
Abstract
Obesity-associated inflammation in white adipose tissue (WAT) is a causal factor of systemic insulin resistance; however, precisely how immune cells regulate WAT inflammation in relation to systemic insulin resistance remains to be elucidated. The present study examined a role for 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) in hematopoietic cells in regulating WAT inflammation and systemic insulin sensitivity. Male C57BL/6J mice were fed a high-fat diet (HFD) or low-fat diet (LFD) for 12 weeks and examined for WAT inducible 6-phosphofructo-2-kinase (iPFK2) content, while additional HFD-fed mice were treated with rosiglitazone and examined for PFKFB3 mRNAs in WAT stromal vascular cells (SVC). Also, chimeric mice in which PFKFB3 was disrupted only in hematopoietic cells and control chimeric mice were also fed an HFD and examined for HFD-induced WAT inflammation and systemic insulin resistance. In vitro, adipocytes were co-cultured with bone marrow-derived macrophages and examined for adipocyte proinflammatory responses and insulin signaling. Compared with their respective levels in controls, WAT iPFK2 amount in HFD-fed mice and WAT SVC PFKFB3 mRNAs in rosiglitazone-treated mice were significantly increased. When the inflammatory responses were analyzed, peritoneal macrophages from PFKFB3-disrputed mice revealed increased proinflammatory activation and decreased anti-inflammatory activation compared with control macrophages. At the whole animal level, hematopoietic cell-specific PFKFB3 disruption enhanced the effects of HFD feeding on promoting WAT inflammation, impairing WAT insulin signaling, and increasing systemic insulin resistance. In vitro, adipocytes co-cultured with PFKFB3-disrupted macrophages revealed increased proinflammatory responses and decreased insulin signaling compared with adipocytes co-cultured with control macrophages. These results suggest that PFKFB3 disruption in hematopoietic cells only exacerbates HFD-induced WAT inflammation and systemic insulin resistance.
Similar content being viewed by others


Introduction
Immune cells regulate the pathogenesis of obesity-associated inflammation in white adipose tissue (WAT), thereby contributing to systemic insulin resistance. As supporting evidence, WAT in mice with diet-induced obesity (DIO) reveals increased macrophage infiltration. The latter is commonly associated with increased systemic insulin resistance [1,2,3]. Moreover, a number of studies in mouse models involving myeloid cell-specific disruption or overexpression of gene(s) in inflammatory signaling and/or metabolic pathways have demonstrated that myeloid cells, in particular macrophages, regulate systemic insulin sensitivity [4,5,6]. This regulation is attributable to, in large part, altering macrophage proinflammatory activation and related paracrine effects of macrophage-derived factors on insulin sensitivity and metabolic responses of key metabolic cells including adipocytes [4,5,6]. Given this, elucidating the mechanisms underlying the activation of immune cells is critical to a better understanding of how inflammation regulates the development and progression of obesity-related metabolic diseases.
Within macrophages, peroxisome proliferator-activated receptor gamma and delta are key transcription factors that stimulate macrophage anti-inflammatory activation [7,8,9]. This is significant because myeloid cell-specific disruption of PPARγ and/or PPARδ increases proinflammatory activation of adipose tissue macrophages and exacerbates obesity-associated insulin resistance [7, 10]. In contrast, the effect of PPARγ activation on reversing high-fat diet (HFD)-induced insulin resistance is mediated, at least in part, by stimulation of anti-inflammatory activation of macrophages in adipose tissue [11]. Macrophage activation status is also regulated by Toll-like receptor 4, c-Jun N-terminal kinase (JNK), and stimulator of interferon genes, such that their myeloid cell-specific disruption protects mice from diet-induced WAT and systemic insulin resistance [5, 12,13,14]. Taken together, these findings demonstrate how the inflammatory status of macrophages governs the outcome of WAT inflammation and systemic insulin sensitivity. However, it remains to be elucidated exactly how nutritional stress induces proinflammatory activation of macrophages, particularly in those found in adipose tissue where increased inflammation plays a causal role in the pathogenesis of insulin resistance and hyperglycemia [1, 10, 15,16,17].
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) is one of the four mammalian PFKFB genes that encode for different isoforms of 6-phosphofructo-2-kinase (PFK2) [18, 19]. As a gene expressed at high abundance in macrophages, PFKFB3 encodes for the inducible isoform of PFK2 (iPFK2). In terms of regulating macrophage activation, PFKFB3 expression is stimulated by lipopolysaccharide (LPS) and associated with macrophage activation status [6, 20]. However, LPS-stimulated PFKFB3 expression appears to be a defensive response that acts to prevent excessive macrophage proinflammatory responses; given that PFKFB3 disruption exacerbates macrophage activation [6]. In contrast, PFKFB3 is needed for indole, a microbiota metabolite, to suppress macrophage proinflammatory activation [6]. In addition, PFKFB3 is also stimulated by activation of adenosine 2A receptor [21], whose activation or disruption respectively suppresses or enhances macrophage proinflammatory activation [4, 22]. In the present study, we examined the expression of PFKFB3/iPFK2 in WAT and in WAT stromal vascular cells (SVC) and related it to WAT inflammation and systemic insulin resistance. We also examined the effect of PFKFB3 disruption in hematopoietic cells on altering diet-induced WAT inflammation and systemic insulin resistance.
Materials and methods
Animal experiments
C57BL/6J mice were originally obtained from Jackson Laboratory (Bar Harbor, ME). Global heterozygous PFKFB3-disrupted (PFKFB3+/−) mice (homozygous PFKFB3 disruption is embryonic lethal) and their wild-type (WT) littermates, as well as myeloid cell-specific PFKFB3-disrupted (Mye-PFKFB3−/−) mice and control (Mye-PFKFB3+/+) mice, were generated and maintained, as described [1, 6, 23]. In brief, mice containing two floxed-PFKFB3 alleles were crossed with LysMCre mice in which a nuclear-localized Cre recombinase was inserted into the first coding ATG of the lysozyme 2 gene to generate LysMCre-PFKFB3F/+ or Mye-PFKFB3+/− mice. The latter were further bred to generate homozygous myeloid cell-specific PFKFB3-disrupted (Mye-PFKFB3−/−) mice and control (Mye-PFKFB3+/+) mice. The resultant mice were used for isolation of bone marrow cells, which were then differentiated into macrophages and used for single cell RNA sequencing analysis described below. All mice were maintained on a 12:12 h light–dark cycle (lights on at 06:00), and fed ad libitum except those that were used for dietary feeding studies. Study 1 (PFKFB3/iPFK2 relevance to WAT inflammation): Male C57BL/6J mice, at 5–6 weeks of age, were fed an HFD (60% fat calories, 20% carbohydrate calories, and 20% protein calories; Research Diets, Inc.) or low-fat diet (LFD, 10% fat calories, 70% carbohydrate calories, and 20% protein calories) for 12 weeks as previously described [1, 24, 25]. Some HFD-fed mice were also treated with rosiglitazone (10 mg/kg body weight per day, orally) for the last 4 weeks. Study 2 (Hematopoietic cell-specific PFKFB3 disruption on WAT inflammation): Chimeric mice in which PFKFB3 was disrupted only in hematopoietic cells were generated using bone marrow transplantation (BMT). In brief, male WT mice, at 5–6 weeks of age, were lethally irradiated and transplanted with bone marrow cells isolated from PFKFB3+/− mice and/or WT mice. After recovery for 4 weeks post-BMT, chimeric mice were fed an HFD for 12 weeks as described in Study 1. After the feeding/treatment period, mice in both studies were fasted for 4 h prior to collection of blood and tissue samples [5, 25]. Epididymal, mesenteric, and perinephric fat depots were dissected and weighted as visceral fat content [26]. After weighing, part of the WAT was either fixed and embedded for histological and immunohistochemical analyses or frozen in liquid nitrogen and then stored at −80 °C for further analyses. Some male mice were fed ad libitum and subjected to isolation of peritoneal macrophages or bone marrow cells as described below. All study protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Texas A&M University.
Isolation of adipose tissue stromal vascular cells
Adipose tissue SVCs were isolated using the collagenase digestion method as described [27]. After digestion and centrifugation, the pelleted cells were collected as SVCs.
Isolation of peritoneal macrophages or bone marrow cells
Both peritoneal macrophages and bone marrow cells were isolated from male PFKFB3+/− mice and its WT littermates, at 8–10 weeks of age as previously described [2, 5, 28]. After erythrocyte lysis with ammonium chloride (StemCell Technologies, Vancouver, Canada), cells were subjected to treatment with LPS, IL-4, or pioglitazone followed by inflammatory assays described below or used for BMT. Some bone marrow cells were induced for differentiation with Iscove’s modified Dulbecco’s medium containing 10% fetal bovine serum and 15% L929 culture supernatant for 8 days. The bone marrow-derived macrophages (BMDMs) were used for the co-culture study.
Single cell RNA sequencing analysis
BMDMs also were prepared from Mye-PFKFB3−/− and Mye-PFKFB3+/+ mice as described [6]. Prior to harvest, BMDMs were treated with LPS (20 ng/mL) for 6 h and further prepared using a 10× Genomics Chromium Controller with v3 Chemistry. The libraries were sequenced using the DNBseq NGS services provided by BGI Americas Corporation (Cambridge, MA). The scRNAseq data were then subjected to bioinformatic analysis of transcriptome: the differential expression analysis was conducted using MAST [29], and the functional enrichment analysis was performed using GSEA [30].
Bone marrow transplantation
BMT was performed as previously described [2, 5]. At 5–6 weeks of age, male WT recipient mice were lethally irradiated and transplanted with bone marrow cells from PFKFB3+/− mice and/or WT littermates. PFKFB3+/− → WT mice, in which PFKFB3 was disrupted only in hematopoietic cells, and WT → WT mice, in which PFKFB3 was intact in all cells, were allowed to recover for 4 weeks. After recovery, the chimeric mice were fed an HFD for 12 weeks and subjected to metabolic assays and tissue collections.
Glucose and insulin tolerance tests
Glucose and insulin tolerance tests were performed as previously described [2]. Mice were fasted for 4 h and intraperitoneally injected with D-glucose (2 g/kg of body weight) or insulin (1 unit/kg of body weight). For glucose tolerance tests, blood samples (5 μL) were collected from the tail vein before and at 30, 60, 90, and 120 min after the glucose bolus injection. Similarly, for insulin tolerance tests, blood samples were collected from the tail vein before and at 15, 30, 45, 60 min after the bolus insulin injection.
Measurement of plasma glucose and insulin
The levels of plasma glucose were measured using glucose assay kit (Sigma and BioVision, Mountain View, CA). The levels of plasma insulin were measured using ELISA kits (Crystal Chem Inc., Downers Grove, IL).
Adipose tissue insulin signaling
After the feeding period, randomly selected mice from Study 2 were fasted for 4 h prior to a bolus injection of insulin (1 unit/kg of body weight) into the portal vein for 5 min. Adipose tissue lysates were examined for the total amount and phosphorylation states of Akt1/2 using Western blot analysis.
Histological and immunohistochemical analyses
Paraffin-embedded mouse WAT blocks were cut into sections of 5 µm thickness and stained with hematoxylin and eosin (H&E) and/or stained for F4/80 expression with rabbit anti-F4/80 antibodies (1:100) (AbD Serotec, Raleigh, NC) [4, 6].
Cell culture and treatment
To analyze macrophage proinflammatory activation status, some peritoneal macrophages were treated with or without LPS (20 ng/mL) for 6 h and analyzed for the expression of TNFα and IL-6. To analyze macrophage anti-inflammatory activation, some peritoneal macrophages were incubated with or without interleukin 4 (IL-4, 10 ng/mL) in the presence or absence of pioglitazone (1 µmol/L) for 48 h and examined for the expression of IL-6 and arginase 1. Additional BMDMs were subjected to adipocyte-macrophage cocultures as described below.
BMDMs prepared from PFKFB3+/− or WT mice were subjected to cocultures with differentiated 3T3-L1 adipocytes. After differentiation for 8 days, adipocytes were co-cultured with BMDMs at a ratio of 10:1 based on the published method [8, 31]. To determine changes in inflammatory signaling, some cells were treated with or without LPS (100 ng/ml) for 30 min before cell harvest. To examine changes in insulin signaling, some cells were treated with or without insulin (100 nmol/L) for 30 min before harvest. Cell lysates were measured for proinflammatory and insulin signaling using Western blot analysis.
Biochemical and molecular assays
The amount of iPFK2 in WAT and the mRNA levels of PFKFB3 in WAT SVC were determined using Western blot analysis and real-time PCR, respectively, as previously described [6, 17]. To determine WAT proinflammatory signaling, lysates of frozen WAT were subjected to Western blot analysis to measure total amount and/or phosphorylation states of NFκB p65 as described [4, 32]. Similarly, to analyze WAT insulin signaling, WAT lysates were examined for the total amount and phosphorylation states of Akt using Western blot analysis [4, 32]. All primary antibodies were from Cell Signaling Technology (Danvers, MA, USA). The maximum intensity of each band was quantified using ImageJ software. To determine gene expression, the total RNA was isolated from cultured/isolated cells and WAT of chimeric mice and subjected to reverse transcription and real-time PCR analysis. Results were normalized to 18 s ribosomal RNA and plotted as relative expression to the average of WT, PBS-treated WT, or WT → WT, which was set as 1.
Statistical methods
Numeric data are presented as means ± standard error (SEM). Statistical significance was determined using unpaired, two-tailed ANOVA or Student’s t tests. Differences were considered significant at the two-tailed P < 0.05.
Results
PFKFB3/iPFK2 expression increases in WAT of DIO mice and in stromal vascular cells of rosiglitazone-treated DIO mice
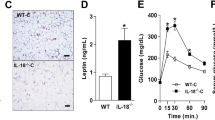
We have previously shown that PFKFB3/iPFK2 plays a critical role in regulating HFD-induced WAT inflammation in mice [1]. However, the extent to which PFKFB3/iPFK2 expression is altered in WAT, in particular immune cells within WAT, in relation to systemic insulin sensitivity remains to be examined. Initially, we examined the amount of iPFK2 in WAT of C57BL/6J mice with DIO. Compared with LFD-fed mice, HFD-fed mice displayed overt obesity (data not shown), which was accompanied with systemic insulin resistance and glucose intolerance (Fig. 1A). Consistently, the area under curve (AUC) calculated from the data of glucose or insulin tolerance tests in HFD-fed mice was significantly greater than that in LFD-fed mice (Supplementary Fig. S1). Meanwhile, HFD-fed mice revealed significant increases in accumulation of F4/80-positive cells in WAT sections and in the phosphorylation states of JNK p46 and NFκB p65 in WAT lysates compared with LFD-fed mice (Fig. 1B, C), indicating increased severity of WAT inflammation. In addition, the amount of iPFK2 in WAT from HFD-fed mice was significantly increased compared with that in WAT from LFD-fed mice. To confirm the relevance of PFKFB3/iPFK2 to WAT inflammation and systemic insulin sensitivity, we examined PFKFB3 mRNAs in SVC of WAT from HFD-fed mice upon treatment with rosiglitazone which is shown to decrease WAT inflammation and improve systemic insulin sensitivity. Compared with those in HFD-fed and control-treated mice, PFKFB3 mRNAs in WAT SVC from HFD-fed and rosiglitazone-treated mice were significantly increased, which was accompanied with a significant decrease in IL-6 mRNAs (Fig. 1D). Together, these results suggest that increased PFKFB3 mRNAs in WAT SVC are associated with decreased WAT inflammation in response to rosiglitazone treatment while iPFK2 amount in WAT is increased and accompanied with increased WAT inflammation.

Male C57BL/6J mice, at 5–6 weeks of age, were fed a high-fat diet (HFD, 60% of fat calories) or low-fat diet (LFD, 10% fat calories) for 12 weeks. Some HFD-fed mice were also treated with rosiglitazone (Rosi, 10 mg/kg/d) or control (phosphate-buffered saline (PBS)) for the last 4 weeks of HFD feeding period. A Glucose tolerance and insulin tolerance tests. After the feeding period, HFD- or LFD-fed mice were fasted for 4 h and subjected to a bolus peritoneal injection of glucose (2 g/kg) or (1 U/kg). B WAT histology. WAT sections were stained with H&E or for F4/80. C Adipose tissue iPFK2 amount and proinflammatory signaling. After harvest, lysates of white adipose tissue (WAT) were measured for iPFK2 amount, as well as p46 and p65 amount and phosphorylation states using Western blot analysis. D The expression of PFKFB3 and IL-6 in WAT stromal vascular cells (SVC). After the feeding/treatment period, WAT of HFD-fed and rosiglitazone- or control-treated mice were digested for collection of SVC. The latter were subjected to measurement of PFKFB3 and IL-6 expression using real-time RT-PCR. For A, C, and D, numeric data are means ± SEM. n = 7–10. *, P < 0.05 and **, P < 0.01 HFD vs. LFD in (A) for the same point and in (C) for the same protein; †, P < 0.05 and ††, P < 0.01 Rosi vs. PBS for the same gene in (D).
PFKFB3 disruption enhances macrophage proinflammatory activation and decreases macrophage anti-inflammatory activation
In previous studies involving BMDMs, PFKFB3 is shown to critically regulate macrophage activation status [6]. A similar finding was observed in the present study using peritoneal macrophages isolated from PFKFB3+/− mice and WT littermates (Fig. 2A, B). Specifically, the mRNA levels of TNFα and/or IL-6 in peritoneal macrophages from PFKFB3+/− mice were significantly higher than their respective levels in peritoneal macrophages from WT mice under either basal or LPS-stimulated condition (Fig. 2C). In addition, under both basal and anti-inflammatory conditions where IL-4, pioglitazone, or both was added, the mRNA levels of IL-6 in PFKFB3+/− macrophages were significantly higher than those in control macrophages (Fig. 2D). In contrast, the mRNA levels of arginase 1 in PFKFB3+/− macrophages were significantly lower than those in control cells under conditions where IL-4, pioglitazone, or both was added (Fig. 2D). Together, these results verify that PFKFB3 disruption increases macrophage proinflammatory activation and decreases macrophage anti-inflammatory activation.

A, B Validation of macrophage PFKFB3 disruption. C, D Macrophage activation. For A–D, peritoneal macrophages were isolated from LFD-fed male PFKFB3+/– mice and wild-type (WT) littermates and measured for the expression of PFKFB3 (A and B) and proinflammatory cytokines (TNFα and IL-6) and/or arginase 1 (C and D) using real-time RT-PCR. For A, representative PCR production of PFKFB3. For B, quantification of PFKFB3 mRNAs. For C, macrophage proinflammatory activation. Prior to harvest, macrophages were treated with or without lipopolysaccharide (LPS, 20 ng/mL) for the last 6 h. For D, macrophage anti-inflammatory activation. Prior to harvest, macrophages were treated with or without interleukin 4 (IL-4, 10 ng/mL) in the presence or absence of pioglitazone (1 µM) for 48 h. For B–D, data are means ± SEM. n = 4–6. *, P < 0.05 and **, P < 0.01 PFKFB3+/- vs. WT in (B), under the same condition in (C), and with these same treatment (IL-4 or IL-4 + Pio) in (D); †, P < 0.05 and ††, P < 0.01 LPS vs. PBS within the same genotype in (C) or treatment with IL-4 + Pio vs. Pio alone for the same genotype (in D).
PFKFB3 disruption increased macrophage expression of genes related to oxidative metabolism and proinflammatory responses
We showed previously that BMDMs from Mye-PFKFB3−/− mice revealed increased proinflammatory responses compared with BMDMs from Mye-PFKFB3+/+ mice [6]. Using the same types of cells from Mye-PFKFB3−/− and Mye-PFKFB3+/+ mice (Supplementary Fig. S2), in the present study we performed an scRNAseq analysis for macrophages to gain insight to how PFKFB3 might regulate macrophage activation. In BMDM samples, global expression profiles of cells from Mye-PFKFB3−/− and Mye-PFKFB3+/+ mice were comparable with each other—no batch effect was present (Fig. 3A). The pooled cells formed two clusters, in which macrophages were identified using highly expressed marker genes including CD68 and Trem2 (Fig. 3B). Among the analyzed cells, various genes were found to be differentially expressed between Mye-PFKFB3−/− and Mye-PFKFB3+/+ macrophages (Fig. 3C). Further analysis by bioinformatics revealed that the expression of member genes in the gene sets related to oxidative phosphorylation (such as ATP5G3, NDUFA6, and ATP6V0D1) and cytokine-cytokine receptor interactions (such as CCL22, CCL7, and CCL2) were more likely to be upregulated in response to PFKFB3 disruption (Fig. 3D). These results suggest that PFKFB3 disruption promotes the proinflammatory activation in macrophages at least through increasing metabolic dysregulation and through enhancing macrophage proinflammatory signaling.

Bone marrow cells from myeloid cell-specific PFKFB3 disrupted (Mye-PFKFB3–/–) mice and control (Mye-PFKFB3+/+) mice were differentiated into macrophages (BMDMs). Prior to harvest, BMDMs were treated with LPS (20 ng/mL) for 6 h and subjected to scRNAseq analysis. A t-SNE representation of cells showing the sample origin: Mye-PFKFB3–/– and Mye-PFKFB3+/+ (WT). B t-SNE representation showing clusters of cells: CD68high-Trem2high and CD68low-Trem2low. C Volcano plot of differentially expressed (DE) genes (fold-changes (FC = log2 transformed Mye-PFKFB3–/– to Mye-PFKFB3+/+ expression ratios) vs. log10-transformed p values) in macrophages. Macrophages with high expression of markers such as CD68 and Trem2 were included in the differential expression analysis. Genes in oval are example genes with expression is upregulated in macrophages from Mye-PFKFB3–/– mice. D Results of GSEA analysis using the ranked list of DE genes, suggesting that the expression of genes related to metabolic and proinflammatory responses is upregulated by PFKFB3 disruption.
Disruption of PFKFB3 in hematopoietic cells does not alter diet-induced adiposity in mice
Myeloid cell-specific PFKFB3 disruption in mice exacerbates diet-induced systemic insulin resistance without altering body weight and adiposity [6]. In the current study, we used BMT as an alternative approach to verify the role of PFKFB3 in hematopoietic cells in regulating diet-induced phenotype in mice. During the feeding period, the body weight of PFKFB3+/− → WT mice did not differ significantly from that of WT → WT mice (Fig. 4A). Also, PFKFB3+/− → WT mice and WT → WT mice consumed comparable amount of food (Fig. 4B). When fat mass was quantified, the amount of epididymal fat mass, mesenteric fat mass, and perinephric fat mass, as well as their combination or abdominal fat mass in PFKFB3+/− → WT mice did not differ significantly from their respective amount in WT → WT mice (Fig. 4C). These results validate that PFKFB3 disruption in hematopoietic cells does not significantly alter the effect of HFD feeding on inducing obesity and adiposity.

Bone marrow cells from PFKFB3+/− mice and/or wild-type (WT) littermates were transplanted into lethally irradiated wild-type mice. After recovery for 4 weeks, chimeric mice were fed an HFD for 12 weeks. PFKFB3+/− → WT mice, WT mice were transplanted with bone marrow cells from PFKFB3+/− mice; WT → WT mice, WT mice were transplanted with WT bone marrow cells. A Body weight of chimeric mice. B Food intake of the chimeric mice. For A and B, body weight was recorded weekly during the feeding period. Also, food amount was monitored weekly and used to calculate food intake. C Visceral fat mass. After the feeding period, mice were fasted for 4 h and subjected to collection of fat pads. Abdominal fat mass was calculated as the sum of epididymal fat, mesenteric fat, and perinephric fat. For A–C, data are means ± SEM. n = 7–10.
Disruption of PFKFB3 in hematopoietic cells exacerbates diet-induced metabolic dysregulation
Myeloid cell-specific PFKFB3 disruption in mice exacerbates diet-induced systemic insulin resistance [6]. Using chimeric mice, the current study verified a role for the PFKFB3 in hematopoietic cells in regulating obesity-associated insulin resistance. Specifically, the severity of HFD-induced hyperglycemia and hyperinsulinemia in PFKFB3+/− → WT mice were significantly greater than that in WT → WT mice (Fig. 5A, B). Consistently, the severity of HFD-induced systemic insulin resistance and glucose intolerance in PFKFB3+/− → WT mice was significantly higher than that in WT → WT mice (Fig. 5C, D). In addition, AUC calculated from the data of glucose or insulin tolerance tests in HFD-fed PFKFB3+/− → WT mice was significantly greater than that in HFD-fed WT → WT mice (Supplementary Fig. S3). These results indicate that PFKFB3 disruption in hematopoietic cells exacerbates the severity of diet-induced systemic metabolic dysregulation. Considering the importance of hematopoietic PFKFB3 in regulating diet-induced WAT inflammation and systemic insulin resistance, we also sought to address the extent to which reconstituting WT hematopoietic cells rescues or alleviates diet-induced phenotype in global PFKFB3-disrupted mice. As demonstrated before [1], PFKFB3+/− mice displayed increased severity of HFD-induced systemic insulin resistance (Supplementary Fig. S4), along with increased WAT inflammation, compared with WT mice. However, after reconstituting WT hematopoietic cells, HFD-fed WT → PFKFB3+/− mice displayed comparable phenotype in WAT inflammation and systemic insulin resistance compared with HFD-fed WT → WT mice (Supplementary Fig. S4). Therefore, hematopoietic PFKFB3 plays a critical role in protecting against HFD-induced WAT inflammation and systemic insulin resistance.

Chimeric (PFKFB3+/− → WT mice and WT → WT) mice, described in Fig. 3, were fed an HFD for 12 weeks. A, B Plasma levels of glucose (A) and insulin (B). Prior to collection of blood samples, HFD-fed mice were fasted for 4 h. C, D Glucose (C) and insulin (D) tolerance tests. After the feeding period, chimeric mice were fasted for 4 h and given a bolus intraperitoneal injection of glucose (2 g/kg) (C) or insulin (1 U/kg) (D) and subjected to the tests. For A–D, data are means ± SEM. n = 10. *, P < 0.05 and **, P < 0.01 PFKFB3+/− → WT vs. WT → WT in (A and B) and for the same time point in (C and D).
Disruption of PFKFB3 in hematopoietic cells exacerbates diet-induced WAT inflammation and decreases WAT insulin signaling
WAT inflammation is key to the development of systemic insulin resistance. We sought to analyze the effect of PFKFB3 disruption in hematopoietic cells on WAT inflammation. Compared with those in HFD-fed WT → WT mice, the sections of WAT in HFD-fed PFKFB3+/− → WT mice displayed significantly more F4/80-postive cells (Fig. 6A), indicating increased WAT macrophage infiltration. Consistently, the phosphorylation states of NFκB p65 in WAT from HFD-fed PFKFB3+/− → WT mice were significantly greater than those in WAT from HFD-fed WT → WT mice (Fig. 6B). Next, we analyzed WAT insulin signaling in HFD-fed chimeric mice. Compared with those in HFD-fed WT → WT mice, insulin-stimulated Akt phosphorylate states in WAT from HFD-fed PFKFB3+/− → WT mice were significantly decreased (Fig. 6C). Therefore, PFKFB3 disruption in hematopoietic cells decreases WAT insulin sensitivity. To analyze adipose tissue bio-physiology, we also examined the mRNA levels of WAT genes related to adipokines and adipose tissue fat metabolism. The mRNA levels of genes for WAT fat mass such as hormone-sensitive lipase (HSL), glycerol kinase (Gyk), and PPARγ in PFKFB3+/− → WT mice were comparable with their respective levels in WT → WT mice (Fig. 6D). In addition, the mRNA levels of adipokines such as leptin, visfatin, and lipocalin2 in WAT from PFKFB3+/− → WT mice did not differ significantly from those from WT → WT mice. However, the mRNA levels of adiponectin were decreased and the mRNA levels of monocyte chemoattractant protein 1 (MCP1), TNFα, and IL-1β were increased in WAT from PFKFB3+/− → WT mice compared with their respective levels from WT → WT mice (Fig. 6D).

Chimeric (PFKFB3+/− → WT mice and WT → WT) mice, described in Fig. 3, were fed an HFD for 12 weeks. A, B WAT inflammation. After harvest, WAT sections were stained with H&E or for F4/80 expression (A). Also, WAT lysates were examined for the phosphorylation states of NFκB p65 (B). C WAT insulin signaling. Prior to harvest, HFD-fed chimeric mice were given a bolus injection of insulin into the portal vein for 5 min. WAT lysates were examined for the total amount and phosphorylation states of Akt. D WAT mRNA levels. For B and C, blots were quantified using densitometry. For D, WAT mRNA levels were quantified using real-time RT-PCR. For bar graphs in (B–D), data are means ± SEM. n = 4 (B and C) or 6 (D). *, P < 0.05 and **, P < 0.01 PFKFB3+/− → WT vs. WT → WT in (B) and in (C) under the same condition (insulin) and in (D) for the same gene.
PFKFB3-disrupted macrophages enhance adipocyte proinflammatory responses and decrease adipocyte insulin signaling
To verify a role for the PFKFB3 in macrophages in regulating adipocyte responses in relation to WAT inflammation and insulin resistance, we performed macrophage-adipocyte cocultures. Under LPS-stimulated conditions, adipocytes co-cultured with PFKFB3+/− BMDMs revealed significantly increased phosphorylation states of JNK p46 and NFκB p65 compared with adipocytes co-cultured with control BMDMs (Fig. 7A). Next, we examined insulin signaling in the cocultures. Compared with adipocytes co-cultured with control BMDMs, adipocytes co-cultured with PFKFB3+/− BMDMs displayed a significant decrease in insulin-stimulated Akt phosphorylation states (Fig. 7B). These results suggest that PFKFB3 disruption promotes macrophage generation of factors that act to enhance adipocyte proinflammatory responses and decrease adipocyte insulin signaling.

Adipocytes, differentiated from 3T3-L1 cells, were co-cultured with BMDMs from PFKFB3+/− mice or WT mice. A Proinflammatory signaling. Prior to harvest, the cocultures were treated with or without LPS (100 ng/mL) or PBS for the last 30 min. B Insulin signaling. Prior to harvest, the cocultures were treated with insulin (100 nM) or PBS for the last 30 min. For A and B, cell lysates were subjected to Western blot analysis. Bar graphs, quantification of blots. Data are means ± SEM. n = 4–6. *, P < 0.05 and **, P < 0.01 cocultures with PFKFB3+/− BMDMs vs. cocultures with WT BMDMs under LPS-stimulated condition in A or insulin-stimulated condition in B; †, P < 0.05 and ††, P < 0.01 LPS vs. PBS for the same cocultures in (A).
Discussion
WAT inflammation is a common characteristic of obesity and contributes to the development and progression of systemic insulin resistance. Within WAT, both adipocytes and immune cells are shown to critically determine the status of obesity-associated WAT inflammation, thereby systemic insulin resistance. As a regulatory gene of glycolysis, PFKFB3 is expressed at high abundance in both adipocytes and macrophages [20, 23] and acts to protect against the proinflammatory responses in both types of cells. In the present study, we demonstrated that PFKFB3 disruption only in hematopoietic cells exacerbated the severity of HFD-induced WAT inflammation and systemic insulin resistance. Consistently, cocultures of adipocytes with PFKFB3-disrupted macrophages revealed increased proinflammatory responses and decreased insulin sensitivity. Therefore, we obtained in vivo and in vitro evidence suggesting a role for PFKFB3 in hematopoietic cells, in particular macrophages, in protecting against obesity-associated WAT inflammation and systemic insulin resistance.
Previous studies have demonstrated that PFKFB3 links nutrient metabolism and the proinflammatory responses in adipocytes, intestinal epithelial cells, and macrophages [6, 17, 33]. Moreover, PFKFB3 is involved in the actions of PPARγ activation on decreasing obesity-associated WAT inflammation and systemic insulin resistance. However, the expression pattern of PFKFB3 in WAT, as well as WAT immune cells, remains not well studied. Here we showed that the amount of iPFK2 in WAT from HFD-WT mice was significantly increased compared with that from LFD-WT mice, which was also accompanied with increased severity of WAT inflammation and systemic insulin resistance. This increase in WAT iPFK2 amount appeared to be a defensive response; as this is supported by two lines of evidence. Firstly, global PFKFB3 disruption in mice exacerbated HFD-induced WAT inflammation [1]. Secondly, iPFK2 amount was increased in both WAT and cultured adipocytes in response to PPARγ activation, which is shown to decrease WAT inflammation and improve systemic insulin sensitivity [17]. In the present study, we validated that PFKFB3 mRNAs in WAT SVC from rosiglitazone-treated mice were significantly increased compared with those from controls, and that the increase in SVC PFKFB3 mRNAs was accompanied decreased IL-6 mRNAs. These findings not only extend the stimulatory effect of PPARγ activation on PFKFB3/iPFK2 expression from adipocytes to WAT immune cells, but more importantly suggest a role for the PFKFB3 in immune cells in protecting against obesity-associated WAT inflammation and systemic insulin resistance. The latter was substantiated by the evidence obtained from the chimeric mice (see below).
PFKFB3 critically regulates macrophage activation status. This has been validated by the findings from a previous study involving BMDMs [6] and by the current study involving peritoneal macrophages. Specifically, PFKFB3 disruption increased LPS-induced macrophage proinflammatory activation and decreased PPARγ activation- and/or IL-4-induced macrophage anti-inflammatory activation. Because iPFK2 functions to promote glycolysis through generating fructose-2,6-bisphosphate as the most powerful activator of glycolytic enzyme 6-phosphofructokinase-1, PFKFB3 regulation of macrophage activation is tied to its property in enhancing glycolysis. Indeed, PFKFB3 disruption decreased macrophage glycolysis rates [6]. As shown in adipocytes, PFKFB3 disruption brought about decreased glycolysis and a compensatory increase in fatty acid oxidation, which accounts for increased generation of reactive oxygen species and the proinflammatory responses [17]. In contrast, iPFK2 overexpression caused significant increases in glycolysis and in the conversion of glucose to fat. This may also be the case in macrophages; however, recent studies have increasingly shown that altered glycolysis in macrophages is also accompanied with changes in metabolic pathways other than glycolysis, e.g., TCA, pentose phosphate shunt, and aspartate-arginosuccinate shunt [34, 35]. The changes are shown to critically regulate macrophage activation status. In the present study, we also validated that PFKFB3 disruption increased macrophage oxidative metabolism as this is suggested by the data upon landscape analysis of macrophage transcriptome. However, future studies are warranted to study the extent to which PFKFB3/iPFK2 is also involved in the regulation of pentose phosphate shunt and aspartate-arginosuccinate shunt in relation to macrophage activation.
PFKFB3 regulation of macrophage activation led us to address the relevance of PFKFB3 in myeloid cells with regards to the regulation of obesity-associated systemic insulin resistance. As reported previously, we validated that PFKFB3 disruption only in myeloid cells exacerbated the severity of HFD-induced systemic insulin resistance. However, the changes in WAT inflammation was not explored. In the present study, we used chimeric mice to analyze HFD-induced WAT inflammation in relation to systemic insulin resistance. Indicated by increases in infiltration of F4/80-positive cells and in the proinflammatory signaling (phosphorylation states of NFκB p65), the severity of diet-induced WAT inflammation in chimeric mice whose PFKFB3 was disrupted in hematopoietic cells was significantly increased compared with that in control mice. Moreover, the increase in WAT inflammation was accompanied with increased severity of HFD-induced systemic insulin resistance. Based on these findings, we propose that the PFKFB3 in hematopoietic cells acts to protect against obesity-associated WAT inflammation, as well as local and systemic insulin resistance. More specifically, macrophage-specific PFKFB3 appeared to account for this role played by PFKFB3 itself. As substantial evidence obtained from adipocyte-macrophage cocultures, we demonstrated that PFKFB3 disruption enabled macrophage generation of factors that function to increase adipocyte proinflammatory responses and decrease adipocyte insulin signaling.
Our finding that PFKFB3 disruption in hematopoietic cells did not alter HFD-induced weight gain and adiposity also suggests that the PFKFB3 found in immune cells and adipocytes functions similarly in terms of protecting against WAT inflammation, but distinctly in terms of regulating adiposity. This notion appears to be true when considering that global PFKFB3 disruption significantly decreased HFD-induced adiposity, but increased WAT inflammation. In contrast, selective overexpression of PFKFB3/iPFK2 in adipocytes significantly increased adiposity, but decreased WAT inflammation [36]. Because of this, it is very likely that the PFKFB3 in immune cells, i.e., hematopoietic cells, acts to protect against WAT inflammation while having no effect on adiposity whereas the PFKFB3 in adipocytes acts to protect against WAT inflammation while promoting adiposity. Consistently, adoptive transfer of PFKFB3-disrupted hematopoietic cells increased the expression of WAT genes related to inflammation and systemic insulin sensitivity MCP1, TNFα, IL-1β and adiponectin, but not the genes related to adiposity such as leptin, HSL, GyK, and PPARγ.
In summary, the present study provides evidence to support a critical role for hematopoietic PFKFB3 in protecting against obesity-associated WAT inflammation and systemic insulin resistance. The notable finding is that PFKFB3 disruption only in hematopoietic cells exacerbated the severity of HFD-induced WAT inflammation, as well as local and systemic insulin resistance. Because macrophage factors generated in response to PFKFB3 disruption acted to enhance adipocyte proinflammatory responses and decrease adipocyte insulin signaling, the PFKFB3 in macrophages is a key player that critically protects against diet-induced metabolic phenotype. As such, the PFKFB3 in macrophages is a likely viable target for managing obesity-associated insulin resistance and human metabolic disease.
References
Huo Y, Guo X, Li H, Wang H, Zhang W, Wang Y, et al. Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J Biol Chem. 2010;285:3713–21.
Xu H, Li H, Woo S-L, Kim S-M, Shende VR, Neuendorff N, et al. Myeloid cell-specific disruption of Period1 and Period2 exacerbates diet-induced inflammation and insulin resistance. J Biol Chem. 2014;289:16374–88.
Pei Y, Li H, Cai Y, Zhou J, Luo X, Ma L, et al. Regulation of adipose tissue inflammation by adenosine 2A receptor in obese mice. J Endocrinol. 2018;239:365–76.
Cai Y, Li H, Liu M, Pei Y, Zheng J, Zhou J, et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology. 2018;68:48–61.
Luo X, Li H, Ma L, Zhou J, Guo X, Woo S-L, et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology. 2018;155:1971–84.
Ma L, Li H, Hu J, Zheng J, Zhou J, Botchlett R, et al. Indole Alleviates Diet-induced Hepatic Steatosis and Inflammation in a Manner Involving Myeloid Cell PFKFB3. Hepatology. 2020;72:1191–203.
Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20.
Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507.
Sag D, Carling D, Stout RD, Suttles J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–41.
Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARδ regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7:485–95.
Stienstra R, Duval C, Keshtkar S, van der Laak J, Kersten S, Muller M. Peroxisome proliferator-activated receptor γ activation promotes infiltration of alternatively activated macrophages into adipose tissue. J Biol Chem. 2008;283:22620–7.
Saberi M, Woods N-B, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10:419–29.
Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo J-L, Naugler W, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–97.
Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339:218–22.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AWJ. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Investig. 2003;112:1796–808. https://doi.org/10.1172/JCI1924612.
Lumeng CN, DeYoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23.
Guo X, Xu K, Zhang J, Li H, Zhang W, Wang H, et al. Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of PPARγ activation in mice. J Biol Chem. 2010;285:23711–20.
Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381:561–79.
Okar DA, Wu C, Lange AJ. Regulation of the regulatory enzyme, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphate. Adv Enzyme Regul. 2004; 44:123–54.
Atsumi T, Nishio T, Niwa H, Takeuchi J, Bando H, Shimizu C, et al. Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/PFKFB3 isoforms in adipocytes and their potential role in glycolytic regulation. Diabetes. 2005;54:3349–57.
Ruiz-García A, Monsalve E, Novellasdemunt L, Navarro-Sabaté A, Manzano A, Rivero S, et al. Cooperation of adenosine with macrophage Toll-4 receptor agonists leads to increased glycolytic flux through the enhanced expression of PFKFB3 gene. J Biol Chem. 2011;286:19247–58.
Csóka B, Selmeczy Z, Koscsó B, Németh ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012;26:376–86.
Chesney J, Telang S, Yalcin A, Clem A, Wallis N, Bucala R. Targeted disruption of inducible 6-phosphofructo-2-kinase results in embryonic lethality. Biochem Biophys Res Commun. 2005;331:139–46.
Woo S-L, Xu H, Li H, Zhao Y, Hu X, Zhao J, et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS ONE. 2014;9:e91111.
Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. 2005;11:183–90.
Wu C, Kang JE, Peng L, Li H, Khan SA, Hillard CJ, et al. Enhancing hepatic glycolysis reduces obesity: Differential effects on lipogenesis depend on site of glycolytic modulation. Cell Metab. 2005;2:131–40.
Stienstra R, Duval C, Keshtkar S, van der Laak J, Kersten S, Muller M. Peroxisome proliferator-activated receptor gamma activation promotes infiltration of alternatively activated macrophages into adipose tissue. J Biol Chem. 2008;283:22620–7.
Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, et al. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–31.
Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Lumeng CN, Deyoung SM, Saltiel AR. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. Am J Physiol Endocrinol Metab. 2007;292:E166–74.
Guo T, Woo S-L, Guo X, Li H, Zheng J, Botchlett R, et al. Berberine ameliorates hepatic steatosis and suppresses liver and adipose tissue inflammation in mice with diet-induced obesity. Sci Rep. 2016;6:22612.
Botchlett R, Li H, Guo X, Qi T, Zhao J, Zheng J, et al. Glucose and palmitate differentially regulate PFKFB3/iPFK2 and inflammatory responses in mouse intestinal epithelial cells. Sci Rep. 2016;6:28963.
Haschemi A, Kosma P, Gille L, Evans Charles R, Burant Charles F, Starkl P, et al. The Sedoheptulose Kinase CARKL Directs Macrophage Polarization through Control of Glucose Metabolism. Cell Metab. 2012;15:813–26.
Jha Abhishek K, Huang Stanley C-C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity. 2015;42:419–30.
Huo Y, Guo X, Li H, Xu H, Halim V, Zhang W, et al. Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. J Biol Chem. 2012;287:21492–500.
Acknowledgements
This work was supported in part by the Hickam Endowed Chair, Gastroenterology, Medicine, Indiana University and the Indiana University Health – Indiana University School of Medicine Strategic Research Initiative (GA, HF, FM), the Senior Research Career Scientist to GA (5I01BX000574) and VA Merit Award and Research Career Scientist to HF (1I01BX003031) from the United States Department of Veteran’s Affairs, Biomedical Laboratory Research and Development Service, and NIH grants DK076898, and DK110035 to GA and SG and DK108959 and DK119421 to HF, Indianapolis, Indiana. In addition, this work was supported in whole or in part by grants from the American Diabetes Association (1-10-BS-76 to CW) and NIH (DK095828 and DK124854 to CW). CW is also supported by the Hatch Program of the National Institutes of Food and Agriculture (NIFA). XG is supported by Fundamental Research Funds of Shandong University (Grant No. 2017TB0028), Young Scholars Program of Shandong University (Grant No. 2018WLJH33), and National Natural Science Foundation of China (Grant No. 81803224). The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs.
Author information
Authors and Affiliations
Contributions
BZ, XG, HX and HL carried out most of experiments involving mice. BZ and HL carried out most of the experiments involving cells. BZ, XG, HX, HL, and TZ collected tissue and cell samples and performed molecular and biochemical assays. HL performed histological assays. JC performed bioinformatic analysis. BJ, YW, QY, JC, SG, FM, HF, and GA contributed to scientific discussion. CW came up the concept of the study and supervised all experiments and wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhu, B., Guo, X., Xu, H. et al. Adipose tissue inflammation and systemic insulin resistance in mice with diet-induced obesity is possibly associated with disruption of PFKFB3 in hematopoietic cells. Lab Invest 101, 328–340 (2021). https://doi.org/10.1038/s41374-020-00523-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-020-00523-z
