
Abstract
Radiomics has potential advantages in the noninvasive histopathological and molecular diagnosis of gliomas. We aimed to develop a novel image signature (IS)-based radiomics model to achieve multilayered preoperative diagnosis and prognostic stratification of gliomas. Herein, we established three separate case cohorts, consisting of 655 glioma patients, and carried out a retrospective study. Image and clinical data of three cohorts were used for training (N = 188), cross-validation (N = 411), and independent testing (N = 56) of the IS model. All tumors were segmented from magnetic resonance (MR) images by the 3D U-net, followed by extraction of high-throughput network features, which were referred to as IS. IS was then used to perform noninvasive histopathological diagnosis and molecular subtyping. Moreover, a new IS-based clustering method was applied for prognostic stratification in IDH-wild-type lower-grade glioma (IDHwt LGG) and triple-negative glioblastoma (1p19q retain/IDH wild-type/TERTp-wild-type GBM). The average accuracies of histological diagnosis and molecular subtyping were 89.8 and 86.1% in the cross-validation cohort, while these numbers reached 83.9 and 80.4% in the independent testing cohort. IS-based clustering method was demonstrated to successfully divide IDHwt LGG into two subgroups with distinct median overall survival time (48.63 vs 38.27 months respectively, P = 0.023), and two subgroups in triple-negative GBM with different median OS time (36.8 vs 18.2 months respectively, P = 0.013). Our findings demonstrate that our novel IS-based radiomics model is an effective tool to achieve noninvasive histo-molecular pathological diagnosis and prognostic stratification of gliomas. This IS model shows potential for future routine use in clinical practice.
Similar content being viewed by others


Introduction
The precise diagnosis of gliomas is essential for personalized healthcare, and will contribute to the improvement of clinical outcomes [1, 2]. Traditionally, the gold standard is diagnosis by pathologists based on representative tumor areas after biopsy or surgical resection [3]. Due to the high inter-observer variability and morphological ambiguity, it is well recognized that histopathology provides inadequate information for precise diagnosis and prognostication [4]. In the latest WHO criteria for brain tumors of the central nervous system, molecular biomarkers were officially incorporated into histopathology for glioma classification [5]. The concept of integrated histological–molecular diagnosis has been widely accepted worldwide and is routinely practiced in several medical centers to overcome the deficiencies of traditional pathological diagnosis [6]. Despite technological advancements, the current diagnostic modality only focuses on a small portion of the tumor, which is not feasible for characterizing the entire tumor due to the high intratumoral cellular and genetic heterogeneity. The potential risk of inconsistencies or discordant diagnoses will always exist if limitations between these focal-panorama connections cannot be resolved [7, 8].
Radiomics aims to mine medical imaging data by extracting a wide range of quantitative features [9, 10]. Based on radiomics methods, WHO grading and several important molecular biomarkers (IDH1/2, 1p19q) in gliomas can be effectively predicted [11, 12]. These previous studies revealed that high-throughput image features contain combined information of both histopathology and genetic alterations. Furthermore, coming from the entire tumor entity, image features are capable of overcoming the limitations of under- and unrepresentative tumor sampling [4].
In this study, we proposed a novel image signature (IS) model to facilitate glioma histological and molecular diagnosis by an automated method. In addition, our IS model can be used to accurately stratify patient survival outcomes and molecular subgroups within specific glioma entities, which cannot be clearly defined according to the current WHO 2016 classification. It is well demonstrated in our three proofs that our IS model shows great potential for the precise diagnosis and prognostication of gliomas.
Materials and methods
Patient cohorts
A total of 655 patients from two hospitals between 2010 and 2017 were enrolled in this study. Overall, 599 patients were diagnosed in Huashan Hospital and the remaining 56 cases were from Shanghai International Medical Center. We conducted the molecular testing of glioma specimen with our research funds, so that the patients did not need to pay to get their molecular testing result. Informed consent for medical data being used for scientific research was achieved from patients. Both Huashan Hospital and Shanghai International Medical Center are affiliated to Fudan University. Our research was approved by the ethics committee in Fudan University, Shanghai, China. The number of the ethic review for our research is HIRB-2010-256-1.
The total of 655 patients were divided into three cohorts. The first cohort, consisting of 188 patients, was used to train deep learning networks for automatic tumor segmentation; hence, it was named deep learning training cohort. In this cohort, glioma cases were included as long as we held their corresponding T1 contrast and T2Flair series of magnetic resonance (MR) images. Once the segmentation network training was complete, the network was used to extract the high-throughput network features of tumors, which we refer to as IS. The second cohort contained 411 patients for the cross-validation of the IS-based histopathological diagnosis and molecular subtyping and was named cross-validation cohort. The third cohort consisted 56 patients that were used for independent testing, and this cohort was therefore named Independent Testing cohort. The first and second cohorts were from Huashan Hospital, and the third cohort was from Shanghai International Medical Center. The inclusion criterion of cross-validation cohort and independent testing cohort was that these cases must contain complete imaging data together with histopathology, molecular biomarkers (IDH1/2, 1p19q, TERTp), and clinical survival follow-up used to investigate the value of the IS model in prognosis stratification. Due to incomplete molecular biomarkers and survival information, the data of the first cohort, which contains MR images and manually labeled tumors, were not suitable for subsequent IS analysis. In our research, two neuropathologists independently reviewed all the histology from the second and third cohorts based on the 2008 WHO criteria. The patient characteristics of deep learning training cohort, cross-validation cohort, and Independent Testing cohort are summarized in Table 1. Preoperative three-dimensional preoperative navigation T1 contrast and T2-FLAIR images were obtained for subsequent research. The imaging parameters, image preprocessing, and molecular testing are described in Appendix Text S1.
Method overview
A deep learning network (3D U-net) was trained to segment the tumors automatically. Once a tumor is accurately segmented, most image features of the tumor will have been stored in the deep learning network. By extracting network features from the 3D U-net, comprehensive image features of the tumor can be obtained. Deep learning training cohort was used to train and validate the 3D U-net segmentation network. After the segmentation network had been built, every case in the cross-validation cohort underwent the following processing steps: tumor segmentation by the established 3D U-net, tumor feature extraction from the 3D U-net, feature dimension reduction and selection, and radiomics model construction. Independent testing cohort was used to further test the radiomics model. The method overview is illustrated in Fig. 1a. Three lines of evidence have been provided to demonstrate the clinical value of the IS model.

a Patients cohorts and flowchart of the method. b Structure of the 3D U-net. c Feature reduction and selection process.
3D U-net training and validation
The structure of the 3D U-net [9] used in our study is shown in Fig. 1b. We randomly divided the deep learning training cohort (188 cases) into a training set (125 cases) and validation set (63 cases) in a 2:1 ratio. The tumors in the training set were manually outlined by two experienced neurosurgeons. The manual segmentation results were used as the ground truth for the 3D U-net training. The MR images of all cases from the deep learning training cohort were skull-stripped and normalized before the training process. In the training process, to obtain the training input of 3D U-net, we randomly extracted 32 × 32 × 32 patches from 125 cases in the training set. We selected 80 patches in the ratio of 10% background, 40% normal brain and 50% tumor from each case, thereby acquiring 10,000 patches, with their corresponding manually segmented labels utilized as the training data for 3D U-net.
In the validation process, we utilized asymmetry detection and a 3D bounding box method proposed in our previous work [13] to obtain the tumor region of interest (ROI). Then, we put the ROI into the trained 3D U-net to obtain element-wise segmentation results.
3D U-net architecture
Our network architecture combines a down-sampling path and an up-sampling path with a concatenation of feature maps at corresponding resolution scales, as shown in Fig. 1b. The path has three blocks and two convolutional layers. Each block includes two consecutive convolutional layers with a filter size of 3 × 3 × 3 and a max pooling layer with a stride of 2 × 2 × 2. By the end of down-sampling path, two consecutive convolutional layers with a filter size of 3 × 3 × 3 are added, with the last layer being the “bottom layer,” where features that are the most representative are integrated. Then, the up-sampling path follows, which also has three blocks. Each of the blocks contains a deconvolution layer with a filter size of 2 × 2 × 2, a concatenation process and two consecutive convolutional layers with a filter size of 3 × 3 × 3. At the end of the network, we implemented a fully connected layer and a softmax function to create an element-wise segmentation structure. In the 3D U-net architecture, we use zero padding in all the convolutional layers to keep the output scales. In addition, the activation function is selected to be rectified linear unit.
In the postprocessing procedure of the segmentation test set, we developed a simple double-scale detection algorithm that combines the tumor bounding details from the 240 × 240 × 32 patch segmentation results and the environmental details from the 320 × 320 × 32 patch segmentation results by calculating their intersection. Then, we restored the segmentation result to its original scale by using the center location information saved previously. Finally, we utilized the largest 3D connection region algorithm to obtain the final automatic glioma segmentation result of each case in the validation set.
Image signature (IS) extraction
Network features were initially extracted from the bottom layer and are highlighted in Fig. 1b. The down-sampling path can also be interpreted as the encoding path, such that the end of the down-sampling path is therefore the best choice for extracting network features. The dimension of the extracted network features (IS) was 40 × 40 × 4 × 256 = 1,638,400.
Details of the diagnostic modeling
Three-step feature selection was used in our histological and molecular subtype prediction. First, Fisher vector clustering was applied to concentrate the large-scale network features; then, a t test with a P threshold value of 0.05 was used to remove statistically insignificant features; finally, sparse representations were used to select the best feature combination for a particular discrimination. To reduce the dimensionality of network features for subsequent analysis, improved Fisher vector encoding [14, 15] was used in our study. The network feature of each tumor was first stretched into a one-dimensional vector, which was then entered into 128 deep filters. For each deep filter, the first- and second-order statistics of Gaussian mixture models with 64 Gaussian components were calculated. The Fisher vector encoding leads to 128 × 64 × 2 = 16,384 features for each tumor, which is described in Appendix Text S2. Then, a t test with a significance threshold set at P = 0.05 was used to select features with statistical significance. After P value selection, the feature dimension was reduced to the range of 1000–3500 for different classification problems. Finally, an improved version of sparse representation [16,17,18] was used to find the optimal feature combination for different classification problems. After a feature combination was selected, sparse representation classification was used to build the radiomics models. The feature selection and classification based on sparse representation is described in Appendix Text S3.
Three radiomics proofs
(1) Noninvasive histopathological diagnosis and molecular subtyping based on IS
IS contains most of the tumor features. However, its direct usage in histological diagnosis and molecular subtyping will lead to overfitting due to its high dimensionality. A three-step feature reduction and selection strategy, as shown in Fig. 1c, was used to establish the diagnostic model. The details of the diagnostic modeling are provided in the previous section. A two-layer binary classification model was used to perform histopathological diagnosis. A three-layer binary classification model was used to determine molecular subtypes. The two binary classification trees are illustrated in Fig. 2.

a Binary classification for histologic type prediction, b classification performances of two binary classifiers, c binary classification for molecular group prediction, d classification performances of four binary classifiers.
(2) Stratification of patient survival outcomes in IDH1/2 wild-type lower-grade glioma (LGG, WHO grade II and III)
In our previous publication, we defined molecular higher-grade glioma, which has poor survival outcomes, such as IDH-wild-type glioblastoma (IDHwt GBM), and molecular lower-grade glioma, which has a good prognosis (GP), such as IDH-mutant LGG (IDHmut LGG) [19]. Thus, we used our IS model to perform prognostic stratification of IDH-wild-type LGG by using IDHwt GBM and IDHmut LGG as two reference groups.
First, the IS clustering centers of the two reference groups were calculated, and then each case in the IDH-wild-type LGG group was divided according to the L1-norm into one of the two cluster centers. In other words, if the IS of a case is closer to the cluster center of IDHmut LGG, it was regarded as clinically prone to IDHmut LGG, otherwise it was labeled as IDHwt GBM. Finally, the prognosis of IDH-wild-type LGG patients was analyzed according to the IS classification. This process is illustrated in Fig. 3a.

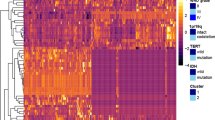
a IS clustering process. b Prognosis stratification based on IS clustering. c Heatmap of IS clustering.
(3) Stratification of patient survival outcomes in triple-negative glioblastoma (GBM, WHO grade IV)
Triple-negative (IDH-wild-type/TERTp-wild-type/1p19q retain) glioblastoma, accounting for ~20% of IDH-wild-type GBM, represents explicit genetic alterations and a relevant median overall survival (mOS) time [20, 21]. We used our IS model to test whether the prognosis of patients with triple-negative glioblastoma could be stratified. Therefore, IDH-wild-type anaplastic astrocytoma (IDHwt AA) and IDH-wild-type/TERTp mutation glioblastoma (IDHwt/TERTpmut GBM) were adopted as two reference groups (IDHwt AA for GP, IDHwt/TERTpmut GBM for poor prognosis). Triple-negative GBM cases were reclassified according to the L1-norm between itself and the IDHwt AA or IDHwt/TERTpmut GBM cluster centers. This process is illustrated in Fig. 4a.

a IS clustering process. b Prognosis stratification based on IS clustering. c Heatmap of IS clustering.
Statistical analysis
For the noninvasive diagnosis of histopathology and molecular subtypes, fivefold cross-validation and independent testing were used. The performance of noninvasive diagnosis was validated by the following quantitative indexes: accuracy, Sensitivity, Specificity, Positive-Predictive Value (PPV), Negative-Predictive Value, and Matthew’s Correlation Coefficient, which are described in Appendix Text S4.
The chi-squared test was used to determine statistical difference of clinical characteristics between the three cohorts. Univariate and multivariate Cox proportional hazards models were applied to evaluate associations between overall survival outcomes and characteristics. Kaplan–Meier curves and the log-rank test were used to estimate the mOS for different histopathological subgroups, molecular subgroups, and IS-based stratified groups. MATLAB 2017 (MathWorks, USA), IBM SPSS statistics 20.0 software (SPSS, Chicago, IL, USA), and R 3.5.1 were used to perform the statistical analysis.
Results
Image segmentation
The image segmentation of the deep learning network (3D U-net) was evaluated by several quantitative indexes, including Dice’s coefficient, PPV and SEN. Definitions of the evaluation indexes are provided in Appendix Text S5. An example of glioma segmentation is illustrated in Fig. S1. The segmentation results are summarized in Table S1.
Noninvasive histopathological diagnosis and molecular subtyping based on IS
The average accuracy values of histological diagnosis and molecular subtyping were 89.8% and 86.1%, respectively, in the cross-validation cohort, while these numbers reached 83.9% and 80.4% in the independent testing cohort. The classification accuracy of each separate binary classifier exceeded 89.2% in the cross-validation cohort. Figure 2 shows all the specific classification results in all tasks. The accuracies of the noninvasive histopathological diagnosis of oligodendroglioma, astrocytoma and glioblastoma are summarized in Fig. 2a, and the classification performance outcomes of the two binary classifiers are shown in Fig. 2b. Meanwhile, molecular subtyping was achieved by assessing the status of IDH1/2 and 1p19q according to the genetic parameters of the 2016 WHO criteria: LGG with IDH1/2 mutant and 1p19q codeletion, LGG with IDH1/2 mutant and 1p19q retained, LGG with IDH1/2 wild type, GBM with IDH1/2 mutant, and GBM with IDH1/2 wild type. The binary classification in molecular subtyping is summarized in Fig. 2c, and the classification performance outcomes of the four binary classifiers are shown in Fig. 2d.
Prognostic stratification of IDH1/2 wild-type LGG based on IS
Several high-profile publications, including our previous paper, identified the genetic heterogeneity of IDH1/2 wild-type LGG with relevant prognosis [19, 22, 23]. EGFR amplification, whole +7/−10 and TERTp mutation serve as hallmarks for IDH1/2 wild-type LGG prognostic stratification. The prognosis of patients harboring EGFR amplification, whole +7/−10 or TERTp mutation is similar to that of patients with IDH-wild type GBM; none of the above three genetic alterations indicate a GP, similar to IDHmut LGG [24]. In our study cohort, the mOS times of IDHmut LGG and IDHwt GBM were 63.87 and 18.50 months, respectively. By using IS clustering analysis in line with two reference groups, all IDH1/2 wild-type LGG cases could be separated into two groups called Group A and Group B, shown as GP group and bad prognosis (BP) group in Fig. 3b. Group A patients had better survival time than Group B patients (48.63 vs 38.27 months, P = 0.023). The imaging features presented obvious differences between Group A and Group B in the clustering heatmap shown in Fig. 3c. Meanwhile, continuous age was used to calculated the age-adjusted survival curves. The result, as shown in Fig. S2, showed that IS significantly associated with overall survival in IDH wild-type LGG. The clinical differences between the two groups are statistically insignificant, as shown in Table 2.
Prognostic stratification of triple-negative GBM based on IS
As the other two reference groups, the mOS times for IDHwt AA and IDHwt/TERTpmut GBM were 33.3 and 17.6 months, respectively. By utilizing the same IS clustering method, triple-negative GBM cases could be stratified into Group C and Group D, shown as GP group and BP group in Fig. 4b. Group C patients had longer mOS times than Group D patients (36.8 vs 18.2 months, P = 0.013). In the multivariate analysis, IS was identified as an independent prognostic factor, as shown in Tables 3 and 4. The IS analysis process of the triple-negative GBM is illustrated in Fig. 4.
Discussion
Preoperative diagnosis and potential risk evaluation have become increasingly important in individualized glioma patient healthcare [1, 2]. In the present study, we developed a novel radiomics model named IS to perform noninvasive histopathological diagnosis and molecular subtyping of glioma and to further explore the heterogeneity of the clinical course. In existing radiomics methods, image features are extracted from MR images based on explicit experiences in image processing, image pattern characteristics, and signal processing. Such methods can hardly guarantee the completeness of the extraction of information from MR images. In the current literature, deep learning-based radiomics outperformed normal radiomics in the prediction of IDH1 using T2-FLAIR MR images in Li’s method [25], suggesting that deep learning-based radiomics is better capable of extracting complete information and features from MR images. Our IS model also utilized a deep learning network to extract image features. Different from existing radiomics methods that explicitly extract image features from MR images, the image features in our research were derived from the image segmentation deep learning network. When the network can accurately perform glioma segmentation, the completeness of the extracted features is assured.
Intratumoral heterogeneity has always posed challenges to prognostication and treatment for glioma patients [8, 26]. We applied IS to designate an accurate and reproducible image clustering strategy to stratify glioma patients into different subtypes with distinct survival times that are not clearly defined in the current diagnostic scheme. To validate the efficacy of this strategy, we selected IDH-wild-type LGG and triple-negative GBM as two representatives because the clinical definition of these two glioma phenotypes is still unclear in the current criteria [5]. IDH-wild-type LGG can be referred to as “diffuse astrocytic glioma, IDH-wild-type, with molecular features of glioblastoma, WHO IV” if the tumor harbors either EGFR amplification, whole +7/−10 or TERTp mutation, with an aggressive clinical course corresponding to IDHwt/TERTpmut GBM [24]. The absence of these three molecular biomarkers indicates that the survival outcomes of IDH-wild-type LGG will be similar to those of IDHmut LGG [27]. This was the theoretical basis on which we developed our strategy of IS clustering reference groups for IDH-wild-type LGG.
Besides, we observed one interesting situation by using our IS stratification system. In Group A, there was a trend toward two potential subgroups based on IS clustering. We speculated if there still existed a specific glioma subtype within IDH wild-type gliomas. At first step, we worked on survival analysis of these two potential IS subgroups (called Group A-1 and Group A-2). It was indicated that Group A-2 patients had better overall survival outcome than that of Group A-1 patient, as shown in Fig. 5. However, there was no statistical significance of such survival diversity. Furthermore, we performed multivariate statistical analysis for Group A-1 and A-2 cases, more A-2 tumors were found located in frontal lobe compared to A-1 tumors, as shown in Table 5. In many previous research reports, tumor location was one of important factors determining patients outcome [28, 29], for example, frontal lobe tumors tend to be good clinical performance, whilst midline tumors being dismal outcome. Such finding may explain why A-2 patients had optimal survival time than A-1 patients did. To our acknowledge, IDH-mutant gliomas preferred to be located in frontal lobe, so we hypothesized that A-2 tumors may have more familiar genetic background with IDH-mutant lower-grade gliomas, and our IS clustering method was capable of revealing this kind of genetic features.

Group A-1 is shown in dotted line and A-2 in solid line.
Triple-negative gliomas are enigmatic tumors with highly heterogeneous backgrounds [30]. Cahill et al. reported the enrichment of BAF complex alterations and activating mutations in genes within the PI3K pathway in IDHwt/TERTpwt GBM. The prognosis for this kind of tumor is better than that of TERTpmut GBM but worse than that of IDHmut GBM [21]. Ichimura et al. also reported the same result and suggested the prognostic value of TERTp mutation, which was impacted by various factors, such as MGMT methylation status [31]. This result demonstrated survival stratification. Hai Yan et al. reported the genomic landscape of triple-negative GBM with two distinct subtypes, namely, IDHwt-ALT and IDHwt-TERTsv [20]. The IDHwt-ALT subtype showed frequent mutations in ATRX and SMARCAL1, similar to TERTpmut GBM in overall survival time. The IDHwt-TERTsv subtype is characterized as activating upstream rearrangement of TERTp with better prognosis than the ALT subtype but inferior prognosis to IDHmut GBM. Therefore, we chose TERTpmut GBM as one of the reference groups for triple-negative GBM. On the other hand, we chose IDHwt AA rather than IDH-mutant GBM as another reference group because IDH-mutant GBM shared a totally different pathogenesis with IDH-wild-type astrocytic glioma. Several articles demonstrated that IDHwt AA is the early developing stage for diffuse glioma evolving into IDH-wild-type GBM, presenting with worse OS than IDH-mutant GBM [22, 32]. This was another theoretical basis for our IS clustering strategy in triple-negative GBM.
There were several limitations in this study. First, this was a retrospective study from only two neurosurgical institutions. Second, we did not perform comprehensive genomic screening of these two representative glioma phenotypes. However, this study provides a novel radiomics-based IS model and clustering strategy for precise noninvasive diagnosis and prognostication and is more adherent to real clinical situations with great potential for future implementation in routine clinical practice.
References
Reifenberger G, Wirsching HG, Knobbe-Thomsen CB, Weller M. Advances in the molecular genetics of gliomas—implications for classification and therapy. Nat Rev Clin Oncol. 2017;14:434–52.
Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, et al. European Association for Neuro-Oncology (EANO) Task Force on Gliomas. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18:315–29.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Cavenee WK, Burger PC, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109.
Quick-Weller J, Tichy J, Harter PN, Tritt S, Baumgarten P, Bähr O, et al. “Two is not enough”—impact of the number of tissue samples obtained from stereotactic brain biopsies in suspected glioblastoma. J Clin Neurosci. 2018;47:311–4.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20.
Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018;136:153–66.
Diamandis P, Aldape KD. Insights from molecular profiling of adult glioma. J Clin Oncol. 2017;35:2386–93.
GLASS Consortium. Glioma through the looking GLASS: molecular evolution of diffuse gliomas and the Glioma Longitudinal Analysis Consortium. Neuro Oncol. 2018;20:873–84.
Kumar V, Gu Y, Basu S, Berglund A, Eschrich SA, Schabath MB, et al. Radiomics: the process and the challenges. Magn Reson Imaging. 2012;30:1234–48.
Aerts HJ, Velazquez ER, Leijenaar RT, Parmar C, Grossmann P, Carvalho S, et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat Commun. 2014;5:4006.
Yu J, Shi Z, Lian Y, Li Z, Liu T, Gao Y, et al. Noninvasive IDH1 mutation estimation based on a quantitative radiomics approach for grade II glioma. Eur Radiol. 2017;27:3509–22.
Jiang C, Kong Z, Liu S, Feng S, Zhang Y, Zhu R, et al. Fusion radiomics features from conventional MRI predict MGMT promoter methylation status in lower grade gliomas. Eur J Radiol. 2019;121:108714.
Ji CH, Yu JH, Wang YY, Chen L, Shi ZF, Mao Y. Brain tumor segmentation in MR slices using improved GrowCut algorithm. In: Seventh International Conference on Graphic and Image Processing (ICGIP 2015) International Society for Optics and Photonics. 2015. https://doi.org/10.1117/12.2228230.
Perronnin F, Mensink T. Improving the fisher kernel for large-scale image classification. In: Proceedings of the 11th European Conference on Computer Vision. 2010. https://doi.org/10.1007/978-3-642-15561-1_11.
Zhang X, Xiong H, Zhou W, Lin W, Tian Q, Picking deep filter responses for fine-grained image recognition. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. 2016. https://doi.org/10.1109/CVPR.2016.128.
Wu G, Chen Y, Wang Y, Yu J, Lv X, Ju X, et al. Sparse representation-based radiomics for the diagnosis of brain tumors. IEEE Trans Med Imaging. 2016;37:893–905.
Wu G, Wang Y, Yu J. Overall survival time prediction for high grade gliomas based on sparse representation framework. In: International MICCAI brainlesion workshop. Cham: Springer; 2017. pp. 77–87.
Zhu X, Suk H, Lee S, Shen D. Subspace regularized sparse multi-task learning for multi-class neurodegenerative disease identification. IEEE Trans Biomed Eng. 2016;63:607–18.
Aibaidula A, Chan AK, Shi Z, Li Y, Zhang R, Yang R, et al. Adult IDH wild-type lower-grade gliomas should be further stratified. Neuro Oncol. 2017;19:1327–37.
Diplas BH, He X, Brosnan-Cashman JA, Liu H, Chen LH, Wang Z, et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat Commun. 2018;9:2087.
Williams EA, Miller JJ, Tummala SS, Penson T, Iafrate AJ, Juratli TA, et al. TERT promoter wild-type glioblastomas show distinct clinical features and frequent PI3K pathway mutations. Acta Neuropathol Commun. 2018;6:106.
Kuwahara K, Ohba S, Nakae S, Hattori N, Pareira ES, Yamada S, et al. Clinical, histopathological, and molecular analyses of IDH-wild-type WHO grade II-III gliomas to establish genetic predictors of poor prognosis. Brain Tumor Pathol. 2019;36:135–43.
Labussière M, Boisselier B, Mokhtari K, Di Stefano AL, Rahimian A, Rossetto M, et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology. 2014;83:1200–6.
Brat DJ, Aldape K, Colman H, Holland EC, Louis DN, Jenkins RB, et al. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018;136:805–10.
Li ZJ, Wang YY, Yu JH, Guo Y, Cao W. Deep Learning based Radiomics (DLR) and its usage in noninvasive IDH1 prediction for low grade glioma. Sci Rep. 2017;7:5467.
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean C, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–93.
Tesileanu CMS, Dirven L, Wijnenga MMJ, Koekkoek JAF, Vincent AJPE, Dubbink HJ, et al. Survival of diffuse astrocytic glioma, IDH1/2-wildtype, with molecular features of glioblastoma, WHO grade IV: a confirmation of the cIMPACT-NOW criteria. Neuro Oncol. 2020;22:515–23.
Tang Q, Lian Y, Yu J, Wang Y, Shi Z, Chen L. Anatomic mapping of molecular subtypes in diffuse glioma. BMC Neurol. 2017;17:183.
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
Chan AK, Mao Y, Ng HK. TP53 and histone H3.3 mutations in triple-negative lower-grade gliomas. N Engl J Med. 2016;375:2206–8.
Arita H, Yamasaki K, Matsushita Y, Nakamura T, Shimokawa A, Takami H, et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol Commun. 2016;4:79.
Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 2010;120:707–18.
Acknowledgements
The authors would like to thank Xinhua Ding from the Department of Neurosurgery, Shanghai International Medical Center and Haixia Chen from the Department of Pathology, Shanghai International Medical Center for providing information on several glioma cases, their corresponding MR images and molecular data in this research.
Funding
The study was supported by the National Natural Science Foundation of China (91959127 and 81702471), National Natural Science Foundation of China (2015CB755500), Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01), and ZJLab, Shanghai Municipal Health Commission Project (2018ZHYL0107).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Luo, H., Zhuang, Q., Wang, Y. et al. A novel image signature-based radiomics method to achieve precise diagnosis and prognostic stratification of gliomas. Lab Invest 101, 450–462 (2021). https://doi.org/10.1038/s41374-020-0472-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-020-0472-x
This article is cited by
-
Multi-parametric MRI-based machine learning model for prediction of WHO grading in patients with meningiomas
European Radiology (2023)
-
Application of radiomics in precision prediction of diagnosis and treatment of gastric cancer
Japanese Journal of Radiology (2022)
