
Abstract
Acute myocardial infarction (AMI) is a common clinical cardiovascular disease, which is the leading cause of death and disability worldwide. Abnormal expression of long noncoding RNAs (lncRNA) is reported to be related to myocardial dysfunctions such as myocardial infarction (MI). In this study, we aimed to investigate the role of lncRNA myocardial infarction-related transcription factors 2 (Mirt2) in AMI and the underlying molecular mechanisms in vivo and in vitro. In vivo AMI model was established by occlusion of the left anterior descending coronary artery. Rats were randomly divided into two groups (five rats per group): the sham group and the AMI group. H9c2 cells were cultured under hypoxia for 4 h and then cultured under normoxia to establish the in vitro hypoxia reoxygenation (H/R) model. Our study shows that the myocardial infarct size and the apoptosis in AMI rats were both significantly increased, indicating that the AMI rat model was successfully established. Additionally, the levels of Mirt2 in AMI rats were increased significantly. Knockdown of Mirt2 by shRNA (shMirt2) had no significant effect on apoptosis and MI in sham rats, but significantly promoted apoptosis and MI in AMI rats. In vitro experiments showed that shMirt2 significantly decreased the level of Mirt2 in H9c2 cells and H9c2 cells treated with H/R. It is worth noting that shMirt2 had no significant effect on H9c2 cells, but significantly increased the levels of oxidative stress markers (malondialdehyde and lactate dehydrogenase), and also increased the number of apoptosis of H/R-treated H9c2 cells. Further mechanistic analysis showed that Mirt2 could protect MI and apoptosis in AMI rats by competitively adsorbing miR-764 and reducing the inhibitory effect of miR-764 on 3-phosphoinositide-dependent kinase 1 (PDK1). More importantly, after overexpression of Mirt2, MI and apoptosis were significantly improved in AMI rats, indicating that Mirt2 showed a protective effect in AMI rats. In summary, these findings suggest that that Mirt2 participated in the regulation of MI through the miR-764/PDK1 axis. Therefore, the current findings provide a theoretical basis for the diagnosis and treatment of clinical MI with changes in Mirt2 levels.
Similar content being viewed by others


Introduction
Heart disease is currently the leading cause of death and disability worldwide [1]. Despite modern reperfusion strategies, there is still a considerable proportion of patients with left ventricular remodeling, leading to heart failure (HF) after myocardial infarction (MI) [2]. Myocardial ischemia, cardiac arrest, or cardiac surgery can cause irreversible damage to the myocardium [3]. However, the integrity of the molecular pathways involved in the biological process of MI is still unknown. Apoptosis is considered the main mechanism leading to MI [4, 5]. Therefore, further study of the mechanism of apoptosis in MI may be helpful to identify effective targeted therapy.
Long noncoding RNAs (lncRNAs) are RNAs longer than 200 nucleotides [6]. Studies have shown that myocardial ischemia-reperfusion injury can cause the abnormal expression of lncRNA [7], suggesting that changes in lncRNA expression may be related to various myocardial dysfunctions and may be a novel marker for the treatment of HF [8, 9]. Zang et al. found that myocardial infarction-related transcripts (Mirt1 and Mirt2) and other lncRNAs were upregulated in mice with MI, but Mirt1 and Mirt2 were negatively correlated with MI area while positively related to left ventricular ejection fraction (LVEF) [10]. Among them, inhibition of Mirt1 can inhibit NF-κB activation and weaken acute myocardial infarction (AMI) [11]. However, the role of Mirt2 in MI has not been reported.
MicroRNAs (miRNAs) play pivotal roles in the pathophysiology of AMI [12]. Abnormal expression of miRNA can lead to changes in several pathological processes of the heart, which are related to changes in cardiac hypertrophy after AMI [13]. Clinical studies have shown that miRNAs, such as miR-1, miR-133a, and miR-499, are significantly related to hsTnT, NT-proBNP, and LVEF, and levels of circulating miR-499 in plasma may help identify patients with increased risk of early death after AMI [14]. As an miRNA molecule, miR-764 has been reported in hepatocellular carcinoma [15]. However, whether miR-764 is implicated in cardiovascular disease remains unknown.
Therefore, the purpose of the current study was to elucidate the role of Mirt2 in MI and the underlying molecular mechanisms in vivo and in vitro. Our findings suggested Mirt2 reduced apoptosis to alleviate MI through regulating miR-764/3-phosphoinositide-dependent kinase 1 (PDK1) axis.
Material and methods
Animal model
Animal experiments were conducted according to the guide for the care and use of National Institutes of Health and approved by the Ethics Committee of Wuhan Third Hospital (Tongren Hospital of Wuhan University). Adult male Spragure–Dawley (SD) rats (12–14 weeks) were acquired from the animal center of Wuhan Third Hospital (Tongren Hospital of Wuhan University) and fed at a stable environment (temperature of 22 ± 2 °C, humidity of 50–55%, and a dark-light cycle of 12 h). Twenty rats were randomly divided into two groups, sham group and AMI group, with 10 rats in each group. AMI model was constructed by occlusion of the left anterior descending (LAD) coronary artery. Briefly, rats were cut into a longitudinal pattern in the left anterior cardiac region. Disconnect the penultimate rib to expose the thorax. Subsequently, the heart was then exposed by removing the pericardium. The LAD branch was permanently ligated 2–3 mm away from the aortic root. The heart with successful ligation could observe the color change of the distal myocardium. After ligation, the heart was repositioned in the chest cavity. Suture the skin and use a syringe to remove air from the chest cavity. After surgery, the rats were individually kept alone in a cage for 24 h to recovery. The rats in the sham operation group only passed under the coronary artery without ligation. Echocardiography (Vevo 770; Visual Sonics) was carried out to assess cardiac function parameter, such as ejection fraction (EF) and fractional shortening (FS). After 10 weeks, all rats were sacrificed by intraperitoneal injection of sodium pentobarbital (200 mg/kg), and samples were collected for examination [16].
Triphenyltetrazolium (TTC) staining
TTC staining was used to detect MI size. The ventricle was transected into 2 mm thick slices. The sections were incubated with 1% TTC at 37 °C for 10 min to locate the infarcted and non-infarcted areas. TTC non-stained area (white) is defined as the infarcted area. The percentage of infarcted area represents the area of infarct.
Measurement of malondialdehyde (MDA) and lactate dehydrogenase (LDH)
The levels of LDH and MDA in H9c2 cells or serum of rats were measured by diagnostic kit (NJBI, China) according to the manufacturer’s instructions.
Terminal deoxynucleotide transferase-mediated dUTP gap terminal (TUNEL) assay
Apoptosis of H9c2 cells or myocardial tissue was detected by TUNEL assay. In brief, tissue sections or H9c2 cells were dewaxed and exposed to 3% H2O2. After that, the samples were digested with pepsin at 37 °C for 30 min, and then subjected to a TUNEL reaction mixture at 25 °C for 60 min and redyed with hematoxylin. The number of apoptotic cells was quantitatively analyzed by TUNEL positive nuclear counting method. Positive cells were assessed by ×100 optical microscopy system.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from myocardial tissue or H9c2 cells and then reversed transcribed into cDNA (Superscript II reverse transcriptase, Life Technologies). qPCR was performed on the CFX96 system (Bio-rad, Nazareth, Belgium) using IQ SYBR Green SuperMix. The mRNA level of target genes was normalized by U6 and β-actin and calculated using CFX Manager2.1 software (Bio-Rad) through relative quantitative method (2−∆∆Ct). All the primers used are as follows:
Mirt2, F, 5′-CCAATCCCGATGCAATGTT-3′, R, 5′-TCAAGCCAACCTGCAACGG-3′;
MiR-764, F, 5′-ACTAACGAAGGCCTACGTC-3′, R, 5′-TCGAACAATGCAACGTACG-3′;
PDK1, F, 5′-GGCAACCATCTTAGAACCG-3′, R, 5′-GGTCCAAACTGAACCTGAC-3′;
U6, F, 5′-CAACGCACTGGTTACCAAT-3′, R, 5′-CATTAGGCATGACATGCAG-3′;
β-actin, F, 5′-TTCCGAATGCTGACGTTGC-3′, R, 5′-CCAACTGCCAAACGTGCTG-3′.
Mirt2 knockdown
Twenty rats were divided into Ad-shRNA group, Ad-shMirt2-1# group, Ad-shMirt2-2# group, and Ad-shMirt2-3# group. The Mirt2-specific shRNAs (shMirt2-1#, shMirt2-2#, and shMirt2-3#) were designed, and the double-stranded oligonucleotide was integrated into the pSIREN-DNR-DsRed-Express Vector (Biomiao, Beijing, China). Then, the donor vector was inserted into a pLP-Adeno-X LP CMV vector (Biomiao, Beijing, China) to construct a recombinant adenovirus (Ad-shMirt2). Adenovirus with shRNA-negative control (NC) was prepared as a control adenovirus (Ad-NC). Adenovirus was intramyocardially injected at the infarcted border as previously reported [17] for in vivo experiments. In vitro, H9c2 cells were infected by adenovirus.
Immunohistochemistry
The expression of caspase 3 in tissue was measured by Immunohistochemical staining. The primary antibody is anti-caspase 3 (#9664, 1:2000). The nuclei were restrained with hematoxylin and examined using image-pro Plus software (version 6.0; Silver Spring, Marland). The positive staining results were analyzed by the semi-quantitative integral optical density value of protein expression index (magnification ×200).
Cell culture and treatment
H9c2 cell line was purchased from the American Type Culture Collection (Manassas, VA) and maintained in DMEM supplemented with 10% (v/v) FBS, 100 U/mL penicillin and 100 mg/mL streptomycin with 5% CO2 in a 37 °C incubator. H9c2 cells were then cultured under hypoxia (5% CO2, 1% O2, and 94% N2) in a hypoxia chamber for 4 h and then cultured under normoxia to construct hypoxia reoxygenation (H/R) model. Experiments were carried out using cells between passages 10 and 14. Subsequently, cells were infected with adenovirus with shRNA or shMirt2 or transfected with miRNA mimics or inhibitors using Lipofectamine 2000 (Invitrogen, USA).
Methyl thiazolyl tetrazolium (MTT) assay
Viability of cells was measured by MTT assay. H/R-treated H9c2 cells (1 × 105 cells/well) were inoculated in 96-well plates. 20 μl MTT solution (5 mg/ml; Sigma-Aldrich) was then mixed with the cells and incubated for 2 h. Thereafter, H9c2 cells were cleared with DMSO and determined at 570 nm using a Microplate reader (Bio-rad, Hercules, CA, USA).
Western blotting
Proteins were isolated from H9c2 cells or myocardium from rats using RIPA buffer (Beyotime Biotechnology, Shanghai, China) containing protease inhibitor. After separating using SDS-PAGE, proteins were transferred onto PVDF membranes. After blocking with skim milk, proteins were hybridized with Bcl-2 (#3498, 1:1000, Cell Signaling Technology, USA), Bax (#5023, 1:1000, CST, USA), cleaved-caspase-3 (#9661, 1:1000, CST, USA), PDK1 (#3062, 1:1000, CST, USA), AKT (#9272, 1:1000, CST, USA), p-AKT (#13038, 1:1000, CST, USA), and GAPDH (#5174, 1:1000, CST, USA) at 4 °C overnight. After washing with TBS, the proteins were hybridized with secondary antibody at 25 °C for 1 h. Finally, proteins were quantified using Image LabTM Software (Bio-Rad, Shanghai, China).
Double luciferase report assay
The relationship between Mirt2 and miR-764, as well as miR-764 and PDK1, was predicted by miRDB (http://mirdb.org/) and Targetscan 7.1 (http://www.targetscan.org/). The Mirt2-WT, Mirt2-MUT, PDK1-WT, and PDK1-MUT 3′-UTR were amplified and integrated into the pGL4.10 luciferase reporter vector, respectively. Cells were transfected with recombinant vectors and mimics (inhibitors) using Lipofectamine 2000 according to the manufacturer’s instructions. After 48 h, Bright-Glo™ luciferin detection system (Promega, USA) was used to measure luciferase activity.
RNA immunoprecipitation (RIP)
The interaction between Mirt2 and miR-764 was confirmed by RIP assays using Magna RNA‐binding protein immunoprecipitation kit (Millipore, Bedford, MA). Simply, H9c2 cells were cleaved and co-cultured with A/G agarose beads containing Ago2 (ab186733; Abcam) or IgG antibody (ab172730; Abcam). Normal rabbit IgG was employed as NC. After washing, proteins of Ago2 or IgG were determined by western blotting.
PDK1 overexpression
The pdk1-overexpressed plasmid and empty vector were purchased from Gene Pharma (Shanghai, China). H9c2 cells were transfected with vectors using Lipofectamine 3000 reagent (Invitrogen).
Statistical analysis
Experimental data are manifested as mean ± standard deviation (SD). Statistical analysis was performed using Graphpad 6.0 software (San Diego, CA). Differences between the two groups were analyzed by Student’s t-test, while differences over three groups were achieved by one-way analysis of variance following hoc post. p value < 0.05 was statistically significant.
Results
The expression of Mirt2 is upregulated in a rat model of AMI
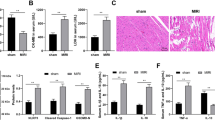
In this study, AMI rat model was constructed by ligation of the permanent anterior descending branch of the left coronary artery and MI was monitored by TTC staining. As illustrated in Fig. 1A, the myocardial infarct size of AMI rats was significantly larger than that of sham-operated rats, indicating that the AMI rat model had been successfully constructed. In addition, we also found that the levels of oxidative stress markers (MDA and LDH) in AMI rats were significantly increased (Fig. 1B). In addition, TUNEL analysis revealed that the number of apoptotic cardiomyocytes in AMI rats was significantly higher than that in sham group (Fig. 1C). Notably, RT-qPCR analysis showed that the level of Mirt2 in AMI rats was significantly higher than that in the sham group, suggesting that the high expression of Mirt2 might be related to AMI (Fig. 1D).

Twenty rats were divided into sham group and AMI group as experimental subjects with 10 rats in each group. A The extent of MI was monitored by TTC staining. B The levels of LDH and MDA in serum of rats were monitored by diagnostic kit. C Apoptosis was monitored by TUNEL assay. D The mRNA level of Mirt2 was monitored by RT-qPCR (**p < 0.01 vs. Sham group).
Knockdown of Mirt2 promoted ischemia-induced myocardial infarction
To further investigate whether the abnormal expression of Mirt2 is related to AMI, three different shRNAs (Ad-shMirt-1#, Ad-shMirt-2#, and Ad-shMirt-3#) were designed to knock down Mirt2. As shown in Fig. 2A, Mirt2 level was reduced in sham and AMI groups after Mirt2 was knocked down. As illustrated in Fig. 2B, C, no significant MI and changes in MDA and LDH levels were observed after Mirt2 knockdown in the sham group. However, in AMI rats, Mirt2 knockdown significantly aggravated AMI-induced MI and increased the levels of oxidative stress markers (MDA and LDH) in serum. Similarly, in the sham group, no significant changes in cardiomyocyte apoptosis and cleaved caspase 3 levels in tissue were observed after the knockdown of Mirt2. In AMI rats, AMI knockdown markedly enhanced AMI-induced apoptosis and cleaved caspase 3 expression (Fig. 2D, E). Taken together, these findings show that knockdown of Mirt2 promoted ischemia-induced MI.

A Twenty rats were divided into Ad-shRNA group, Ad-shMirt2-1# group, Ad-shMirt2-2# group, and Ad-shMirt2-3# group as experimental subjects. A The mRNA level of Mirt2 was monitored by RT-qPCR (**p < 0.01 vs. Ad-shRNA group). B–E The rats were randomly divided into four groups (five rats/group): Sham + Ad-shRNA group, Sham + Ad-shMirt2 group, AMI + Ad-shRNA group, and AMI + Ad-shMirt2 group. B The extent of MI was monitored by TTC staining. C The levels of oxidative stress markers (LDH and MDA) in serum of rats were monitored by diagnostic kit. D Apoptosis was monitored by TUNEL assay. E The expression of cleaved caspase 3 was monitored by IHC (**p < 0.01 vs. Sham + Ad-shRNA group or AMI + Ad-shRNA group).
Knockdown of Mirt2 exacerbated H/R-induced cardiomyocyte injury
This study expounded the effect of Mirt2 on H/R-induced H9c2 cell injury. As shown in Fig. 3A, after knockdown of Mirt2 by shRNA, the mRNA level of Mirt2 in the control group and H/R group was reduced. Moreover, this study found that knockdown of Mirt2 had no significant effect on the viability, MDA and LDH levels, and apoptosis of H9c2 cells. It is worth noting that the downregulation of Mirt2 profoundly inhibited the viability of H9c2 cells and reduced the levels of oxidative stress markers (MDA and LDH) in the cells after H/R treatment (Fig. 3B–D). At the same time knockdown of Mirt2 also aggravated H/R-induced H9c2 cell apoptosis (Fig. 3E). Western blotting showed that downregulation the expression of Mirt2 elevated the protein levels of pro-apoptotic proteins (Bax and cleaved caspase 3), while reduced the protein levels of antiapoptotic protein Bcl-2 (Fig. 3F). Taken together, our findings show Mirt2 knockdown exacerbated H/R-induced cardiomyocyte injury.

H9c2 cells were cultured under hypoxia for 4 h and then cultured under normoxia conditions to establish H/R model. H9c2 cells or H/R-treated H9c2 cells were infected with adenovirus containing shRNA or shMirt2. A The mRNA level of Mirt2 was monitored by RT-qPCR. B Viability of H9c2 cells was monitored by MTT assay. C, D The levels of LDH and MDA in cells were monitored by diagnostic kit. E Apoptosis was monitored by TUNEL assay. F The protein levels of Bcl-2, Bax, Cleaved caspase 3 were monitored by western blotting (**p < 0.01 vs. control group or Ad-shRNA group).
Mirt2 negatively regulated miR-764 expression as a molecular sponge
Recent studies have shown that lncRNAs act as miRNA sponges in hypoxia-induced injury. Similarity, our study exhibited that miR-764 was a suppositional target of Mirt2 by using miRDB (http://mirdb.org) (Fig. 4A). To clarify the targeting relationship between Mirt2 and miR-764, RIP assay and luciferase reporter assay were performed. The results revealed that miR-764 mimics specifically decreased the luciferase activity of Mirt2-WT, while miR-764 inhibitors specifically increased the luciferase activity of Mirt2-WT (Fig. 4B). In addition, RIP assay showed that both Mirt2 and miR-764 in H9c2 cells were rich in Ago2 IP, that is, Mirt2 directly interacted with miR-764 in H9c2 cells (Fig. 4C, D). As shown in Fig. 4E, knockdown of Mirt2 significantly upregulated the mRNA level of miR-764 compared with the Ad-shRNA group. Overall, these results demonstrated that Mirt2 negatively regulated miR-764 expression as a molecular sponge in H9c2 cells.

A The speculated target of Mirt2 was predicted by using miRDB. B The target relationship between Mirt2 and miR-764 was confirmed by double luciferase report assay (**p < 0.01 vs. miR-NC group or NC inh group). C, D The target relationship between Mirt2 and miR-764 was confirmed by RNA immunoprecipitation (RIP) (**p < 0.01 vs. anti-IgG group). E Dividing the 20 rats into Ad-shRNA group, Ad-shMirt2-1# group, Ad-shMirt2-2# group, and Ad-shMirt2-3# group as experimental subjects. The mRNA level of miR-764 was monitored by RT-qPCR (**p < 0.01 vs. Ad-shRNA group).
Mirt2 upregulated PDK1 expression by adsorbing miR-764
To explore whether Mirt2 acts as a competitive endogenous RNA in hypoxia-induced injury, TargetScan (http://www.targetscan.org/vert_72/) was employed to define the relationship of miR-764 and phosphatidylinositol-dependent protein kinase 1 (PDK1) (Fig. 5A). To verify the targeting relationship between miR-764 and PDK1, we performed a luciferase reporter assay. The results confirmed that miR-764 mimics obviously reduced the luciferase activity of PDK1-WT, while the luciferase activity of PDK1-MUT hardly changed. In contrast, miR-764 inhibitors increased the luciferase activity of PDK1-WT, while the luciferase activity of PDK1-MUT hardly changed (Fig. 5B), indicating that PDK1 directly binds to miR-764. In addition, miR-764 mimics significantly downregulated the mRNA and protein levels of PDK1, while miR-764 inhibitors showed the opposite effect (Fig. 5C, D). Moreover, PDK1 expression was significantly decreased after Mirt2 was silenced (Fig. 5E). In summary, Mirt2 upregulated PDK1 expression by adsorbing miR-764.

A The speculated target of Mirt2 was predicted by using TargetScan. B The target relationship between miR-764 and PDK1 was confirmed by double luciferase report assay (**p < 0.01 vs. miR-NC group or NC inh group). C The mRNA level of PDK1 was monitored by RT-qPCR (**p < 0.01 vs. miR-NC or NC inh group). D The protein level of PDK1 was monitored by western blotting (**p < 0.01 vs. miR-NC or NC inh group). E The protein level of PDK1 was monitored by western blotting (**p < 0.01 vs. Ad-shRNA + NC inh or Ad-shMirt2 + NC inh group).
Mirt2 modulated H/R-induced cardiomyocyte injury through PDK1
To confirm whether Mirt2 regulates H/R-induced H9c2 cell injury through PDK1, PDK1 was overexpressed. As shown in Fig. 6A, PDK1 overexpression obviously reversed shMirt2-induced decrease of H9c2 cells activity in the H/R group. PDK1 overexpression also reversed shMirt2-induced the increase of MDA and LDH levels in H/R group (Fig. 6B, C). Furthermore, PDK1 overexpression reversed apoptosis in the H/R group caused by shMirt2 (Fig. 6D). Western blotting analysis showed that PDK1 overexpression suppressed the protein levels of Bax and cleaved caspase 3 and promoted the protein levels of PDK1 and Bcl-2 (Fig. 6E). Taken together, these results show that Mirt2 regulated H/R-induced cardiomyocyte injury through PDK1.

H/R-treated H9c2 cells were infected with adenovirus containing shMirt2 or/and PDK1. A Viability of H9c2 cells was measured by MTT assay. B, C The levels of oxidative stress markers (LDH and MDA) in cells were monitored by diagnostic kit. D Apoptosis was monitored by TUNEL assay. E The protein levels of PDK1, Bcl-2, Bax, Cleaved caspase 3 were monitored by western blotting (*p < 0.05, **p < 0.01 vs. control group or H/R group or H/R + Ad-shMirt2 group).
Mirt2 overexpression showed a protective effect in AMI rats
Based on previous studies, this study examined the effect of Mirt2 overexpression on AMI. As shown in Fig. 7A, after the adenovirus Ad-Mirt2 infected the rats in sham group and AMI group, the level of Mrit2 was significantly increased indicating that Mirt2 was successfully overexpressed in the rats. Then, this study explored the effects of Mirt2 overexpression on MI, MDA and LDH, and apoptosis. As shown in Fig. 7B, Mirt2 overexpression had no significant effect on infarction size in rats in the sham group, whereas obviously improved infarction size in AMI rats. In addition, Mirt2 overexpression reduced the levels of oxidative stress markers (MDA and LDH) in the serum of AMI rats, but had no significant effect on the levels of oxidative stress markers (MDA and LDH) in the serum of sham-operated rats (Fig. 7C, D). Moreover, Mirt2 overexpression inhibited cardiomyocyte apoptosis in AMI rats, but had no significant effect on cardiomyocyte apoptosis in the sham operation group (Fig. 7E). In addition to these pathological improvements, echocardiography showed that exogenous Mirt2 overexpression in AMI rats significantly improved cardiac function (e.g., increased EF and FS), but had no significant effect to rats in the sham operation group (Fig. 7F). Furthermore, we found that exogenous overexpression of Mirt2 in AMI rats significantly increased the level of PKD1 and promoted the phosphorylation of downstream AKT (Fig. S1). In summary, these results suggest that Mirt2 showed protective effect in AMI rats.

Dividing the 20 rats into Sham + Ad-NC group, Sham + Ad-Mirt2 group, AMI + Ad-NC group, and AMI + Ad-Mirt2 group as experimental subjects. A The mRNA level of Mirt2 was monitored by RT-qPCR (**p < 0.01 vs. Sham + Ad-NC group or AMI + Ad-NC group). B The extent of MI was monitored by TTC staining. C, D The levels of oxidative stress markers (LDH and MDA) in serum of rats were monitored by diagnostic kit. E Apoptosis was monitored by TUNEL assay. F Cardiac function parameter (EF and FS) was measured using electrocardiograph (**p < 0.01 vs. Sham + Ad-NC group or AMI + Ad-NC group).
Discussion
AMI is a common clinical cardiovascular disease, which is characterized by myocardial cell ischemia and hypoxia damage caused by coronary plaque rupture, thrombosis, lumen stenosis, or occlusion [18]. Long-term myocardial ischemia can lead to apoptosis of cardiomyocytes and severe MI [19]. In the ischemic and hypoxic environment, cardiomyocytes will trigger oxidative stress reactions, and a large amount of oxygen free radicals will be generated in a short period of time, which will damage the cardiomyocyte membrane [20]. In this study, an AMI rat model was constructed by permanent ligation of the LAD coronary artery, and MI, MDA and LDH levels, and apoptosis were measured in AMI rats. The results showed that AMI rats had a significant MI accompanied by increased levels of oxidative stress markers (MDA and LDH). Apoptosis of cardiomyocytes in AMI rats was also increased significantly. These results indicate that AMI rats have been successfully established in this study.
In recent years, it is believed that lncRNAs are related to various myocardial dysfunctions and may be new markers for treating HF [21]. A study by Wang et al. [22] revealed that lncRNA CHAST is an independent predictor of cardiac contractile function in early AMI and can be used as a candidate biomarker for cardiac remodeling. The expression of myocardial infarction-associated transcript (Mirt1 and Mirt2) is upregulated in MI mice, but the expression of Mirt1 and Mirt2 is negatively related to the area of MI and positively related to the LVEF [10, 11]. Among them, inhibition of lncRNA Mirt1 can inhibit NF-κB activation and attenuate AMI [11]. Unfortunately, the effect of Mirt2 on MI and underlying molecular mechanism remains unclear. Previous studies have shown that the expression of Mirt2 was upregulated in LPS-stimulated PC12 cells and serum samples of SCI patients. However, overexpression of Mirt2 has protective effect on PC12 cell injury induced by LPS [23]. Mirt2 is considered to have negative feedback regulation in various cellular processes, which is characterized by the significant increase in Mirt2 expression under LPS stimulation and it effectively alleviated LPS-induced inflammatory response by inhibiting TRAF6 oligomerization and autoubiquitination [24]. Similar to the above results, this study found that the level of Mirt2 was significantly increased after ischemia-reperfusion stimulation, which prevented cardiomyocyte apoptosis and MI, and protected myocardial tissue from oxidative damage. Knockdown of Mirt2 aggravated MI and cardiomyocyte apoptosis in AMI rats. In addition, in vitro studies showed that knockdown of Mirt2 decreased the viability and the level of MI markers (MDA and LDH) in H/R-H9c2 cells, and promoted apoptosis of H9c2 cells induced by H/R. As expected, overexpression of Mirt2 significantly reduced MI and inhibited cardiomyocyte apoptosis in AMI rats. In conclusion, our study showed that Mirt2 also plays a negative feedback role in MI and cardiomyocyte apoptosis. Further studies on the molecular mechanism showed that Mirt2 reduced the inhibitory effect of miR-764 on PDK1 expression by competitive adsorption of miR-764, thereby showing protective effects on AMI rats and H/R-treated H9c2 cells in vitro and in vivo.
Recent studies have shown that miRNAs are a promising new therapeutic target that can affect multiple genes through different signaling pathways [25]. The miRNAs therapeutic regulation is used for atherosclerosis, AMI, cardiovascular and pulmonary hypertension, and ischemic injury [26]. MiRNAs in patients with AMI were positively correlated with NT-proBNP and hsTnT, and negatively correlated with LVEF [14]. miR-764 is one of miRNAs and has been reported in diseases [15]. Previous studies have shown that miR-764 could target Ninsurin 2 (NINJ2) to regulate neuronal cell death induced by hydrogen peroxide [27]. However, there is very little study on miR-764 in AMI. We sought to reveal whether miR-764 is involved in regulating MIRT2 in AMI rats and H/R-induced cardiomyocyte injury. The results showed after knocking down Mirt2, the levels of miR-764 in the cells were significantly increased. More interestingly, Mirt2 acted as a molecular sponge for miR-764, and PDK1 was found to be one of the targets of miR-764. Similarly, miR-764 overexpression can inhibit H/R-induced cardiomyocyte injury. These findings demonstrate that downregulation of miR-764 may be a control factor for MI.
PDK1 plays an indispensable role in the phosphatidylinositol 3 kinase (PI3K) PDK1-Akt pathway [28]. The activation of PI3K leads to the production of phosphatidylinositol-3,4,5-triphosphate. Subsequently, PDK1 and Akt are recruited to the plasma membrane, and then the membrane-bound PDK1 phosphorylates Akt at Thr308, thereby activating Akt [29], which participated in cardiovascular pathological processes such as atherosclerosis, myocardial hypertrophy, and vascular remodeling [30, 31]. PDK1 plays a key role in regulating cardiac function and tumor metastasis by interfering with the microenvironment [32]. In addition, PDK1 is also an important regulator in arterial thrombosis [33]. Notably, this study found that PDK1 is a target for miR-764. MiR-764 overexpression significantly reduced PDK1 levels. However, PDK1 overexpression promoted the viability of H9c2 cells after H/R induction, reduced the levels of oxidative stress markers (MDA and LDH), and further inhibited the apoptosis of H9c2 cells induced by H/R. Taken together, current study revealed that PDK1 is involved in the implication of Mirt2 and miR-764 on MI.
In conclusion, this study shows that Mirt2 expression was upregulated in MI models, and knockdown of Mirt2 aggravated MI and cardiomyocyte apoptosis. In vitro studies have shown that Mirt2 can sponge-adsorb miR-764 and promote PDK1 expression, thereby inhibiting cardiomyocyte apoptosis and alleviating MI. Mirt2 knockdown can aggravate MI and cardiomyocyte apoptosis in vitro and in vivo. The current findings provide a theoretical basis for the diagnosis and treatment of clinical MI due to changes in Mirt2 levels.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Townsend N, Wilson L, Bhatnagar P, Wickramasinghe K, Rayner M, Nichols M. Cardiovascular disease in Europe: epidemiological update 2016. Eur Heart J. 2016;37:3232–45.
Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Executive summary: heart disease and stroke statistics-2012 update: a report from the American Heart Association. Circulation. 2012;125:188–97.
Joliat GR, Halkic N, Pantet O, Ben-Hamouda N. Ischemic stroke and ST-elevation myocardial infarction revealing infective endocarditis. Eur Rev Med Pharmacol Sci. 2017;21:4640–1.
Alqahtani F, Aljohani S, Tarabishy A, Busu T, Adcock A, Alkhouli M. Incidence and outcomes of myocardial infarction in patients admitted with acute ischemic stroke. Stroke. 2017;48:2931–8.
Adameova AD, Bhullar SK, Elimban V. Activation of β1-adrenoceptors may not be involved in arrhythmogenesis in ischemic heart disease. Rev. Cardiol. Med. 2018;19:97–101.
Qiu L, Zhao Q, Dai L, Zhu A, Xu X, Zhao S, et al. Long non-coding RNA DANCR alleviates hypoxia-caused H9c2 cells damage through up regulation of HIF-1alpha. Artif Cells Nanomed Biotechnol. 2020;48:533–41.
Sun T, Cheng YT, Yan LX, Krittanawong C, Qian W, Zhang HJ. LncRNA MALAT1 knockdown alleviates myocardial apoptosis in rats with myocardial ischemia-reperfusion through activating PI3K/AKT signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23:10523–31.
Ong SB, Katwadi K, Kwek XY, Ismail NI, Chinda K, Ong SG. Non-coding RNAs as therapeutic targets for preventing myocardial ischemia-reperfusion injury. Expert Opin Ther Targets. 2018;22:247–61.
Cong L, Su Y, Wei D, Qian L, Xing D, Pan J, et al. Catechin relieves hypoxia/reoxygenation-induced myocardial cell apoptosis via down-regulating lncRNA MIAT. J Cell Mol Med. 2020;24:2356–68.
Zangrando J, Zhang L, Vausort M, Maskali F, Marie PY, Wagner DR, et al. Identification of candidate long non-coding RNAs in response to myocardial infarction. BMC Genomics. 2014;15:460.
Li X, Zhou J, Huang K. Inhibition of the lncRNA Mirt1 attenuates acute myocardial infarction by suppressing NF-kappaB activation. Cell Physiol Biochem. 2017;42:1153–64.
Yan Y, Dang H, Zhang X, Wang X, Liu X. The protective role of MiR-206 in regulating cardiomyocytes apoptosis induced by ischemic injury by targeting PTP1B. Biosci Rep. 2020;40:BSR20191000.
Sygitowicz G, Maciejak-Jastrzebska A, Sitkiewicz D. MicroRNAs in the development of left ventricular remodeling and postmyocardial infarction heart failure. Pol Arch Intern Med. 2020;130:59–65.
Hromadka M, Cerna V. Prognostic value of MicroRNAs in patients after myocardial infarction: a substudy of PRAGUE-18. Dis Markers. 2019;2019:2925019.
Chen Y, Chen J, Liu Y, Li S, Huang P. Plasma miR-15b-5p, miR-338-5p, and miR-764 as biomarkers for hepatocellular carcinoma. Med Sci Monit. 2015;21:1864–71.
Daliang Z, Lifang Y, Hong F, Lingling Z, Lin W, Dapeng L, et al. Netrin-1 plays a role in the effect of moderate exercise on myocardial fibrosis in rats. PloS ONE. 2019;14:e0199802.
Hu H, Xuan Y, Wang Y, Xue M, Suo F, Li X, et al. Targeted NGF siRNA delivery attenuates sympathetic nerve sprouting and deteriorates cardiac dysfunction in rats with myocardial infarction. PloS ONE. 2014;9:e95106.
Xing X, Guo S. miR-26a-5p protects against myocardial ischemia/reperfusion injury by regulating the PTEN/PI3K/AKT signaling pathway. 2020;53:e9106.
Gao J, Guo Y, Liu Y, Yan J, Zhou J, An X, et al. Protective effect of FBXL10 in myocardial ischemia reperfusion injury via inhibiting endoplasmic reticulum stress. Respir Med. 2020;161:105852.
Huang H, Qing X, Li H. Isoflurane preconditioning protects the myocardium against ischemia and reperfusion injury by upregulating GRM1 expression. Curr Neurovasc Res. 2020;17:171–6.
Shi H, Sun H, Li J, Bai Z, Wu J, Li X, et al. Systematic analysis of lncRNA and microRNA dynamic features reveals diagnostic and prognostic biomarkers of myocardial infarction. Aging. 2020;12:945–64.
Wang X, Wang L, Ma Z, Liang W, Li J, Li Y, et al. Early expressed circulating long noncoding RNA CHAST is associated with cardiac contractile function in patients with acute myocardial infarction. Int J Cardiol. 2020;302:15–20.
Li H, Xu Y, Wang G, Chen X, Liang W, Ni H. Long non-coding RNA Mirt2 relieves lipopolysaccharide-induced injury in PC12 cells by suppressing miR-429. J Physiol Biochem. 2019;75:403–13.
Du M, Yuan L, Tan X, Huang D, Wang X, Zheng Z, et al. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat Commun. 2017;8:2049.
Wei Q, Zhu R, Zhu J, Zhao R, Li M. E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC cells. Oncol Res. 2019;27:827–34.
Wojciechowska A, Braniewska A, Kozar-Kaminska K. MicroRNA in cardiovascular biology and disease. Adv Clin Exp Med. 2017;26:865–74.
Jing D, Yinzhu L, Jinjing P, Lishuang L, Guozhuan Z. Targeting ninjurin 2 by miR-764 regulates hydrogen peroxide (H(2)O(2))-induced neuronal cell death. Biochem Biophys Res Commun. 2018;505:1180–8.
Choi JH, Yang YR, Lee SK, Kim SH, Kim YH, Cha JY, et al. Potential inhibition of PDK1/Akt signaling by phenothiazines suppresses cancer cell proliferation and survival. Ann N Y Acad Scie. 2008;1138:393–403.
Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell. 2000;103:185–8.
Abeyrathna P, Su Y. The critical role of Akt in cardiovascular function. Vascul Pharmacol. 2015;74:38–48.
Moon G, Kim J, Min Y, Wi SM, Shim JH, Chun E, et al. Phosphoinositide-dependent kinase-1 inhibits TRAF6 ubiquitination by interrupting the formation of TAK1-TAB2 complex in TLR4 signaling. Cell Signal. 2015;27:2524–33.
Qian XJ, Li XL, Xu X, Wang X, Feng QT, Yang CJ. alpha-SMA-Cre-mediated excision of PDK1 reveals an essential role of PDK1 in regulating morphology of cardiomyocyte and tumor progression in tissue microenvironment. Pathol Biol. 2015;63:91–100.
Chen X, Zhang Y, Wang Y, Li D, Zhang L, Wang K, et al. PDK1 regulates platelet activation and arterial thrombosis. Blood. 2013;121:3718–26.
Author information
Authors and Affiliations
Contributions
FZ and QL designed the study, supervised the data collection, and analyzed the data; JL interpreted the data and prepare the paper for publication; BL and DL supervised the data collection, analyzed the data, and reviewed the draft of the paper. All authors have read and approved the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Animal experiments were undertaken according to the guide for the care and use of National Institutes of Health and approved by the Ethics Committee of Wuhan Third Hospital (Tongren Hospital of Wuhan University).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhu, F., Li, Q., Li, J. et al. Long noncoding Mirt2 reduces apoptosis to alleviate myocardial infarction through regulation of the miR-764/PDK1 axis. Lab Invest 101, 165–176 (2021). https://doi.org/10.1038/s41374-020-00504-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-020-00504-2

{kind=link}