.
Jin B, Jin H, Wu HB, Xu JJ, Li B. Long non-coding RNA SNHG15 promotes CDK14 expression via miR-486 to accelerate non-small cell lung cancer cells progression and metastasis. J Cell Physiol. 2018;233:7164–72.
Liu K, Hou Y, Liu Y, Zheng J. LncRNA SNHG15 contributes to proliferation, invasion and autophagy in osteosarcoma cells by sponging miR-141. J Biomed Sci. 2017;24:46.
Yuan J, Tan JTM, Rajamani K, Solly EL, King EJ, Lecce L, et al. Fenofibrate rescues diabetes-related impairment of ischemia-mediated angiogenesis by PPARα-independent modulation of thioredoxin-interacting protein. Diabetes. 2019;68:1040–53.
Dunn LL, Simpson PJ, Prosser HC, Lecce L, Yuen GS, Buckle A, et al. A critical role for thioredoxin-interacting protein in diabetes-related impairment of angiogenesis. Diabetes. 2014;63:675–87.
Patella F, Leucci E, Evangelista M, Parker B, Wen J, Mercatanti A, et al. MiR-492 impairs the angiogenic potential of endothelial cells. J Cell Mol Med. 2013;17:1006–15.
Kapusta A, Feschotte C. Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications. Trends Genet. 2014;30:439–52.
Jiang Y, Wu W, Jiao G, Chen Y, Liu H. LncRNA SNHG1 modulates p38 MAPK pathway through Nedd4 and thus inhibits osteogenic differentiation of bone marrow mesenchymal stem cells. Life Sci. 2019;228:208–14.
Liu Y, Lau J, Li W, Tempel W, Li L, Dong A, et al. Structural basis for the regulatory role of the PPxY motifs in the thioredoxin-interacting protein TXNIP. Biochem J. 2016;473:179–87.
Zhang P, Wang C, Gao K, Wang D, Mao J, An J, et al. The ubiquitin ligase itch regulates apoptosis by targeting thioredoxin-interacting protein for ubiquitin-dependent degradation. J Biol Chem. 2010;285:8869–79.
Tseng PC, Kuo CF, Cheng MH, Wan SW, Lin CF, Chang CP, et al. HECT E3 ubiquitin ligase-regulated Txnip degradation facilitates TLR2-mediated inflammation during group A Streptococcal infection. Front Immunol. 2019;10:2147.
Huy H, Song HY, Kim MJ, Kim WS, Kim DO, Byun J-E, et al. TXNIP regulates AKT-mediated cellular senescence by direct interaction under glucose-mediated metabolic stress. Aging Cell. 2018;17:e12836–e12836.
Zhao Y, Li X, Tang S. Retrospective analysis of the relationship between elevated plasma levels of TXNIP and carotid intima-media thickness in subjects with impaired glucose tolerance and early Type 2 diabetes mellitus. Diabetes Res Clin Pract. 2015;109:372–7.
Chen J, Hui ST, Couto FM, Mungrue IN, Davis DB, Attie AD, et al. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 2008;22:3581–94.
Funding
This work was supported by the Major Science and Technology Project in Medical and Health of Zhejiang Province (co-constructed Project by Province and the Ministry, 2020380400, WKJ-ZJ-2003) and the Key Program of Natural Science Foundation of Zhejiang Province (LZ21H020001).
Author information
Authors and Affiliations
Contributions
Dong-lin Li and Hong-kun Zhang performed development of methodology and writing, review and revision of the paper; Tian-chi Chen, Xun Wang, and Lu Tian performed investigation; Zi-heng Wu, Xiao-hui Wang, Yun-yun He, and Yang-yan He provided visualization and supervision; Tao Shang and Yi-lang Xiang performed writing-reviewing and editing; Qian-qian Zhu and Ming-chun Lai contributed to the conception of the study, performed the experiments and wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
All animal experiments were approved by the Ethics Committee of the The First Affiliated Hospital, School of Medicine, Zhejiang University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhu, Qq., Lai, Mc., Chen, Tc. et al. LncRNA SNHG15 relieves hyperglycemia-induced endothelial dysfunction via increased ubiquitination of thioredoxin-interacting protein. Lab Invest 101, 1142–1152 (2021). https://doi.org/10.1038/s41374-021-00614-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-021-00614-5
This article is cited by
-
Circular RNA ZBTB46 depletion alleviates the progression of Atherosclerosis by regulating the ubiquitination and degradation of hnRNPA2B1 via the AKT/mTOR pathway
Immunity & Ageing (2023)
-
Ubiquitin-specific peptidase 25 exacerbated osteoarthritis progression through facilitating TXNIP ubiquitination and NLRP3 inflammasome activation
Journal of Orthopaedic Surgery and Research (2023)
-
The role of TXNIP in cancer: a fine balance between redox, metabolic, and immunological tumor control
British Journal of Cancer (2023)
-
Ultrastructural features mirror metabolic derangement in human endothelial cells exposed to high glucose
Scientific Reports (2023)
-
LINC00460 Promotes Cutaneous Squamous Cell Carcinoma Progression Through Stabilizing ELAVL1 Protein
Molecular Biotechnology (2023)
