
Abstract
Lipid droplet (LD) accumulation in cancer results from aberrant metabolic reprograming due to increased lipid uptake, diminished lipolysis and/or de novo lipid synthesis. Initially implicated in storage and lipid trafficking in adipocytes, LDs are more recently recognized to fuel key functions associated with carcinogenesis and progression of several cancers, including prostate cancer (PCa). However, the mechanisms controlling LD accumulation in cancer are largely unknown. EPHB2, a tyrosine kinase (TKR) ephrin receptor has been proposed to have tumor suppressor functions in PCa, although the mechanisms responsible for these effects are unclear. Given that dysregulation in TRK signaling can result in glutaminolysis we postulated that EPHB2 might have potential effects on lipid metabolism. Knockdown strategies for EPHB2 were performed in prostate cancer cells to analyze the impact on the net lipid balance, proliferation, triacylglycerol-regulating proteins, effect on LD biogenesis, and intracellular localization of LDs. We found that EPHB2 protein expression in a panel of human-derived prostate cancer cell lines was inversely associated with in vivo cell aggressiveness. EPHB2 silencing increased the proliferation of prostate cancer cells and concurrently induced de novo LD accumulation in both cytoplasmic and nuclear compartments as well as a “shift” on LD size distribution in newly formed lipid-rich organelles. Lipid challenge using oleic acid exacerbated the effects on the LD phenotype. Loss of EPHB2 directly regulated key proteins involved in maintaining lipid homeostasis including, increasing lipogenic DGAT1, DGAT2 and PLIN2 and decreasing lipolytic ATGL and PEDF. A DGAT1-specific inhibitor abrogated LD accumulation and proliferative effects induced by EPHB2 loss. In conclusion, we highlight a new anti-tumor function of EPHB2 in lipid metabolism through regulation of DGAT1 and ATGL in prostate cancer. Blockade of DGAT1 in EPHB2-deficient tumors appears to be effective in restoring the lipid balance and reducing tumor growth.
Similar content being viewed by others
Introduction
EPHB2, a member of the ephrin family of tyrosine kinase receptors (RTK) has been identified as a tumor suppressor gene in prostate cancer with somatic inactivating mutations present in ~10% of sporadic tumors [1, 2]. Using nonsense mediated RNA decay inhibition in combination with array CGH, the EPHB2 gene was found to be completely inactivated (biallelic inactivation) in the prostate cancer metastatic cell line DU145 [1]. The exact function of the gene is still unknown, but introduction of wild-type EPHB2 significantly reduced clonogenic growth of DU145 cells suggesting that EPHB2 may act as a tumor suppressor gene. Decreased EPHB2 gene expression is also frequently found in prostate cancer tissues [3]. The clinical and/or biological consequences of EPHB2 loss in prostate cancer initiation and or progression is unclear.
Limited studies have shown that dysregulation of RTK signaling can result in aberrant metabolism through disruption of critical metabolic pathways. For example, decreased ephrin A1 (EFNA1) signaling can lead to increased glutamine degradation generating α-KG that can be further metabolized to citrate and donate acetyl-CoA groups for lipid biosynthesis. The resultant lipid accumulation has been shown to enhance tumor growth in a mouse model of breast cancer [4]. In obesity, reduction of adipose ephrin ligand EFNB1 may accelerate the vicious cycle with inflammatory cells involved in adipose tissue inflammation through the release of free fatty acid (FA) and adipocytokines [5]. Whether these effects on lipid metabolism are conserved in EPHB2 and/or other members of the ephrin family or involve triacylglycerol (TAG) pathways has not been previously studied.
To sustain the constant demand of energy associated with a proliferative phenotype, cancer cells undergo metabolic re-programming by increasing their rate of fatty acid (FA)s synthesis and storage in lipid droplets (LD) [6]. LDs are cytoplasmic lipid-enriched organelles composed of a monolayer of phospholipids covering a hydrophobic core of neutral lipids, mostly TAG and cholesteryl esters (CEs) [7]. LD formation known as LD biogenesis or lipogenesis is a common response to a lipid overload environment to dampen the potential damaging surge of lipids (lipotoxicity). Other types of stress factors including nutrients and lipid deprivation may also stimulate the accumulation of LD [8]. It is becoming increasingly evident that imbalances in energy and/or redox metabolism can promote LD formation in cancer cells [6]. High rate of FA turnover is maintained, in part, by modulating a very tight balance between key enzymes in lipogenesis (TAG) and lipolysis- regulating proteins including, but not limited to, diacylglycerol transferases (DGATs), adipose triglyceride lipase (ATGL) and pigment epithelium-derived factor (PEDF) [9]. The molecular mechanisms that control the balance between lipogenesis and lipolysis ratio in prostate cancer cells is largely unknown and whether EPHB2 is a regulator of lipid content or these proteins has not been studied.
There is growing evidence suggesting that height and greater body mass index based on the measure of central adiposity, confer a higher risk of developing advanced or lethal prostate cancer [10, 11]. We and others have shown previously the functional consequences of LD accumulation in prostate cancer progression [12, 13]. In addition to increasing the proliferation of prostate cancer cells, LDs can also regulate cell motility through depletion of the non-centrosomal microtubule-organizing center GM130 [14]. Recently we demonstrated that V-ATPase can regulate LD velocity, a novel metabolic feature associated with aggressive prostate cancers especially in an acidic tumor microenvironment [15].
In this study we describe a novel role of EPHB2 in lipid metabolism and its biological effects on prostate cancer. Downregulation of EPHB2 in prostate cancer cells had a strong positive effect on de novo LD formation and subsequent accumulation of LDs in both cytoplasmic and nuclear cellular compartments. Loss of EPHB2 was associated with increased prostate cell proliferation. We show that mechanisms for the EPHB2-mediated metabolic re-programming involved altering enzymes controlling the rate of lipogenesis/lipolysis turnover in tumor cells.
Materials and methods
Cell culture and reagents
Tumor initiated prostate epithelial cell line BPH1 and its carcinoma derivative CAFTD1 cells (from our stocks) were cultured in RPMI 1640 with 10% Cosmic Calf Serum (CCS; GE Lifesciences, Marlborough, MA), and 1% antibiotic-antimycotic as previously described [16]. Prostate cancer cells PC3 (ATCC® CRL-1435™) and LNCaP (ATCC® CRL-1740™) were obtained from the ATCC. LNCaP cells were cultured in RPMI 1640 (Gibco, Grand Island, NY) with 10% Fetal Bovine Serum (FBS; Sigma Aldrich St. Louis, MO) and 1% antibiotic-antimycotic (Gibco, Grand Island, NY) and PC3 cells were grown in F12K/DMEM media (ThermoFisher Scientific, Waltham, MA) supplemented with 10% FBS and 1% antibiotic-antimycotic. Benign prostate epithelial cells BHPrE cells were cultured in DMEM/F12 supplemented with 5% FBS, 1% Insulin-Transferrin Selenium-X (ITS; Gibco, Grand Island, NY), 0.4% bovine pituitary extract (BPE; Hammond Cell Tech, Windsor CA), 1% antibiotic-antimycotic, and 1:1000 10 ng/ml epidermal growth factor (EGF; Sigma Aldrich, St- Louis, MO) [17]. When noted, experimental conditions included exposure to 714 µl per 10 ml Oleic Acid (OA, Cat No. O3008, Sigma Aldrich, St-Louis, MO) and/or 1 μM DGAT1 inhibitor A-922500 (Cat. No. 10012708, Cayman Chemical, Ann Arbor, MI for at least 48 h [14].
RNA silencing
Inhibition of EPHB2 expression was achieved via reverse transfection using Lipofectamine® RNAiMAX Reagents (Invitrogen life technologies, Carlsbad, CA) following manufacturer’s recommendations. Prior to transfection, cells were cultured and passaged upon reaching 80% confluence with 0.25% trypsin-EDTA (ThermoFisher Scientific, Waltham, MA). About 500 k cells were transfected and plated in each well of a six-well plate as follows: Negative Control no. 1 silencerTM (Cat. No. AM4611 ThermoFisher Scientific Waltham, MA), EPHB2 siRNA (Cat. No AM51331, ThermoFisher Scientific Waltham, MA), and three EPHB2 DsiRNA (13.1, 13.2, and 13.3) included in the TriFECTa DsRNA kit (Cat. No hs.EPHB2.13; Integrated DNA Technologies Coralville, IA). After 24 h cells were exposed to different experimental conditions (OA and/or inhibitors) before they were harvested for downstream applications.
Western blot analysis
Protein lysates were isolated with RIPA buffer (Cat. No. J63306, Alfa Aesar Ward Hill, MA) combined with 100× Protease Inhibitor cocktail (Cat No. 1862209 Thermo Fisher, Waltham, MA) and 100× Phosphatase inhibitor cocktail (Cat. No P5726 Sigma, St. Louis, MO).
Protein concentration was quantified using PierceTM BCA Protein assay kit (Cat. No. 23225, ThermoFisher Waltham, MA). Twenty micrograms of protein were run using Mini-PROTEAN® TGX Stain-Free™ Precast 4–20% Gels (Bio-Rad Laboratories, Inc. California, USA) in Tris/Glycine/SDS Buffer at 200 V for 40 min. Samples were then transferred unto a 0.2 μm polyvinylidene difluoride membrane in the Trans-Blot® Turbo™ Blotting system for 10 min at a constant 1.3 A up to 25 V. (Bio-Rad Laboratories, Inc. California, USA). Membranes were blocked with 3% BSA and 1% Tween 20 in PBS for 30 min followed by overnight incubation (constant rocking) at 4 °C with each primary antibody. The source and dilution of antibodies are indicated in Table 1. The next day samples were then incubated for 1 h at room temperature (constant rocking) with appropriate HRP-conjugated secondary antibodies (Cell Signaling Technologies Inc. at 1:5000 dilution). Prior to visualization blots were exposed to Clarity™ Western ECL Substrate kit (Bio-Rad Laboratories, Inc. California, USA). Images were acquired with the ChemiDoc Touch (BioRad) using a stain-free technology for gel, membrane, and blot images to allow for total protein normalization [18].
Lipid droplets staining
Cells were grown on glass coverslips coated with Poly-L-Lysine (Cat No. P8920, Sigma Aldrich, St- Louis, MO). After culture and treatment, cells were washed with PBS (×3), fixed in 10% formalin (20 min at room temperature) followed by a second PBS wash (×3), permeabilized with 0.1% Triton X-100 for 10 min and washed with PBS (×3). After blocking for 30 min in 5% normal horse serum, cells were stained using 4 μg/ml Nile Red diluted in 100% Propylene Glycol (from a 1 mg/ml Nile Red stock in Acetone) and washed in PBS (×3). After staining, coverslips were mounted on glass slides using ProLongTM Gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA, USA) and sealed with Permaslip mounting medium (Newcomer Supply). Z-stacks of the cells were done using the Nikon Eclipse Ti inverted microscope equipped with Xcite 120 LED light source and Zyla sCMOS camera equipped with NIS-Element version 4 software. Lipid droplet quantification was done on FIJI using the addon and protocol (https://www.jlr.org/content/60/7/1333.short) [19].
When indicated, cells were stained with Oil-Red-O (ORO, Cat no. 1277B; Newcomer Supply, Middleton, WI, USA) to visualize neutral lipids. Similar to NileRed staining, cells were grown on Poly-L-Lysine-coated coverslips, washed three times with PBS and fixed in 10% formalin for 20 min at room temperature before stained with ORO. The coverslips were mounted on glass slides before sealing with Permaslip. Pictures from representative fields (n = 4) were taken for each treatment using a ×100 objective for the quantification of the number of LDs per cell using an Olympus BH-2 microscope.
Lipid droplet quantification by flow cytometry
Cells were harvested with 0.25% trypsin-EDTA (ThermoFisher Scientific, Waltham, MA, USA) and washed with PBS (×3), resuspended in PBS, and kept on ice prior to FACS analysis. After setting both side scatters (SSC) and forward scatters (FSC), the voltages and compensation between scatters were set to determine the scale of control samples. Once optimized, three different gates were generated to assess lipid content and granularity as previously described [20].
RNA and qRT-PCR
Total RNA was extracted from cells that were exposed to EPHB2 silencing using a RNeasy® Mini Kit (Qiagen, Cat#74106) following manufacturer’s recommendation. For cDNA synthesis 1 μg total RNA was added to a reaction mix using an iScriptTM cDNA Synthesis Kit (Bio-Rad, Hercules, California, USA). For real-time semiquantitative PCR 1 μl cDNA template was added to IQ RealTime SYBR Green PCR Supermix (Bio-Rad). Relative quantitation was calculated by the ΔΔCt method normalized to GAPDH. The EPHB2 primers Eph B2-Forward (ATGATCCGCAATCCCAACAGC) and Eph B2-Reverse (CATCATCTGAGACACGACGTC) used were purchased from Integrated DNA Technologies (Coralville, IA).
Immunofluorescence
Cells were grown and treated as described above on glass coverslips. At the end of the experiment, cells were washed with PBS (×3), fixed in 4% paraformaldehyde for 20 min, and permeabilized with 0.1% triton X-100 for 15 min. Cells were incubated for 20 min in 5% normal horse serum followed by 1:1000 dilution of mouse anti-Ki-67 (ab16667 Abcam) for 1 h at room temperature, wash with PBS (×3), and incubation with AlexaFluor 488 anti-mouse secondary antibody for 1 h. After staining, coverslips were mounted on glass slides using ProLong Gold antifade reagent with DAPI (Invitrogen, Carlsbad, C, USA) and sealed with Permaslip mounting medium (Newcomer Supply).
Statistical analysis
Data are presented as the mean ± standard deviation (SD) representing three independent experiments performed in triplicate. One-way ANOVA and Dunnett’s multiple-comparisons test were used to determine the significance of difference between groups. Differences were considered statistically significant when P < 0.05. To determine differences between two groups, we used Student’s t-test, where the differences were considered statistically significant when P < 0.05. This analysis was performed using GraphPad Prism, version 7.03.
Results
EPHB2 silencing increases the proliferation of prostate cancer cells
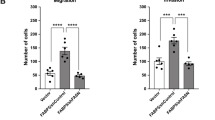
Decreased EPHB2 expression has been recognized in prostate cancer tissues and the gene is completely inactivated in the metastatic prostate cancer cell line DU145, suggesting a possible association with disease progression [1, 3]. To better understand the basal levels in prostate cancer cell lines commonly used in research, we decided to assess EPHB2 protein expression in a panel of cell lines representing different stages of prostate cancer from a benign state (BHPrE1), premalignant (BPH1), organ confined (CAFTD1) up to metastatic disease (LNCaP and PC3). Western blots show the presence of several isoforms in all cells analyzed, except PC3 cells showing only one distinct band (Fig. 1A). Quantification of protein expression (normalized to total protein) demonstrated an inverse correlation with prostate cancer cells aggressiveness (Fig. 1B) with higher expression in benign BHPrE1 and low tumorigenic prostate cancer cells BPH1 and CAFTD1 and lower expression in more aggressive cell lines LNCaP and PC3 cells. These results align with the notion that EPHB2 might serve as a tumor suppressor and highlights its potential role during tumor progression. Restoration of EPHB2 levels in DU145 cells was shown to impair cell proliferation and colony formation [1]. To determine whether downregulation of EPHB2 expression affects the proliferation of prostate cancer cells, we performed RNA silencing experiments in prostate epithelial cell lines representing a premalignant state (BPH1) and metastatic LNCaP cells. To knockdown EPHB2 we compared two methods, a traditional 21-mer RNA duplexes and several (n = 3) 27-mer dicer-substrate RNA duplexes (DsiRNA) optimized for dicer processing and increasing siRNA potency [21]. Compared to scramble controls, the EPHB2-siRNA (exon 3 target) showed more than 50% reduction of mRNA (65% reduction) and protein (72% reduction) expression in BPH1 cells (Fig. 1B, C Left panels). Both EPHB2-DsiRNA 13.1 and 13.3 targeting exons 14 and 5 respectively led to a significant decrease in EPHB2 mRNA (>75%) and protein (>90%) expression. The EPHB2-DsiRNA 13.2, a 3'UTR target led to a minimal but statistically significant reduction (Fig. 1B, C Right panels). The transcriptional changes observed with the qRT-PCR results were associated with translational changes in EPHB2 protein expression. For validation purposes we decided to use DsiRNA 13.3 and the siRNA with similar target regions in subsequent experiments. EPHB2 silencing significantly increased the percentage of Ki-67+ staining in high EPHB2-expressing BPH1 (>2 fold) and CAFTD1 (1.2 fold) cells. Interestingly, further EPHB2 downregulation in low EPHB2-expressing LNCaP cells was also able to significantly increase Ki-67+ cells (1.2 fold) (Fig. 1D). These results suggest that loss of EPHB2 expression may have a role in prostate cancer progression by increasing the rate of proliferation.

A Western blot analysis of EPHB2 protein levels in prostate cancer cell lines representing benign (BHPrE1), low tumorigenic prostate cancer cells (BPH1 and CAFTD1) and more aggressive prostate cancer cell lines (LNCaP and PC3). Three distinct bands around 110 kDa corresponding to 3 EPHB2 isoforms are visualized. All cells display at least two distinct isoforms with benign and low tumorigenic cancer cells expressing a strong expression of a dominant band (Left). Protein expression was analyzed and normalized against total protein using the BioRad ImageLab software (Right). B EPHB2 mRNA expression was quantitated after BPH1 cells were knockdown with either siRNA (Left) or three different DsiRNA (Right) approaches. C EPHB2 protein expression analysis of BPH1 cells exposed to siRNA (Left), DsiRNA 13.1, and DsiRNA 13.3 (Right) compared to scramble control cells. D Dual Ki67 (green) and DAPI (blue) immunofluorescence of BPH1, CAFTD1, and LNCaP cells with EPHB2 knockdown compared to scramble control cells (Top). The number of Ki67 positive cells over the total number of cells was quantified (Bottom). *p < 0.05.
EPHB2 deficiency promotes intracellular lipid accumulation
Accumulation of LDs in prostate cancer cells can increase their pro-tumorigenic (proliferation and motility) potential [12, 14]. Loss of tumor suppressor genes can alter metabolic circuitry and stimulate the accumulation of lipids in cancer cells [22,23,24,25]. To determine whether loss of EPHB2, a putative tumor suppressor gene in PCa, can affect LD, we measure lipid content using a simple method for the detection and quantification of fat accumulation by flow cytometry in BPH1 cells with/without EPHB2 siRNA silencing. This FACS approach has been widely used in adipocytes and non-adipocytes providing a simple, sensitive, quantitative, and reliable method to rapidly assess lipid accumulation and differentiation in single cells [26, 27]. Baseline conditions for lipid content in BPH1 cells were initially determined by comparing Scr Ctrl cells before and after exposure to OA (Fig. 2A, B leftmost flow cytometry panels). We detected increased cytoplasmic granularity (SSC-H axis), a measure of lipid content, in cells with lower levels of EPHB2. Using the scramble control cells population as a reference, three arbitrary gates representing BPH1-cell-specific regions of basal (LDD Low) and increased lipid accumulation (LDD Mid and LDD High) were selected (Fig. 2A). A significant decrease in LDD Low levels were observed in BPH1-siRNA (89%) and BPH1-DsiRNA13.3 (79%) compared to BPH1-Ctrl (96%). There was a simultaneous increase in LDD Mid in EPHB2-silenced cells BPH1-siRNA (10%) and BPH1-DsiRNA13.3 (21%) vs. BPH1-Ctrl (96%). To test whether loss of EPHB2 affects the response to a lipid-rich environment, we exposed these cells to OA for 48 h and lipid content quantified by flow cytometry. Exposure to OA significantly increased the percentage of LDD Mid and High in EPHB2 knock-down vs BPH1-Ctrl cells (Fig. 2B). To determine whether these observations were cell-specific, we performed a similar set of experiments in the prostate cancer cell line CAFTD1. Interestingly in the absence of OA, CAFTD1 and LNCaP cells show increased LDD Mid in all groups compared to BPH1 groups (Supplementary Fig. 1). However, the degree of LDD changes in these two cells was lower with OA addition compared to the effects on BPH1 cells. These results suggest a novel role of EPHB2 in the formation and/or accumulation of LD in prostate epithelial cells.

A EPHB2 was knockdown in BPH1 cells and culture in the presence/absence of OA before harvested for flow cytometry analysis. Dot plot of side scatter (SSC; x-axis) versus forward scatter (FSC; y-axis) of BPH1 cells show the granularity distribution divided in three gates that included: (a) the LDD Low region representing the majority of BPH1 cells under basal conditions (blue edges of the contour plot), (b) LDD mid that included control BPH1 with the highest granularity values for Scr Ctrl cells and (c) LDD high regions (Left). Analysis of lipid content based on the quantitation of the percentage of cells/gate (Right). B LNCaP EPHB2 knockdown and Scr Ctrl cells were exposed lipid overload (OA) before LDD flow cytometry analysis of granularity distribution (Left). Bar graph shows the quantitation of the percentage of LNCaP cells in each gate is (Right). *p < 0.05.
EPHB2 loss alters cytoplasmic and nuclear LD size and density in prostate epithelial cells
To better understand the specific changes in LD content exerted by EPHB2 loss in prostate epithelial cells, we decide to stain cells with Oil-Red-O, a diazo dye used for the visualization of LD using 2D conventional bright-field microscopy [14]. As shown in Fig. 3A, downregulation of EPHB2 increased the number of LDs represented by lipid droplet density (LDD) in BPH1-siRNA (1.7 fold) compared to BPH1-Ctrl cells. Interestingly, LNCaP-siRNA cells demonstrated a higher increase in LDD (>2 fold) than LNCaP-Ctrl cells (Fig. 3A, Right panel). To quantify LD more accurately, we used a recently developed ImageJ-based system (ALDQ) based on fluorescence microscopy staining [19]. This method allows for better LD cluster resolution and 3D quantitation of the number of LDs and a more accurate assessment of LD size in an automated manner. Figure 3B (Left panel) shows a representative image of cells co-stained with Nile Red and nuclear DAPI. Analysis of LDD (the number of LDs/cell) shows significant de novo accumulation of LD in cells with EPHB2 silencing (2.5 fold) compared to BPH1-Ctrl cells. (Fig. 3B Right panel). The addition of OA further increased LDD in all groups with the highest effects observed in cells with EPHB2 knock down. Recent studies have shown that LD size matters in cancer biology and its change is associated with cancer development and other disease conditions [28, 29]. To determine whether loss of EPHB2 has an effect on LD size, LDs were quantified with ALDQ. Figure 3C shows the number of LD (y-axis) based on size (x-axis). Compared to LDs in the BPH1-Ctrl group (green dots), EPHB2 knockdown skewed LD distribution curve to the right with an increased number of larger LDs in both EPHB2-siRNA (red dots) and EPHB2-DsiRNA13.3 (blue dots) cells. Exposure to OA significantly increased the size of LDs in all groups with the highest effects in cells with EPHB2 deficiency (Fig. 3C, Right panel). Similar observations were found in CAFTD1 cell lines (Supplementary Fig. 2A). However, there was a uniform distribution of LDs in CAFTD1-Ctrl cells, different from BPH1-Ctrl. EPHB2 downregulation by DsiRNA13.3 led to a significant accumulation of small LDs (Supplementary Fig. 2B). In contrast to BPH1, the size changes of LDs in response to OA were more pronounced, especially in CAFTD1 cells with EPHB2 knock down (Supplementary Fig. 2C). Recent studies identified LDs inside the nucleus, however their functional significance is still under investigation and the role in cancer is unknown [30, 31]. Confocal 3D images of nuclear LD in Fig. 3D revealed the presence of larger and more abundant LDs in EPHB2-silenced cells compared to Scr Ctrl cells aligned near the cytoplasm/nuclear interphase (inner panels; front and side view) and sparse within the nucleus, especially in cells with DsiRNA13.3 knock down (Left panel). ALDQ quantification shows a significant LD accumulation in EPHB2-silenced cells compared to control cells (Fig. 3D Right panel). Similar results were observed in CAFTD1 cells (data not shown).

A BPH1 and LNCaP engineered cells expressing EPHB2 siRNA and their corresponding Scr Ctrl were stained with Oil-Red-O to identify LDs (Left panel). LD density per cell in BPH1 and LNCaP cells was quantified and compared between knockdown and control groups. B To determine the 3D content of LD, NileRed (green) and DAPI (blue) dual staining was performed, imaged with confocal microscopy and analyzed using the LD quantification software ALDQ (Left panel). ALDQ quantification of NileRed LDs in control and knockdown cells in the presence/absence of OA (Right panel). C Scatter dot plot of LD size distribution obtained from ALDQ analyzed images in EPHB2 siRNA (red), DsiRNA13.3 (blue) knock down cells compared to controls cells (green) (Left panel). Analysis of LD size of silenced EPHB2 and Scr Ctrl cells exposed to OA was compared to basal (OA-) levels (Right panel). D Confocal 3D images of nuclear LD revealed the presence LDs aligned near the cytoplasm/nuclear interphase (inner panels; front and side view) and sparse within the nucleus (Left panel). ALDQ quantification of nuclear LD in EPHB2 knockdown cells was compared to Scr Ctrl cells (Right panel). *p < 0.05.
These data provide evidence of a potential role of EPHB2 on LD accumulation and size increase suggesting that the mechanisms involved in LD growth are affected by alterations in EPHB2 ephrin signaling.
Loss of EPHB2 increases lipogenesis/lipolysis ratio in prostate cancer cells
Lipid metabolism is controlled by a number of genes that regulate the tight balance between the formation and storage (lipogenesis) as well as the degradation (lipolysis) of LDs [7]. Alteration of the signaling induced by ephrin-A1, a member of the ephrin ligand subfamily, has been linked to glutamine degradation and subsequent lipid accumulation in breast cancer cells [4, 5]. To determine whether loss of EPHB2 is involved in the regulation of lipid-regulating genes, western blot analysis of molecules that regulate key aspects of lipogenesis and lipolysis were assessed. Compared to Scr Ctrl cells, downregulation of EPHB2 in BPH1 cells led to a significant increased expression of a number of lipogenesis-regulating genes including DGAT1, DGAT2, FITM, GPAT4, and PLIN2 (Fig. 4A). EPHB2 knock-down also decreased the expression of several lipolysis-regulating genes including ATGL, PEDF, CGI-58, and HSL when compared to Scr Ctrl group. The presence of physiological levels of LDs is controlled by a tight balance between the rate of lipogenesis and lipolysis exerted by these genes. Altered expression of these molecules has direct consequences on the rate of LD accumulation and storage, therefore we measured the contribution of each gene on the lipogenesis/lipolysis ratio net effect. The relative expression of each lipogenesis gene against each lipolysis gene was calculated in Scr Ctrl cells to determine basal conditions and set to a value of one. Similar calculations were performed for each of the EPHB2 knockdown groups, and then compared to the Scr Ctrl cells. As shown in Fig. 4B (Right panel), loss of EPHB2 favored lipogenesis in 22 (yellow to red) out of 30 comparisons. Values below one are presented in green. The highest ratio was observed with DGAT1, DGAT2, and PLIN2 genes when compared to key lipolysis-regulating genes ATGL and PEDF (Fig. 4B, left) [14]. Similar observations were seen in CAFTD1 cells with DGAT1, DGAT2, PLIN2, and FITM showing the highest lipogenesis/lipolysis ratio (Supplementary Fig. 3). These results suggest that loss of EPHB2 may play a critical inhibitory role in the regulation of genes responsible for lipogenesis and lipolysis homeostasis overall controlling lipid storage in LD.

A Immunoblot showing the expression of molecules that regulate lipogenesis and lipolysis in EPHB2 knock down and control BPH1 cells (Left panels). Western blot analysis shows a significantly increased protein expression of key enzymes involved in lipogenesis (Upper right panel) and simultaneous decreased expression of those associated with lipolysis (Lower right panel). B Quantitation of the lipogenesis/lipolysis ratio of lipogenesis genes (LG) over lipolysis-related ATGL and PEDF and comparison between Scr Ctrl and EPHB2-silenced groups. (Left panels). The complete comparison of lipogenesis and lipolysis genes showing the highest ratios shaded in dark red and the lowest numbers in dark green (Right). *p < 0.05.
DGAT1 inhibition reduces lipid accumulation induced by EPHB2 loss
Upon EPHB2 loss, we observed the highest effect on lipogenesis-related genes DGAT1 and DGAT2 (Fig. 4), therefore we decided to test whether targeting DGAT1 alone could abrogate these effects in EPHB2 knock-down cells. Using the DGAT1-specific inhibitor A 922500 (devoid of activity against DGAT2, ACAT1, or ACAT2), we saw a significant decreased DGAT1 expression in both EPHB2 knock-down cells (EPHB2-siRNA and EPHB2-DsiRNA13.3) with no significant effects on DGAT2 and other genes associated with lipogenesis (Fig. 5A). Interestingly DGAT1 inhibition led to a slight increased expression of ATGL, ATGL-binding protein PEDF, and ATGL activator CGI-58. As a result of DGAT1 inhibition and changes in lipolysis-related genes, there was decreased lipogenesis (Fig. 5B) and increased activation of ATGL (lipolysis) by CGI-58 compared to EPHB2-silenced cells in the absence of the DGAT1 inhibitor (Fig. 5C). Next, to determine whether DGAT1 inhibition of EPHB2 knockdown cells translated in biological effects on LD formation/accumulation, NileRed staining was performed and quantified with ALDQ. As shown in Fig. 5D, there was a significantly reduced density of cytoplasmic and nuclear LDs in EPHB2-siRNA and EPHB2-DsiRNA13.3 cells after DGAT1 inhibition. Compared to untreated cells, this decrease was more pronounced in the formation of small LDs in both groups exposed to DGAT1 inhibitor (red and dark blue dots in Fig. 5E). We have shown previously that DGAT inhibition can influence in vitro and in vivo tumor growth in highly aggressive and bone metastatic prostate cancer cell line PC3. PC3 cells are known to express high levels of DGAT1 [14]. Therefore, we decided to measure the effects of DGAT1 suppression in cells with EPHB2 deficiency on proliferation. Addition of a DGAT inhibitor significantly decreased the number of proliferative cells assessed by Ki67 staining of both EPHB2-siRNA and EPHB2-DsiRNA13.3 cells compared to untreated cells (Fig. 5F). These results suggest that targeting DGAT1 can reduce lipogenesis/lipolysis ratio and lipid changes (LD formation/accumulation) associated with the loss of the tumor suppressor EPHB2 in prostate cancer cells.

A EPHB2 engineered BPH1 cells were exposed to a DGATi for 48 h before protein isolation. Western blot images show a clear effect of DGAT1 inhibition on the protein levels. The expression of key lipogenesis and lipolysis-related genes were assessed. B Quantitation of DGAT1/lipolysis genes including ATGL, PEDF, and CGI58 in EPHB2-silenced cells exposed to the DGAT1 inhibitor compared to the absence of DGAT1 inhibition. C The CGI-58/ATGL ratio based on protein expression was analyzed in EPHB2 knock down cells treated with DGAT1 inhibitor and compared to untreated cells. D NileRed staining of EPHB2 knock-down BPH1 cells in the presence/absence of DGAT1 inhibitor (Left). Cells exposed to the DGAT1 inhibitor exhibited a significant decrease in LD density compared to untreated cells (Right). E Dot plot analysis of LD distribution shows a clear reduction of newly formed (smaller size) LDs in EPHB2 knock down cells after treated with DGAT1 inhibitor (dark blue and red dots). F Dual Ki67 (red) and DAPI (blue) immunofluorescence of BPH1 silenced cells with EPHB2 siRNA or DsiRNA13.3 in the presence/absence of a DGAT1 inhibitor (Left). Quantitation of Ki67 positive cells exposed to DGAT1 inhibitor was compared to control untreated cells (Right). *p < 0.05.
Discussion
Loss of tumor suppressor function has been associated with altered metabolic changes in cancer cells; however, the mechanisms for this association are not clear [22,23,24,25]. Here, we established a link between EPHB2, an RTK with potential tumor suppressor function, with LD biology in PCa cells. EPHB2 gene expression is low in PCa tissue samples compared to normal prostate cells with somatic inactivating mutations accounting for ~10% of sporadic tumors [1, 2]. We assessed the expression of EPHB2 in a panel of prostate cancer cell lines ranging from benign BHPrE1 to highly aggressive (and metastatic) PC3 cells, and found that EPHB2 expression inversely correlated with prostate cancer cell aggressiveness. Western blot shows three distinct isoforms around 110 kDa. BHPrE, BPH1, and CAFTD1 cells expressed similar isoforms (two upper bands), LNCaP did not express one of them and in addition, showed a lower third band. PC3 cells only expressed one of them (Fig. 1A). Although the origin and function of these distinct isoforms are currently unknown, there was a distinct loss of EPHB2 expression in all isoforms in more aggressive cells. These results align with a previous work showing that a biallelic inactivation in the prostate cancer metastatic cell line DU145 [1] may support a tumor suppressive role of EPHB2 during tumor progression. Further support of this concept was confirmed when restoration of EPHB2 in DU145 (with nonfunctional EPHB2) cells decreased its clonogenic capacity in soft agar [1]. In our current study of several different PCa cell lines, EPHB2 silencing had a significant positive impact on cell proliferation in prostate cancer.
Metabolic dysregulation in obesity drives ectopic LD accumulation in non-adipose tissues, and this can increase the risk profile for insulin-resistance, cardiovascular disease, and cancer [32]. Excess of free FA and other adipose-derived lipids observed in obesity may enhance cancer susceptibility by altering the intracellular lipid metabolism and promoting an inflammatory tumor microenvironment [33]. Lipogenesis dominates during the anabolic processes in cancer cells, where FA synthesis is a critical step during the generation of building blocks in complex lipids. The pathways that trigger tumor lipogenesis can lead to the accumulation and storage of newly formed LD. Recent evidence in lipid biology supports an intimate relationship between tumor development and activation of lipogenic and lipolytic enzymes suggesting that targeting lipid mobilization from LDs may be an appropriate and effective tumor suppressive tool for cancer therapy.
Prostate cancer is characterized by a slow glycolysis and may be more reliant on both intra and extracellular FA oxidation to provide ATP as a source of energy to sustain cell proliferation [33, 34]. We and others have recently shown that prostate cancer cells exhibit increased LD (number and size) in high-grade carcinomas and enhanced rates of de novo FA synthesis [13, 15, 35]. The underlying mechanisms that facilitate the accumulation of LDs in prostate cancer are unknown.
A limited number of studies have shown that alterations of ephrin ligand A-1 (EFNA1) signaling increase glutaminolysis, a critical metabolic pathway that leads to lipid accumulation, enhancing tumor growth in both mouse and human models of breast cancer [4]. Targeting de novo FA synthesis with Orlistat (a FASN inhibitor) suppresses lipid accumulation and the proliferative phenotype observed in ephrin-deficient breast cancer cells [4]. EPHB2, an RTK member of the ephrin receptor subtype, binds to several ephrin ligands in the following order of affinity: EFNB2, B1, A5, A2, A4, A1, and A3 [36]. Whether different members of the ephrin family (receptors and or ligands) can affect lipid metabolism in different types of cancers has not been studied. Here, we showed that silencing EPHB2 in prostate epithelial cells increased the accumulation of de novo LDs as well as the response to a lipid overload to simulate an obese environment. The increased LD density was accompanied by changes in LD size with the accumulation of larger LD in the cytoplasm of EPHB2 knockdown cells. It is believed that the number of LDs reflects the energy storage capacity and their adaptation for demanding environments [37]. Our observations suggest that loss of EPHB2 tumor suppressor function allows cells to acquire and store more energy in LDs to sustain the increased proliferation capacity.
Interestingly, in addition to increased LD density in the cytoplasm, EPHB2-deficient cells showed LD in the nuclear/cytoplasmic interphase with a significant number inside the nucleus. LDs are considered ubiquitous organelles that after emerging from the endoplasmic reticulum (ER) in the cytoplasm, are stored in this cellular compartment for later use as a source of energy, membrane biogenesis or to support organelles [38, 39]. The origin and functional significance of LDs in the nucleus is currently under intense investigation and signaling pathways controlling the localization of LDs have not been defined. Similar to their cytoplasmic counterparts, nuclear LDs display, overall, a similar morphology. However, some recent evidence shows distinct differences in their protein and neutral lipid composition suggesting an innovative role for this organelle in nuclear protein regulation and nuclear lipid signaling [40]. It remains to be determined whether the EPHB2-mediated increase in nuclear LDs reflects a new role in nuclear signaling or reflects a nuclear-to-cytoplasmic shuttling process. For example, in hepatocytes, under ER stress nuclear LD formation correlate with apolipoprotein B100 free luminal LDs as a result of increased microsome triglyceride transfer protein activity [41]. These nuclear LDs recruit CTP:phosphocholine cytidylyl-transferase α (CCTα), the rate-limiting enzyme of the Kennedy pathway for phosphatidylcholine synthesis, allowing for relocation into the nucleus through defects in type I nucleoplasmic reticulum [41]. Also, nuclear relocation of DGAT2 and CCTα in close association with the promyelocytic leukemia isoform II has been shown to induce nuclear LD formation [42]. Future studies will determine the contribution of these potential inducers for the accumulation of nuclear LD and establish the role in the nucleus in prostate cells with EPHB2 loss.
LD biogenesis in the ER is initiated with the synthesis of neutral lipids, mainly TAG and CE, by acylation of their respective precursors; diacylglycerol (DAG) and sterols, mainly ergosterol and zymosterol in yeast, and cholesterol in mammals. Under normal conditions, the presence of LDs is tightly controlled by the balance of a number of key enzymes involved in the formation (lipogenesis) and destruction (lipolysis) of these organelles [7]. Key rate-limiting enzymes of the lipogenesis/lipolysis ratio such as DGAT1, DGAT2, and ATGL are considered essential for TAG metabolism and LD accumulation. Both DGAT1 and DGAT2 are overexpressed in several types of cancer tissues including liver, colon, breast bladder, ovarian, prostate, and pancreatic tumors [43].
DGAT1 knockdown in LNCaP cells has been shown to decrease cell growth, reduce colony formation and affect autophagy [12]. Interestingly, the inability to break down TAG to liberate FA in LNCaP can also impair cell invasion and growth [44]. ABDH5, an ATGL-activating coenzyme markedly decreased in metastatic castration-resistant PCa, acts as a metabolic tumor suppressor preventing epithelial to mesenchymal transition and the Warburg effect [44]. Castration-resistant C4-2B and PC3 cells showed reduced palmitate-induced apoptosis due to a high rate of FA oxidation and TAG synthesis [34]. We observed increased expression of DGAT1 and DGAT2 and decreased ATGL in EPHB2-silenced cells compared to controls. Quantitation of the degree in protein expression alteration showed increased lipogenesis/lipolysis ratio that could explain the accumulation of LDs. Further exposure to a DGAT1-specific inhibitor (A-922500), inhibited the formation of LD induced by EPHB2 loss in prostate cancer cells. Blocking LD accumulation has direct implications on prostate cancer growth as a result of increased energy availability due to changes in lipid metabolism.
The loss of an RTK ephrin family member EPHB2 is critical in prostate cancer cells by regulating key enzymes and proteins in the TAG pathway (Fig. 6). Loss of EPHB2, a gene with potential tumor suppressor function, increased de novo LD lipogenesis and the response to an obesogenic environment inducing prostate cancer cell proliferation. These results have implications for tumor progression, especially in the setting of obesity where there is more ectopic lipid in non-adipocytes and higher extracellular lipid availability that further enhance FA flux inside cancer cells. LD morphology and compartmentalization appears to be sensitive to altered expression of EPHB2 and the discovery of intranuclear LDs requires additional studies to identify other nuclear signaling partners. In EPHB2-deficient tumors, the addition of a DGAT1 inhibitor to traditional therapy could be beneficial in restoring normal lipid turnover and reducing tumor growth.

Left. Under normal EPHB2 signaling conditions, the rate of lipogenesis/lipolysis is balanced. Middle. Loss of EPHB2 receptor leads to activation of the TAG pathway with increased expression of genes associated with lipogenesis such as DGAT1 and DGAT2 and simultaneous decreased levels of the lipolysis-regulating enzyme ATGL. These changes ultimately tip the lipogenesis/lipolysis balance toward the formation of LDs in the endoplasmic reticulum (ER) and subsequent accumulation in both nuclear and cytoplasmic compartments. This de novo accumulation of LD increases the density and size of newly formed LDs creating a high energy state that can foment the proliferation of prostate cancer cells. Right. Targeting DGAT1 reduces lipogenesis. With DGAT1 inhibition there is a simultaneous increase in lipolysis-regulating ATGL. These changes affect LDs accumulation by increasing lipolysis and releasing FFA. The effects of restoring lipid content have a negative impact on cell proliferation in prostate cancer cells with EPHB2 deficiency (Right). M cell membrane, C cytoplasm, N nucleus, ER endoplasmic reticulum, TAG triacylglyceride, LDs lipid droplets, Rx DGAT1-specific inhibitor, and FFA free fatty acids.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Huusko P, Ponciano-Jackson D, Wolf M, Kiefer JA, Azorsa DO, Tuzmen S, et al. Nonsense-mediated decay microarray analysis identifies mutations of EPHB2 in human prostate cancer. Nat Genet. 2004;36:979–83.
Robbins CM, Hooker S, Kittles RA, Carpten JD. EphB2 SNPs and sporadic prostate cancer risk in African American men. PloS one. 2011;6:e19494.
Kim AH, Nguyen J, Schluchter M, Wang B, Nguyen C. MP29-18 The eph family of receptor tyrosine kinases as tumor suppressors in african-americans with prostate cancer. The J Urol. 2018;199:e372.
Youngblood VM, Kim LC, Edwards DN, Hwang Y, Santapuram PR, Stirdivant SM, et al. The ephrin-A1/EPHA2 signaling axis regulates glutamine metabolism in HER2-positive breast cancer. Cancer Res. 2016;76:1825–36.
Mori T, Maeda N, Inoue K, Sekimoto R, Tsushima Y, Matsuda K, et al. A novel role for adipose ephrin-B1 in inflammatory response. PloS one. 2013;8:e76199.
Petan T, Jarc E, Jusovic M. Lipid droplets in cancer: guardians of fat in a stressful world. Molecules. 2018;23:1946.
Jackson CL. Lipid droplet biogenesis. Curr Opin Cell Biol. 2019;59:88–96.
Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100:3077–82.
Senga S, Kobayashi N, Kawaguchi K, Ando A, Fujii H. Fatty acid-binding protein 5 (FABP5) promotes lipolysis of lipid droplets, de novo fatty acid (FA) synthesis and activation of nuclear factor-kappa B (NF-kappaB) signaling in cancer cells. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863:1057–67.
Cao Y, Ma J. Body mass index, prostate cancer-specific mortality, and biochemical recurrence: a systematic review and meta-analysis. Cancer Prev Res. 2011;4:486–501.
Genkinger JM, Wu K, Wang M, Albanes D, Black A, van den Brandt PA, et al. Measures of body fatness and height in early and mid-to-late adulthood and prostate cancer: risk and mortality in The Pooling Project of Prospective Studies of Diet and Cancer. Ann Oncol. 2020;31:103–14.
Mitra R, Le TT, Gorjala P, Goodman OB Jr. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer. 2017;17:631.
Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19:393–406.
Nardi F, Franco OE, Fitchev P, Morales A, Vickman RE, Hayward SW, et al. DGAT1 inhibitor suppresses prostate tumor growth and migration by regulating intracellular lipids and non-centrosomal MTOC protein GM130. Sci Rep. 2019;9:3035.
Nardi F, Fitchev P, Brooks KM, Franco OE, Cheng K, Hayward SW, et al. Lipid droplet velocity is a microenvironmental sensor of aggressive tumors regulated by V-ATPase and PEDF. Lab Investig. 2019;99:1822–34.
Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61:8135–42.
Jiang M, Strand DW, Fernandez S, He Y, Yi Y, Birbach A, et al. Functional remodeling of benign human prostatic tissues in vivo by spontaneously immortalized progenitor and intermediate cells. Stem Cells. 2010;28:344–56.
Posch A, Kohn J, Oh K, Hammond M, Liu N. V3 stain-free workflow for a practical, convenient, and reliable total protein loading control in western blotting. J Vis Exp. https://doi.org/10.3791/50948, 50948 (2013).
Exner T, Beretta CA, Gao Q, Afting C, Romero-Brey I, Bartenschlager R, et al. Lipid droplet quantification based on iterative image processing. J Lipid Res. 2019;60:1333–44.
Lee YH, Chen SY, Wiesner RJ, Huang YF. Simple flow cytometric method used to assess lipid accumulation in fat cells. J Lipid Res. 2004;45:1162–7.
Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23:222–6.
Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–48.
Keckesova Z, Donaher JL, De Cock J, Freinkman E, Lingrell S, Bachovchin DA, et al. LACTB is a tumour suppressor that modulates lipid metabolism and cell state. Nature. 2017;543:681–6.
Maimouni S, Issa N, Cheng S, Ouaari C, Cheema A, Kumar D, et al. Tumor suppressor RARRES1- A novel regulator of fatty acid metabolism in epithelial cells. PloS one. 2018;13:e0208756.
Parrales A, Iwakuma T. p53 as a regulator of lipid metabolism in cancer. Int J Mol Sci. 2016;17:2074.
Kraus NA, Ehebauer F, Zapp B, Rudolphi B, Kraus BJ, Kraus D. Quantitative assessment of adipocyte differentiation in cell culture. Adipocyte. 2016;5:351–8.
Campos V, Rappaz B, Kuttler F, Turcatti G, Naveiras O. High-throughput, nonperturbing quantification of lipid droplets with digital holographic microscopy. J Lipid Res. 2018;59:1301–10.
Suzuki M, Shinohara Y, Ohsaki Y, Fujimoto T. Lipid droplets: size matters. J Electron Microsc . 2011;60:S101–116.
Cohen BC, Shamay A, Argov-Argaman N. Regulation of lipid droplet size in mammary epithelial cells by remodeling of membrane lipid composition-a potential mechanism. PloS one. 2015;10:e0121645.
Farese RV Jr., Walther TC. Lipid droplets go nuclear. J Cell Biol. 2016;212:7–8.
Barbosa AD, Siniossoglou S. New kid on the block: lipid droplets in the nucleus. FEBS J. https://doi.org/10.1111/febs.15307 (2020).
Long J, Zhang CJ, Zhu N, Du K, Yin YF, Tan X, et al. Lipid metabolism and carcinogenesis, cancer development. Am J Cancer Res. 2018;8:778–91.
Corbet C, Feron O. Emerging roles of lipid metabolism in cancer progression. Curr Opin Clin Nutr Metab Care. 2017;20:254–60.
Balaban S, Nassar ZD, Zhang AY, Hosseini-Beheshti E, Centenera MM, Schreuder M, et al. Extracellular fatty acids are the major contributor to lipid synthesis in prostate cancer. Mol Cancer Res. 2019;17:949–62.
Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Instit. 2009;101:519–32.
Himanen JP, Chumley MJ, Lackmann M, Li C, Barton WA, Jeffrey PD, et al. Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nat Neurosci. 2004;7:501–9.
Yu J, Li P. The size matters: regulation of lipid storage by lipid droplet dynamics. Sci China Life Sci. 2017;60:46–56.
Pol A, Gross SP, Parton RG. Review: biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J Cell Biol. 2014;204:635–46.
Tirinato L, Pagliari F, Limongi T, Marini M, Falqui A, Seco J, et al. An overview of lipid droplets in cancer and cancer stem cells. Stem Cells Int. 2017;2017:1656053.
Layerenza JP, Gonzalez P, Garcia de Bravo MM, Polo MP, Sisti MS, Ves-Losada A. Nuclear lipid droplets: a novel nuclear domain. Biochim et Biophys Acta. 2013;1831:327–40.
Soltysik K, Ohsaki Y, Tatematsu T, Cheng J, Fujimoto T. Nuclear lipid droplets derive from a lipoprotein precursor and regulate phosphatidylcholine synthesis. Nat Commun. 2019;10:473.
Ohsaki Y, Kawai T, Yoshikawa Y, Cheng J, Jokitalo E, Fujimoto T. PML isoform II plays a critical role in nuclear lipid droplet formation. J Cell Biol. 2016;212:29–38.
Hernandez-Corbacho MJ, Obeid LM. A novel role for DGATs in cancer. Adv Biol Regul. 2019;72:89–101.
Chen G, Zhou G, Aras S, He Z, Lucas S, Podgorski I, et al. Loss of ABHD5 promotes the aggressiveness of prostate cancer cells. Sci Rep. 2017;7:13021.
Acknowledgements
We want to thank Yana Filipovich for helping us with the culture of cell lines used in this project and Nicholas Mehta for his help with western blot.
Funding
This work was supported by the US National Institutes of Health/National Cancer Institute (RO1 CA24920), Department of Defense (PC190755) and the Rob Brooks Fund for Personalized Cancer Care.
Author information
Authors and Affiliations
Contributions
OF and SC perform study concept and design. AM, FN performed cell culture and western blot experiments, MG provided acquisition and analysis of LD data, VG performed immunofluorescence experiments. OF and AM performed writing and paper preparation, SC and SH performed evaluation of experiments and review of the paper prior submission. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval and consent to participate
This study did not require ethical approval.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Morales, A., Greenberg, M., Nardi, F. et al. Loss of ephrin B2 receptor (EPHB2) sets lipid rheostat by regulating proteins DGAT1 and ATGL inducing lipid droplet storage in prostate cancer cells. Lab Invest 101, 921–934 (2021). https://doi.org/10.1038/s41374-021-00583-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-021-00583-9
This article is cited by
-
Identifying prognostic markers in spatially heterogeneous breast cancer microenvironment
Journal of Translational Medicine (2023)
-
Distinct features of two lipid droplets types in cell nuclei from patients with liver diseases
Scientific Reports (2023)
-
Lipid droplet biogenesis and functions in health and disease
Nature Reviews Endocrinology (2023)
