
Abstract
Odd-skipped related 1 (Osr1) is a novel tumor suppressor gene in several cancer cell lines. Non-alcoholic steatohepatitis (NASH) is considered as a high-risk factor for hepatocellular carcinoma (HCC). This study is aimed to investigate the novel role of Osr1 in promoting the progression of hepatic steatosis to NASH. Following 12 weeks of diethylnitrosamine (DEN) and high-fat diet (HFD), wildtype (WT) and Osr1 heterozygous (Osr1+/−) male mice were examined for liver injuries. Osr1+/− mice displayed worsen liver injury with higher serum alanine aminotransferase levels than the WT mice. The Osr1+/− mice also revealed early signs of collagen deposition with increased hepatic Tgfb and Fn1 expression. There was overactivation of both JNK and NF-κB signaling in the Osr1+/− liver, along with accumulation of F4/80+ cells and enhanced hepatic expression of Il-1b and Il-6. Moreover, the Osr1+/− liver displayed hyperphosphorylation of AKT/mTOR signaling, associated with overexpression of Bcl-2. In addition, Osr1+/− and WT mice displayed differences in the DNA methylome of the liver cells. Specifically, Osr1-responsible CpG islands of Ccl3 and Pcgf2, genes for inflammation and macrophage infiltration, were further identified. Taken together, Osr1 plays an important role in regulating cell inflammation and survival through multiple signaling pathways and DNA methylation modification for NAFLD progression.
Similar content being viewed by others


Introduction
Over the past few decades, the prevalence of non-alcoholic fatty liver disease (NAFLD) has rapidly increased and affects 70–80% of the obese population. Non-alcoholic steatohepatitis (NASH) is a more severe type of NAFLD, characterized by hepatic steatosis and inflammation, with or without fibrosis [1,2,3]. Patients with NASH have an increased risk of developing hepatic fibrosis, which can progress to hepatocellular carcinoma (HCC) and cirrhosis [1,2,3]. Currently, NASH has become the leading cause of chronic liver disease in developed countries. In addition, NAFLD is also a risk factor for cardiovascular disease, independent of other metabolic factors [1,2,3]. As a consequence, it is classified as a medical condition with high therapeutic needs.
The pathogenesis of NASH is considered to be multifactorial with several pathways involved, and can be described by a “two-hit” hypothesis. The “first hit” is classified by enhanced lipogenesis and lipolysis due to insulin resistance, which can then progress to lipotoxicity accompanied by oxidative stress, lipid peroxidation, mitochondrial dysfunction, and increased hepatic inflammation, comprising the “second hit” [4,5,6]. Several signaling pathways have been found to contribute to the onset and progression of NASH. The insulin, MAPK, and JNK signaling pathways contribute to hepatic fat accumulation and cell injury in the pathogenesis of NAFLD in the rat [7,8,9,10]. The AMPK and mTOR signaling pathways are known to participate in altering metabolic homeostasis [11, 12]. In addition, the Toll-like receptor, JNK, and Nuclear factor-κB (NF-κB) signaling pathways promote the production of pro-inflammatory cytokines and chemokines, driving the innate immune system to mediate steatosis, inflammation, and fibrosis [13,14,15].
The Osr1 gene encodes a putative transcription factor containing four C2H2-type zinc finger motifs [16]. It was first reported to regulate the proliferation of cardiac precursors during embryonic heart development, and to mediate apoptosis during urogenital development [17,18,19,20,21]. An Osr1 deletion results in embryonic lethality before E14.5 [17, 19, 21]. Epigenetic regulation was found to control the expression of Osr1. Hypermethylated CpG islands associated with Osr1 transcription have been reported in lung and gastric carcinoma, and renal tumor cells [20, 22, 23]. Osr1 has recently been reported as a novel tumor suppressor gene [22, 24,25,26,27,28], as well as a potential prognostic biomarker in gastric cancer [24]. However, the role of Osr1 in the progression of NAFLD towards HCC development has yet to be determined.
In this study, a high-fat (HF) diet plus diethylnitrosamine (DEN) induced NASH model on Osr1 heterozygous male mice was adapted. To understand the role of Osr1 in NAFLD/NASH progression, hepatic fat accumulation and liver injury, along with the associated molecular differences of the Osr1 heterozygous mice versus the wildtype were evaluated.
Materials and methods
Chemicals, antibodies, and reagents
Diethylnitrosamine (DEN) was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Antibodies against IRS1, phospho-IRS-1(Ser1101), NF-κB, phospho-NF-κB, phospho-AKT(Ser473), phospho–AKT (Thr308), JNK, phospho-SAPK/JNK (Thr183/Tyr185), phospho-p38MARK (Thr180/Tyr182), phospho-p44/42 MARK (Erk1/2) (Thr202/Tyr204), mTOR, phospho-mTOR, and GAPDH were purchased from Cell Signaling Technology (USA). The bicinchoninic acid (BCA) protein assay kit and the phosphatase inhibitor tablets were purchased from Thermo Fisher Scientific (USA).
Animals
Male Osr1+/− and WT mice (C57BL/6 background), purchased from Jackson Laboratory, were treated with either chow diet (CD) or DEN and HFD from weaning to 12 weeks of age. Only male mice were included in the study due to their increased sensitivity to HFD treatment [29,30,31]. The mice were then humanely euthanized, and blood and liver samples were collected for experiments. All mouse experiments were completed according to a protocol reviewed and approved by the Institutional Animal Care and Use Committee of Texas A&M University, in compliance with the USA Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Diet and treatments
Diets were purchased from Research Diets, LLC (New Brunswick, NJ, USA). The HFD (Cat. no. D12492) had an energy density of 5.157 kcal/g (60% fat, 20% carbohydrate, and 20% protein). The fat source is composed of 92% lard and 8% soybean oil. The concentrations of vitamins, minerals, and proteins were modified to ensure that these nutrients in the HF diet were equivalent to those in the chow diet on a per kilocalorie basis. Mice were injected intraperitoneally (i.p.) with 20–25 µg/g of DEN once at 21 days of age.
Intraperitoneal injected glucose tolerance test (IPGTT)
At the end of week 12, offspring mice from each experimental group were fasted overnight for 12 h and then subjected to an IPGTT early the next morning. Glucose tolerance tests were conducted with a dose of 2.0 g/kg body weight. The tail vein blood glucose levels were measured with an automated glucometer (Bayer, Elkhart, IN) at baseline and 15, 30, 60, and 120 min after the injection.
Analysis of hepatic triglyceride (TG), serum alanine aminotransferase (ALT) activity and insulin concentration
Serums were collected and stored at −80 °C until measured. Serum ALT activity was measured using an ALT Activity Assay kit (MAK052, Sigma-Aldrich, USA) according to the manufacturer’s instructions. The plasma concentrations of insulin were determined using an insulin enzyme-linked immunosorbent assay (ELISA) commercial kit (RayBiotech, USA) according to the manufacturer’s instructions. The hepatic TG content was measured using the Triglyceride Assay Kit (Abcam, Cambridge, UK) according to the manufacturer’s instructions.
Histopathology evaluation
Mouse livers were collected, fixed in 10% buffered formalin for 48 h, and then were subjected to histopathological processing, followed by paraffin embedding. Sections were taken at 5 microns thick and at least five serial sections per sample were collected for histological analysis. Sections were subjected to hematoxylin-eosin staining for granuloma counting and steatosis evaluation, and Masson trichrome staining (Newcomer supply 9179A, USA) for collagen fiber observation.
For Immunofluorescent (IF) staining and Immunohistochemistry (IHC) staining, the slides of liver sections with 5 microns in thickness were deparaffinized with xylene before staining. First antibody Osr1, purchased from Santa Cruz (sc-376529, California) were used at concentration of 1:50, and F4/80 from ThermoFisher Scientific were used at 1:200. Secondary antibodies of IF staining used were: F(ab’)2-Goat anti-Mouse IgG-Alexa 594 (Catlog#A11020, 1:800 dilution) and Goat anti-Rabbit IgG-Alexa 488 (Catlog#A31628, 1:800 dilution) from Thermo Fisher Scientific. Chemical reagent used were DAPI (Catlog#F6057) from Sigma-Aldrich (Oakville, ON, Canada). Primary antibody incubation took place in a humidified box overnight at 4 °C. Secondary antibody incubation took place at room temperature for 30 min in a dark box. Incubation was stopped by washing the slides with PBS. After staining by DAPI, IF slides were observed by fluorescence microscope under corresponding wave length. For IHC staining, slides were treated followed the protocol of VECTASTAIN ABC-HRP Kit for IHC (Fisher NC9213702). Slides were reacted with ImmPACT DAB Peroxidase substrate (Fisher NC9567138) at the final step and scanned by ScanScope FL from Leica.
RNA extraction and RT-PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, USA), and the concentration was determined in triplicate using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Total mRNA (1ug) was amplified and reverse transcribed using ReadyScript®cDNA Synthesis Mix (Sigma-Aldrich, USA), and qPCR was performed using CFX384TM Real-Time System (BIO-RAD, USA) with the CFX Manager 3.1software. Primers used for RT-PCR analysis are shown in Supplementary Table 1.
Reduced-representation bisulfite sequencing (RRBS)
RRBS was performed on the liver samples of three WT and Osr1+/− littermate mice exposed to HFD and DEN for 12 weeks, according to the manual’s instruction of Ovation RRBS Methyl-Seq System 1–16 (NuGEN Technologies, Inc., San Carlos, CA). Briefly, liver genomic DNA was extracted using Quick-DNA Universal Kit (ZYMO Research, USA). Genomic DNA was digested with MspI for 1 h, and ligated to methylated adapters with ligation adaptor mixes and ligation enzyme mix. The reaction was then subjected to final repair using the final repair enzyme mix at 60 °C for 10 min, followed by 10 min at 70 °C. Bisulfite conversion was performed using QIAGEN EpiTect Fast DNA Bisulfite Kit (QIAGEN, USA). RRBS libraries were amplified by PCR with the amplification enzyme mix and subjected for sequencing on an Illumina HiSeq 4000 at Texas A&M AgriLife Genomics and Bioinformatics Core (College Station, TX).
Bioinformatics analysis of RRBS
Bisulfite sequencing reads from RRBS were aligned and CpG methylation was analyzed using Bismark (v0.16.3) [32]. Differential methylation regions (DMR) were analyzed using an in-house program that was developed based on machine learning algorithm, mean shift, and regularized t-test. The mean shift algorithm was used to locate the regions with densely methylated CpG sites. This customized mean shift algorithm was constructed on kernel density estimation (KDE). A Gassuain kernel with 50 bp bandwidth was used to quantify the weight of nearby points. The program then iteratively shifts to the next CpG site for identifying the nearest peak on the KDE curve. Differential methylation was assessed by a regularized t-test method as defined below.
where mt and mc are the means for the treatment and control groups, respectively, and se is the estimated standard error which is calculated as follows:
where sc2 and st2 are the estimated standard deviations for the control and treatment groups, respectively.
Differentially methylated regions (DMRs) located within 5 kb upstream to 1 kb downstream of transcription start sites were considered for further analysis for their associations with gene expression.
Statistical analysis
Differences between the WT and Osr1+/− groups were analyzed by one-way ANOVA. For the longitudinal data, such as body weight and food consumption, a linear mixed model was used for the analysis of repeated measures with each individual mouse as a random effect. All analyses were carried out by using SAS JMP software (SAS Institute Inc., Cary, NC, USA) and R statistical programming language.
Results
Osr1 is expressed in hepatocytes and macrophages in liver
Osr1 is expressed in the developing heart and the intermediate mesoderm during mouse embryonic development. Its mRNA expression is lower, but detectable in the liver [24]. Osr1 expression in the liver of WT mice was observed by IHC staining. Osr1 staining was prominently detected in two types of cells. The cytosol of hepatocytes displayed weak, but positive staining for Osr1 in a scattered format (Fig. 1a, white arrowhead), while spindle shaped, non-hepatocytes, suspected to be macrophages, displayed a strong expression of Osr1 (Fig. 1a, black arrowhead).Thus, co-IF staining was performed using anti-F4/80 and anti-Osr1. The colocalization of F4/80 and Osr1 indicated that Osr1 is highly expressed in liver macrophages (Fig. 1b).

a IHC staining of Osr1 on WT liver tissue. White arrow head indicates weak Osr1 expression in the hepatocytes. Black arrow head indicates strong Osr1 expression in liver macrophages. b Co-IF staining of Osr1, F4/80 and DAPI on WT liver tissue.
Hepatic expression of Osr1 in the Osr1 +/− mouse is about half that in the WT mouse
Western blots were performed on the liver samples of the Osr1+/− and WT male mice (Fig. 2a and b). Western blot confirmed Osr1 expression in the liver of the WT mice. As expected, the Osr1 expression level in Osr1+/− mice was only half of that in the WT liver.

a Western blot results of Osr1 in the livers of Osr1+/− and WT mice. b Quantification of Osr1 expression in the liver measured by western blots. c Body weight of Osr1+/− and WT mice during the 12-week treatment period. d Serum insulin level of Osr1+/− and WT mice exposed to HFD and DEN treatment for 12-weeks. e IPGTT results tested at 12 weeks of age, upon HFD and DEN treatment. f Area under the curve of IPGTT. Data is presented as Mean ± SE, n = 8. *P < 0.05, **P < 0.01,***P < 0.001 vs. WT group.
Osr1 downregulation neither affected body weight gain, nor glucose tolerance
The average body weight was not different between the WT and the Osr1+/− groups when offered the chow diet or the HFD and DEN treatment. However, both the WT and Osr1+/− groups continuously gained weight faster on the HFD and DEN treatment than the groups on CD (Fig. 2c). After a 12-week treatment on HFD and DEN, the serum insulin levels of the Osr1+/− group showed an increasing trend versus the WT groups, although the difference is not statistically significant (Fig. 2d). The IPGTT was also performed at the end of week 12. Consistent with the insulin levels, the fasting glucose levels of the Osr1+/− group was similar to the WT mice no matter which diet was provided (Fig. 2e). Both the Osr1+/− and WT groups responded alike to the glucose challenge (Fig. 2e, f).
Upon HFD and DEN treatment, the Osr1 +/− mice had more severe liver injury compared to the WT mice
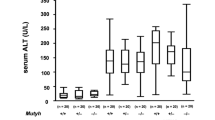
On the CD, both the Osr1+/− and WT mice displayed normal liver histology at 12 weeks (Fig. 3a, e). However, lipid drop accumulation in the livers of both the Osr1+/− and WT mice was observed following the HFD and DEN treatment (Fig. 3). The Osr1+/− mice displayed more severe liver injury, likely a borderline NASH, evidenced by obvious hepatic steatosis, a moderate number of foci, hepatocellular ballooning, and the presence of visible glycogenated nuclei (Fig. 3i). Interestingly, these histopathological changes were not observed in the WT liver (Fig. 3a–d). Hepatic triglyceride (TG) content of Osr1+/− mice was higher than the WT mice with marginal significance (P = 0.068) (Fig. 3j). Serum ALT levels were measured and found to be significantly higher in the Osr1+/− mice (Fig. 3k). In addition, important genes involved in lipogenesis, such as Chrebp and Scd-1 (Fig. 3l), and genes involved in phospholipid synthesis, such as Agpat1, were expressed more prominently in the Osr1+/− mice (Fig. 3m). These results suggest that Osr1+/− mice experienced more severe liver damage when compared to the WT mice upon HFD and DEN.

a–h Histology of the liver (HE staining). i Visible glycogen nuclei were only observed in the Osr1+/− liver upon HFD and DEN treatment. j Hepatic TG content of Osr1+/− and WT livers upon HFD and DEN treatment. k Serum ALT level of Osr1+/− and WT livers upon HFD and DEN treatment was measured as described in Methods. l and m Expression of key genes involved in lipid metabolism was measured by RT-PCR in Osr1+/− and WT livers upon HFD and DEN treatment. Data is presented as Mean ± SE, n = 4. *P < 0.05, **P < 0.01,***P < 0.001 vs. WT group.
Although obvious NASH was not observed after the 12-week treatment of HFD and DEN, a NASH CRN Scoring System was used to evaluate the NAFLD progression in the Osr1+/− and WT mice (Table 1) [33]. From 9 Osr1+/− mice 5 (56%) scored higher than “2” and 2 (22%) scored higher than “4”, while only 1 out of 8 WT mice scored higher than “2” or “4” (12.5%). The Osr1+/− mice scored marginally higher than the WT group (2.89 vs. 1.13, P = 0.0767). When one WT mouse scored “5”, considered an outlier, was removed from the group, the WT group was given an average score of 0.57, which was significantly lower than the 2.89 score of the Osr1+/− group (P = 0.0080). To be noted, the lobular inflammation score and the ballooning score of the Osr1+/− group were both significantly higher than those of the WT group.
Upon HFD and DEN treatment, the Osr1 +/− mice displayed early signs of fibrogenesis
Trichrome staining did not display any sign of fibrosis in the WT mice, while the Osr1+/− mice showed deposition of collagen fibers along perisinusoids or between the central vein and portal tracts, indicating the initiation of fibrosis (Fig. 4a). The Osr1+/− mice also displayed significantly elevated expression of Tgfβ and Fn1 (Fig. 4b). These results suggest that reduced Osr1 expression, along with a high-fat diet and DEN treatment, promotes liver injury and fibrosis onset.

a Masson-Trichrome staining of the Osr1+/− and WT livers upon HFD and DEN treatment. b Expression of key genes involved in fibrogenesis was measured by RT-PCR in Osr1+/− and WT livers upon HFD and DEN treatment. Data are presented as Mean ± SE, n = 4. *P < 0.05 vs. WT group.
Upon HFD and DEN treatment, the Osr1 +/− mice showed increased hepatic inflammation
Although the Osr1+/− mice did not respond abnormally to the glucose challenge, the relationship between the severely injured liver and desensitized insulin signaling remains in question. Insulin signaling was detected by measuring the expression levels of Irs-1 and p-Irs1in the liver (Fig. 1S). However, the expression of Isr1 nor p-Isr1 at the Ser301, Ser612, and Ser636/639 residues were affected in Osr1+/− mice in comparison to the WT.
The association of Osr1 levels and AMPK activity in the liver was then examined. Similar to the insulin signaling results, (Fig. 2S) no expression changes of AMPK-α, -β, and Acc, nor phosphorylation alterations of these subunits were observed in the Osr1+/− mice.
According to the “two hit” hypothesis, hepatic inflammation triggers the “second hit” for NAFLD progression [34]. Compared to the WT mice under HFD and DEN treatment, the Osr1+/− mice displayed increased hepatic macrophage infiltration, which could be observed by F4/80 staining (Fig. 5a) and evaluated by counting the number of F4/80 positive cells (Fig. 5b). The relationship between Osr1 and the regulation of inflammatory signaling pathways, such as NF-κB and JNK activity, was then measured in the liver. (Fig. 5c). Osr1 deficiency did not change the hepatic expression of p-JNK54/46, NF-κB and p-NF-κB under chow diet (Fig. 5c–e). However, the Osr1+/− mice significantly enhanced hepatic phosphorylation levels of both p54 JNK and p46 JNK when compared to the WT controls exposed to HFD and DEN (Fig. 5c, d). The Osr1+/− mice also exhibited a higher level of phosphorylated NF-κB (Fig. 5c–e). Consistently, the hepatic expression of Il-1β and Il-6 was significantly higher in the Osr1+/− mice exposed to HFD and DEN (Fig. 5f). There was no observable difference between the expression of Tnf-α, Ccl-2, Il-8 and Il-10 in the Osr1+/− and the WT mice.

a IF staining of F4/80 on liver tissues showed increased macrophage accumulation in the liver of Osr1+/− mice. b There were more F4/80+ cells accumulation in the Osr1+/− liver than the WT liver upon HFD and DEN treatment. c–e Western blot of p-JNK (46/54) and p-NF-κB showed higher expression in Osr1+/− mice exposed to HFD and DEN. f Expression of key genes involved in inflammation was measured by RT-PCR in Osr1+/− and WT livers upon HFD and DEN treatment. Data is presented as Mean ± SE, n = 4. *P < 0.05, **P < 0.01,***P < 0.001 vs. WT group.
Osr1 +/− mice displayed enhanced hepatic Akt/m-TOR activity and altered expression of apoptotic genes
Osr1 has been previously reported as a tumor repressor gene in gastric cancer [24]. Thus, the current study, adapting a HFD and DEN treatment, was designed to induce abnormal cell proliferation and apoptosis. Although this study failed to induce hepatocellular carcinoma due to short-term treatment, the correlation of Osr1 expression levels with cell proliferation and survival, in concurrence with the HFD and DEN treatment, was still worth investigating. Activation of the Akt/mTOR signaling pathway, which is known to regulate cell death [35,36,37], was then detected (Fig. 6). Osr1 deficiency did not change the hepatic expression of Akt, p-Akt, mTOR and p-mTOR under chow diet (Fig. 6a–c). However, exposed to HFD and DEN, the Osr1+/− mice displayed hyperphosphorylation of Akt at Ser473, but not at Ser308, without basal Akt protein level changes (Fig. 6a, b). The Osr1+/− mice also displayed increased expression of both mTOR and phosphorylated mTOR, suggesting overactivation of the Akt/mTOR signaling pathway (Fig. 6a, c).

a–c Western blot of liver tissues showed a higher expression of p-Akt at Ser473 in Osr1+/− mice, but not at Ser308, without effecting basal levels of Akt (6a & b). Higher protein expression of mTOR and p-mTOR in Osr1+/− mice exposed to HFD and DEN were detected by western blot (6a, c, & d). d Expression of key genes involved in apoptosis was measured by RT-PCR in Osr1+/− and WT livers upon HFD and DEN treatment. Expression of the anti-apoptotic gene, Bcl-2, was up-regulated in Osr1+/− mice, and pro-apoptotic genes, Bid and Casp8, were down-regulated. e–f Knockdown of Osr1 using Osr1-siRNA on HEK293T cells caused an up-regulation of Bcl-2, but not Bid or Casp8. g Expression of key genes involved in cell proliferation was measured by RT-PCR in Osr1+/− and WT liver upon HFD and DEN treatment. None of the cell proliferation-related genes were found to have expression alterations between Osr1+/− and WT mice. Data of figures is presented as Mean ± SE, n = 4. *P < 0.05 vs. WT group.
Next, the mRNA levels of the key apoptotic genes, including Bcl2, Casp3, Bid, Trp53, Casp8, and Casp9, were measured by real-time PCR. The anti-apoptotic gene, Bcl2, was markedly increased in the Osr1+/− mice, while the pro-apoptotic genes, Bid and Casp8, were downregulated (Fig. 6d). The expression of Osr1 was then knocked down in HEK293 cells by RNAi (Fig. 6e). With the lower Osr1 expression, there was significantly elevated mRNA levels of Bcl-2, while the expressions of Bid and Casp8 remained unchanged. These results suggest that Osr1 heterozygous mice display an enhanced ability of cell survival (Fig. 6f).
The effect of the diminished expression of Osr1 on cell proliferation was then addressed. The mRNA levels of genes involved in cell proliferation, such as Atm, Cdk2, Cdk4, Cdk6, Chk1, Chk2, Pai, Pten, Cip1, Cycb1, and Cycd2, were measured. The expression of these cell proliferation genes in the livers of the Osr1+/− and WT mice showed no significant changes (Fig. 6g).
Osr1 +/− mice displayed differences in the DNA methylome of their hepatocytes
Because expression of Osr1 is controlled by epigenetic regulation and hypermethylation of CpG islands [22], the role of Osr1 as a regulator of DNA methylation was explored. To address this question, both WT and Osr1+/− liver methylomes were generated at single-base resolution by RRBS. As expected, most of the hypomethylated CpGs resided in the CpG islands proximal to the transcription start sites (TSSs). The methylation sites located between 5kbp upstream of the TSS and 5kbp downstream of the transcription end sites (TESs) were subjected to bioinformatic analysis. The RRBS data identified 233 genes with 144 and 109 hyper- or hypomethylated genomic sites (CpG islands) in the Osr1+/− and WT livers, respectively. These genes were categorized according to their biological role of either metabolism, inflammation, proliferation, programmed cell death, or other by Gene Ontology (GO) analysis. The majority of the genes, 51.9% hypermethylated and 44.9% hypomethylated, were related to metabolism (Fig. 7a, b). Approximately 5.9%, 10.2%, and 8.4% hypomethylated genes were characterized as inflammatory, proliferative, and apoptotic genes, respectively (Fig. 7a). Roughly 3.81%, 17.56%, and 9.16% hypermethylated genes were classified as inflammatory, proliferative, and apoptotic, respectively (Fig. 7b).

a, b Liver methylomes were generated at single-base resolution by RRBS in both WT and Osr1+/− mice. The RRBS data identified 233 genes with 144 and 109 hyper- or hypomethylated genomic sites (CpG island) in the Osr1+/− and WT livers, respectively. These genes were categorized by the biological processes of metabolism, inflammation, proliferation, programmed cell death, and other by Gene Ontology (GO) analysis. c With RRBS analysis, several CpG sites within 5 kb upstream of TSS and 1 kb downstream of TES of Dnmt3a were found (6C). d There was a marginally significant increase of Dnmt3a expression in the Osr1+/− liver compared to the WT measured by RT-PCR. e, f Several methylation sites of Ccl3 and Pcgf2 were identified by RRBS. g Expression of Ccl3 and Pcgf2 was detected by RT-PCR. Data of figures is presented as Mean ± SE, n = 4. *P < 0.1, **P < 0.05 vs. Wildtype group.
Dnmt3a encodes for DNA (cytosine-5)-methyltransferase 3A, which catalyzes the transfer of methyl groups to CpG sites in DNA. With RRBS analysis, several CpG sites within the 5 kb upstream of TSS and 5 kb downstream of TES were found (Fig. 7c). Among these sites, a hypomethylated CpG site (P = 0.0165) located 488 bp downstream of TSS was identified. The methylation level at the site was 29.52% lower in the Osr1+/− liver (Table 2). There was a marginally significant increase of Dnmt3a expression in the Osr1+/− liver compared to the WT (Fig. 7d, P < 0.1).
DNA methylation on Ccl3 and Pcgf2 was associated with Osr1 levels
Among these genes, expression changes of Ccl3 and Pcgf2 between the WT and Osr1+/− livers were found to be associated with the methylation changes within their transcription regulatory regions. Several methylation sites of both genes were identified by RRBS (Fig. 7e, f). One genomic locus of Ccl3, located 740 bp downstream of the TSS, displayed a 24.55% decrease in the methylation level (Fig. 7e and Table 2). The expression of Ccl3 exhibited a 1-fold increase in the Osr1+/− group when compared to the WT (Fig. 7g, P < 0.1 with marginal significance). The methylation level of Pcgf2 at 1843bp downstream of the TSS was 23.53% higher in the Osr1+/− group (Fig. 7f and Table 2), which is consistent to its decreased expression in the Osr1+/− liver (Fig. 7g, P < 0.05).
Discussion
In the past three years, a new role of Osr1 as a novel tumor suppressor has been reported in various types of cancer, including lung, gastric and renal carcinoma [22, 24,25,26,27,28]. It has also been found to be a potential prognostic biomarker in gastric cancer [24]. However, its role in HCC onset and progression remains unknown. NASH is considered a high-risk factor for HCC. DEN is an acute hepatotoxin and carcinogen studied in mouse models. A single application of DEN at 10–90 mg/kg body weight is sufficient to induce hepatic tumors in rodents [38, 39]. In the current study, a single dose of DEN at 25 µg/g body was given to the C57BL/J6 mice at weaning. This protocol successfully induced premalignant lesions after 24 weeks and HCC-like tumors after 42 weeks on C57BL/J6 mice [39], demonstrating the efficiency for DEN to cause liver injury. In addition to the DEN treatment, a 60% HFD was provided to the mice for 12 weeks to promote NASH development. In general, at least 12-week of HFD treatment is required to induce severe steatohepatitis on C57BL/J6 mice [40]. In the current study, obvious pathological changes of NASH were not present, although the Osr1+/− mice displayed borderline NASH diagnostic potential (score = 2.89). This was evidenced by hepatic steatosis, with inflammation and ballooned hepatocytes [33]. In this study, the Osr1+/− mice displayed more advanced fatty liver disease, featured with more severe steatosis and hepatocellular ballooning, as well as increased macrophage infiltration. In addition, increased Tgfb and Fn1 expression and deposition of collagen fibers between the central vein and portal tracts in the Osr1+/− mice suggested the active progression towards NASH. This study provided novel evidence on the important role of Osr1 on NAFLD progression.
Development of HCC is known to undergo sequential histopathological changes, denoted as the hepatic inflammation-fibrosis-cancer (IFC) axis [41]. Numerous studies have identified NF-κB as a potential master orchestrator of the IFC axis through the regulation of immune, fibrogenic, and oncogenic mediators. Powerful activation of NF-κB signaling induces the secretion of proinflammatory cytokines, including TNF-α and IL-6, which then promotes fibrosis in the hepatic immune system [41, 42]. Strong activation of JNK has also been observed in the liver of mice on a high-fat diet (HFD), and JNK knockout mice were found to be protected from HFD-induced hepatocyte injury and steatosis [43, 44]. Similar to NF-κB signaling, the activation of the JNK pathway contributes to the production of inflammatory cytokines, such as TNF-α, IL-1, and IL-6, which further induces hepatocellular injury, inflammation, and fibrosis [45, 46]. Osr1 deficiency does not affect the NF-κB and JNK signaling under physiological conditions. However, macrophage infiltration, associated with the activation of NF-κB and JNK signaling, along with an overexpression of Tnfα and Il-1β were found in the Osr1+/− mice exposed to HFD and DEN. These mice also displayed early signs of collagen deposition, suggesting an active progression of hepatic inflammation to fibrosis. These results suggest that Osr1 participates in the regulation of hepatic inflammation via NF-κB and JNK signaling. Interestingly, a recent study on tongue squamous cell carcinoma reported that Osr1 inhibited tumor cell migration and invasion by obstructing the NF-κB pathway [25, 26]. To be noted, strong Osr1 expression was observed in the macrophages of the liver, highly suggesting an important role of Osr1 in the regulation of hepatic inflammation. Future studies using a transgenic mouse model specifically deleting Osr1 in the macrophages will disclose this potential role of Osr1 in NASH progression.
The PI3K/AKT/mTOR pathway is a major intracellular signaling pathway that regulates multiple cellular functions, including cell proliferation and apoptosis. Activation of this pivotal signaling pathway, resulting in the inhibition of apoptosis, is found in 30–50% of HCC cases [47]. Although the treatment of our study did not induce HCC due to a short experimental period, it is known to be an effective method to cause HCC and over activate oncogenic signaling pathways [38]. A universal precancerous feature is enhanced cell survival and proliferation. The transactivation of anti-apoptotic genes, such as Bcl2, provides an explanation for the inhibition of programmed cell death by NF-kB signaling [48]. An intricate crosstalk between the NF-κB and JNK signaling pathways influences the cell’s decision between life and death [48, 49]. In this study, the hyperactivation of the PI3K/AKT/mTOR, NF-κB, and JNK signaling pathways in the Osr1+/− mice indicated an increase in cell survival upon DEN and HFD treatment. These findings support the evidence that decreased Osr1 expression results in elevated expression of anti-apoptotic genes, such as Bcl2 and Bid. Being protected from programmed cell death, the injured cells may undergo self-fixation and eventually become malignant [50]. Future studies will adapt the same model with a longer experimental period to disclose the importance of Osr1 for cell survival though the regulation of multiple signaling pathways, including the PI3K/AKT/mTOR, NF-κB, and JNK, during HCC development.
Expression of Osr1 is controlled by epigenetic regulation. Methylation of CpG islands in Osr1 coding regions has been documented in multiple cancer cell lines [22, 51]. As a tumor suppressor gene, its role in epigenetic regulation may be elucidated. In this study, a marginally increased expression of Dnmt3a was reported, and a total of 253 CpG methylation sites were identified by RRBS analysis in the Osr1+/− liver. These CpG sites are located within the regulatory region of 233 different genes that play important roles in regulating cell metabolism, inflammation, proliferation, and apoptosis. In addition, two important genes, Ccl3 and Pcgf2, with their CpG methylation sites potentially under the regulation of Osr1, were identified. CCL3, also known as macrophage inflammatory protein-1α, is responsible for liver immune cell infiltration and therefore mediates experimental liver fibrosis [52]. The increased expression and hypomethylation of Ccl3 is consistent with the increased macrophage infiltration and inflammation in the Osr1+/− liver. The Pcgf2 gene negatively regulates the expression of different cytokines, chemokines, and chemokine receptors, and plays a role in decreasing immune cell migration [53]. In this study, expression of Pcgf2 was downregulated in the Osr1+/− liver, and the hypermethylated CpG site was identified. These results suggest that Osr1 influences DNA methylation, and might act as a key regulator of the mechanism driving cancer development.
In summary, Osr1 plays an important role in regulating cell survival, cell inflammation, and macrophage migration in liver. Accordingly, this study has identified Osr1 as a novel repressor gene in the progression of NAFLD/NASH.
References
Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10:330–44.
Armstrong MJ, Adams LA, Canbay A, Syn WK. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology. 2014;59:1174–97.
Lim S, Oh TJ, Koh KK. Mechanistic link between nonalcoholic fatty liver disease and cardiometabolic disorders. Int J Cardiol. 2015;201:408–14.
Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–5.
Liu W, Baker RD, Bhatia T, Zhu L, Baker SS. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life Sci. 2016;73:1969–87.
Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology. 2016;150:1769–77.
Kodama Y, Brenner DA. c-Jun N-terminal kinase signaling in the pathogenesis of nonalcoholic fatty liver disease: multiple roles in multiple steps. Hepatology. 2009;49:6–8.
Tarantino G, Caputi A. JNKs, insulin resistance and inflammation: a possible link between NAFLD and coronary artery disease. World J Gastroenterol. 2011;17:3785–94.
Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–101.
Czaja MJ. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol Metab. 2010;21:707–13.
Zeng L, Tang WJ, Yin JJ, Zhou BJ. Signal transductions and nonalcoholic fatty liver: a mini-review. Int J Clin Exp Med. 2014;7:1624–31.
Caron A, Richard D, Laplante M. The roles of mTOR complexes in lipid metabolism. Annu Rev Nutr. 2015;35:321–48.
Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–35.
Miura K, Seki E, Ohnishi H, Brenner DA. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic Fatty liver disease. Gastroenterol Res Pract. 2010;2010:362847.
Kaidashev IP. NF-kB activation as a molecular basis of pathological process by metabolic syndrome. Fiziol Zh. 2012;58:93–101.
Coulter DE, Swaykus EA, Beran-Koehn MA, Goldberg D, Wieschaus E, Schedl P. Molecular analysis of odd-skipped, a zinc finger encoding segmentation gene with a novel pair-rule expression pattern. Embo J. 1990;9:3795–804.
Wang Q, Lan Y, Cho ES, Maltby KM, Jiang R. Odd-skipped related 1 (Odd 1) is an essential regulator of heart and urogenital development. Dev Biol. 2005;288:582–94.
Xie L, Hoffmann AD, Burnicka-Turek O, Friedland-Little JM, Zhang K, Moskowitz IP. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev Cell. 2012;23:280–91.
Zhang KK, Xiang M, Zhou L, Liu J, Curry N, Heine Suner D, et al. Gene network and familial analyses uncover a gene network involving Tbx5/Osr1/Pcsk6 interaction in the second heart field for atrial septation. Hum Mol Genet. 2016;25:1140–51.
Zhang Z, Iglesias D, Eliopoulos N, El Kares R, Chu L, Romagnani P, et al. A variant OSR1 allele which disturbs OSR1 mRNA expression in renal progenitor cells is associated with reduction of newborn kidney size and function. Hum Mol Genet. 2011;20:4167–74.
Zhou L, Liu J, Olson P, Zhang K, Wynne J, Xie L. Tbx5 and Osr1 interact to regulate posterior second heart field cell cycle progression for cardiac septation. J Mol Cell Cardiol. 2015;85:1–12.
Zhang Y, Yuan Y, Liang P, Guo X, Ying Y, Shu XS, et al. OSR1 is a novel epigenetic silenced tumor suppressor regulating invasion and proliferation in renal cell carcinoma. Oncotarget. 2017;8:30008–18.
Zhao J, Liang Q, Cheung KF, Kang W, Lung RW, Tong JH, et al. Genome-wide identification of Epstein-Barr virus-driven promoter methylation profiles of human genes in gastric cancer cells. Cancer. 2013;119:304–12.
Otani K, Dong Y, Li X, Lu J, Zhang N, Xu L, et al. Odd-skipped related 1 is a novel tumour suppressor gene and a potential prognostic biomarker in gastric cancer. J Pathol. 2014;234:302–15.
Chen W, Wu K, Zhang H, Fu X, Yao F, Yang A. Corrigendum to “Odd-skipped related transcription factor 1 (OSR1) suppresses tongue squamous cell carcinoma migration and invasion through inhibiting NF-kappaB pathway” [Eur. J. Pharmacol. 15 (2018) 33-39]. Eur J Pharmacol. 2019;847:158.
Chen W, Wu K, Zhang H, Fu X, Yao F, Yang A. Odd-skipped related transcription factor 1 (OSR1) suppresses tongue squamous cell carcinoma migration and invasion through inhibiting NF-kappaB pathway. Eur J Pharmacol. 2018;839:33–9.
Du E, Lu C, Sheng F, Li C, Li H, Ding N, et al. Analysis of potential genes associated with primary cilia in bladder cancer. Cancer Manag Res. 2018;10:3047–56.
Wang Y, Lei L, Zheng YW, Zhang L, Li ZH, Shen HY, et al. Odd-skipped related 1 inhibits lung cancer proliferation and invasion by reducing Wnt signaling through the suppression of SOX9 and beta-catenin. Cancer Sci. 2018;109:1799–810.
Imajo K, Yoneda M, Kessoku T, Ogawa Y, Maeda S, Sumida Y, et al. Rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Int J Mol Sci. 2013;14:21833–57.
Zhong F, Zhou X, Xu J, Gao L. Rodent models of nonalcoholic fatty liver disease. Digestion. 2020;101:522–35.
Ballestri S, Nascimbeni F, Baldelli E, Marrazzo A, Romagnoli D, Lonardo A. NAFLD as a sexual dimorphic disease: role of gender and reproductive status in the development and progression of nonalcoholic fatty liver disease and inherent cardiovascular risk. Adv Ther. 2017;34:1291–326.
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–2.
Puri P, Sanyal AJ. Nonalcoholic fatty liver disease: definitions, risk factors, and workup. Clin Liver Dis. 2012;1:99–103.
Byrne CD, Olufadi R, Bruce KD, Cagampang FR, Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clin Sci. 2009;116:539–64.
Feng N, Luo J, Guo X. Silybin suppresses cell proliferation and induces apoptosis of multiple myeloma cells via the PI3K/Akt/mTOR signaling pathway. Mol Med Rep. 2016;13:3243–8.
Fu YF, Liu X, Gao M, Zhang YN, Liu J. Endoplasmic reticulum stress induces autophagy and apoptosis while inhibiting proliferation and drug resistance in multiple myeloma through the PI3K/Akt/mTOR signaling pathway. Oncotarget. 2017;8:61093–106.
Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk Lymphoma. 2018;59:2524–34.
Tolba R, Kraus T, Liedtke C, Schwarz M, Weiskirchen R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab Anim. 2015;49:59–69.
Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut. 2012;61:1733–43.
Zhou Y, Xie L. High fat diet mouse model in the study of nonalcoholic fatty liver disease and hepatocellular carcinoma. Am J Digest Dis. 2015;2:6.
Elsharkawy AM, Mann DA. Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology. 2007;46:590–7.
Luedde T, Schwabe RF. NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–18.
Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci USA. 2006;103:16454–9.
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–6.
Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009;137:1467–77 e5.
Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–97.
Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–83. 83 e1-11
Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7.
Wullaert A, Heyninck K, Beyaert R. Mechanisms of crosstalk between TNF-induced NF-kappaB and JNK activation in hepatocytes. Biochem Pharmacol. 2006;72:1090–101.
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87.
Daugaard I, Dominguez D, Kjeldsen TE, Kristensen LS, Hager H, Wojdacz TK, et al. Identification and validation of candidate epigenetic biomarkers in lung adenocarcinoma. Sci Rep. 2016;6:35807.
Heinrichs D, Berres ML, Nellen A, Fischer P, Scholten D, Trautwein C, et al. The chemokine CCL3 promotes experimental liver fibrosis in mice. PLoS One. 2013;8:e66106.
Wang W, Yuasa T, Tsuchiya N, Ma Z, Maita S, Narita S, et al. The novel tumor-suppressor Mel-18 in prostate cancer: its functional polymorphism, expression and clinical significance. Int J Cancer. 2009;125:2836–43.
Acknowledgements
This study is supported by grants from the National Institutes of Health (NIDDK 1R01DK112368-01 to LX and KKZ). This work is supported by the USDA National Institute of Food and Agriculture, [Hatch] project [1010406] to LX.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhou, Y., Liu, Z., Lynch, E.C. et al. Osr1 regulates hepatic inflammation and cell survival in the progression of non-alcoholic fatty liver disease. Lab Invest 101, 477–489 (2021). https://doi.org/10.1038/s41374-020-00493-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-020-00493-2
