
Abstract
Epithelial-to-mesenchymal transition (EMT) of epithelium and airway epithelial cell proliferation disorder are key events in idiopathic pulmonary fibrosis (IPF) pathogenesis. During EMT, epithelial cell adhesion molecules (EpCAM, such as E-cadherin) are downregulated, cytokeratin cytoskeletal transforms into vimentin-based cytoskeleton, and the epithelial cells acquire mesenchymal morphology. In the present study, we show abnormal upregulation of tumor protein p63 (TP63) and downregulation of miR-184 in IPF. Transforming growth factor beta 1 (TGF-β1) stimulation of BEAS-2B and A549 cell lines significantly increased the protein levels of Tp63, alpha-smooth muscle actin (α-SMA), and vimentin, but decreased EpCAM protein levels, and promoted viability of both BEAS-2B and A549 cell lines. TP63 knockdown in BEAS-2B and A549 cell lines significantly attenuated above-described TGF-β1-induced fibrotic changes. miR-184 targeted TP63 3′-UTR to inhibit Tp63 expression. miR-184 overexpression within BEAS-2B and A549 cell lines also attenuated TGF-β1-induced fibrotic changes. miR-184 overexpression attenuated bleomycin-induced pulmonary fibrosis in mice. Moreover, TP63 overexpression aggravated TGF-β1-stimulated fibrotic alterations within BEAS-2B and A549 cells and significantly reversed the effects of miR-184 overexpression, indicating miR-184 relieves TGF-β1-stimulated fibrotic alterations within BEAS-2B and A549 cells by targeting TP63, while TP63 overexpression reversed miR-184 cellular functions. In conclusion, the miR-184/TP63 axis modulates the TGF-β1-induced fibrotic alterations in epithelial cell lines and bleomycin-induced pulmonary fibrosis in mice. Therefore, these results confirm that the miR-184/TP63 axis is involved in IPF progression.
Similar content being viewed by others

Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrosing interstitial pulmonary disease associated with a high burden of disease and early death, characterized by progressive scarring of the pulmonary parenchyma that leads to progressive loss of lung function and respiratory failure and death [1]. Usual interstitial pneumonia (UIP) is one of the pathological changes of the pulmonary tissues in IPF patients, in which different cellular events have been involved, including disordered proliferation and epithelial-to-mesenchymal transition (EMT) of epithelium [2, 3]. Thus, the important role of the lung epithelium as the primary cell target of injury has been proposed [4,5,6].
The main features of EMT are decreased expression of epithelial cell adhesion molecules (EpCAM, such as E-cadherin), cytokeratin cytoskeletal transforming into vimentin-based cytoskeleton, and the acquisition of mesenchymal morphology [7, 8]. Through EMT, epithelial cells lose cell polarity, lose their attachment to the basement membrane, and at the same time become more migrative, invasive, and antiapoptotic [7, 8]. In the process of EMT and the pathogenesis of IPF, numerous profibrotic cytokines secreted via macrophages, alveolar epithelial cells, fibroblasts, and/or myofiblasts play a critical role, in particular, transforming growth factor β1 (TGF-β1) [9]. TGF-β1 induces myofibroblast phenotype, increases the collagen synthesis, and reduces matrix metalloproteinase 1 synthesis [9]. Notably, a previous study demonstrated a unique expression signature within TGF-β1-mediated airway epithelium, showing 20 specifically upregulated genes, such as tumor protein p63 (TP63, ΔNp63α as the dominant isoform), jag 1 (jagged 1) and the genes of structural proteins [10]. Inflammatory factors such as TGF-β1 stimulation could elicit p63 expression as one of the early molecular events of IPF epithelial cell abnormalities [10]. p63 expression within IPF squamous metaplasia showed to be consistent with the previous immunohistochemical study of IPF lungs which demonstrated ΔNp63 is expressed at abnormal proliferation site at the junction of bronchioles and alveoli including squamous metaplasia [11]. Thus, we speculate that p63, encoded by TP63, plays a key role in IPF pathogenesis. Searching for agents inhibiting abnormal upregulation of TP63 might be a promising strategy of improving IPF.
As previously reported, various miRNAs (microRNAs) exert an important effect on both EMT and IPF [12,13,14]. For example, let-7d expression showed to be downregulated while HMGA2 expression showed to be upregulated within TGF-β1-induced A549 alveolar epithelial cells [15]. Another miRNA, miR-200c, which belongs to the miR-200 family, could target the transcription factor ZEB1, subsequently modulating E-cadherin expression [16, 17]. miR-26a targeted the HMGA2 gene and inhibited its expression to regulate EMT, therefore, reducing the occurrence of IPF [18]. Since the partial or perfect complementarity between miRNAs and target mRNAs causes repression of translation or mRNA degradation [19], herein, the study performed bioinformatics analysis to search for miRNAs that might target TP63 to exert an effect on EMT and IPF pathogenesis.
Herein, we validated the TP63 expression by bioinformatics and experimental analyses and examined the specific functions of TP63 on TGF-β1-stimulated normal bronchial epithelial cell line (BEAS-2B) and cancerous alveolar epithelial cell line (A549). Next, we used online tools to predict miRNAs that might target TP63; among the candidate miRNAs, miR-184 has been regarded as one of the differentially expressed miRNAs in IPF. We validated the putative miR-184 binding to TP63, and determined the specific roles of miR-184 in TGF-β1-stimulated fibrotic alterations. Finally, the dynamic effects of miR-184 and TP63 were examined in TGF-β1-stimulated BEAS-2B and A549 cell lines. In summary, we provide a mechanism of miR-184/TP63 axis modulating TGF-β1-induced fibrotic changes in normal bronchial epithelial cell line (BEAS-2B) and alveolar epithelial cell line (A549).
Materials and methods
Tissue sampling
Twenty human IPF lung tissue samples obtained from the patients who underwent surgical biopsy in Hunan Province People’s Hospital. IPF was diagnosed in the patients based on clinical–radiological criteria according to guidelines and histologic confirmation of UIP pattern [20]. The healthy control tissue samples were collected from autopsy. All clinical sampling was performed under the approval of the institutional review board and ethics committee of Hunan Province People’s Hospital. All experiments were conducted in accordance with the approved guidelines. Written informed consents were obtained from all subjects. Tissues were formalin-fixed and paraffin-embedded after sampling.
Hematoxylin & eosin (H&E) and Masson staining
Tissue samples were fixed with 10% formalin and embedded in paraffin and then stained with H&E and Masson’s trichrome to examine the degree of fibrosis. The collagen was stained blue on a pale red background.
RNA fluorescence in situ hybridization (FISH)
A Specific DIG-labeled miR-184 probe was obtained from Servicebio tech (Wuhan, China). After digested with proteinase K, the tissue sections were prehybridized with a hybridization solution and then incubated with miR-184 probes in hybridization buffer overnight at 37 °C. After blocking with BSA, cells were incubated with anti-DIG-HRP for 1 h at 37 °C. Then, cells were incubated with FITC-TSA reagent for 5 min at room temperature. Cell nuclei were stained with DAPI for 5 min at room temperature. The tissue sections were imaged by fluorescence microscopy (Olympus).
Cell lines
A human bronchial epithelial cell/human pulmonary epithelial cell ling, BEAS-2B, was obtained from ATCC (CRL-9609™; Manassas, VA, USA) and cultured in BEGM kit from Lonza (CC-3170; Basel, Switzerland). A human lung adenocarcinoma cancer cell line (epithelioid), A549, was obtained from ATCC (CCL-185™) and cultured in ATCC-formulated F-12K Medium (30-2004, ATCC) supplemented with 10% FBS (Invitrogen, Carlsbad, CA, USA). All cells were cultured at 37 °C in 5% CO2.
Cell transfection
The exogenous overexpression or inhibition of miR-184 was achieved by transfection of miR-184 mimic or miR-184 inhibitor (GenePharma, Shanghai, China). Tp63 knockdown was achieved by the transfection of si-Tp63 1/2/3 (GenePharma). Tp63 overexpression was achieved by the transfection of Tp63-overexpressing vector (GenePharma). All transfections are performed using transfection agent Lipofectamine 3000 (Invitrogen).
Immunofluorescence (IF) staining for lung tissues
For the detection of alpha-smooth muscle actin (α-SMA) and Tp63 in lung tissues, tissue samples were fixed with 10% formalin and embedded and sectioned, then blocked with 100 mM ammonium chloride buffer for 10 min to minimize autofluorescence. Nonspecific binding sites were blocked with PBS containing 10% horse serum followed by an incubation with the primary antibody specific to α-SMA (14395-1-AP, Proteintech, Wuhan, China) and Tp63 (bs-0723R, BIOSS, Woburn, MA, USA) overnight at 4 °C, diluted in blocking solution. Then, the sections were incubated with Alexa Fluor® 488-conjugated or Cy5-conjugated goat antirabbit secondary antibody (Abcam) for 2 h at room temperature. DAPI (Beyotime, China) was used to stain the nucleus before capturing images. The images were acquired using a fluorescence microscope (Nikon, Japan). The green fluorescence indicates α-SMA, the red fluorescence indicates Tp63 expression, and the blue fluorescence indicates nucleus.
For the detection of α-SMA in cells, cells were seeded onto coverslips overnight and fixed with 4% paraformaldehyde PBS and incubated with anti-α-SMA (14395-1-AP, Proteintech) at 4 °C overnight. Cy5-conjugated goat antirabbit IgG H&L secondary antibody (Abcam) was incubated at room temperature for 1 h. The nuclei were stained with DAPI (Beyotime). Images were taken with a fluorescent microscope. The red fluorescence indicates α-SMA expression, and the blue fluorescence indicates nucleus.
Immunoblotting analysis for protein levels
Total proteins were extracted from transfected and/or treated cells, loaded (50 μg per lane) on 10% sodium dodecyl sulfate-polyacrylamide gels, and transferred onto PVDF membranes (Thermo Fisher Scientific), which were blocked for 2 h at 37 °C with 5% nonfat milk in Tris-buffered saline with Tween 20 (TBST) and then incubated overnight at 4 °C with primary antibodies listed below: α-SMA (14395-1-AP, Proteintech), Tp63 (bs-0723R, BIOSS), vimentin (60330-1-lg, Proteintech), EpCAM (ab71916, Abcam), Tap63 (618901, BioLegend, USA), ΔNp63 (ZA-0483, ZSGB-Bio, China), and GAPDH (T0004; Affinity, Changzhou, China). Then, the membranes were incubated with an HRP-conjugated secondary antibody for 1 h at 37 °C and then coated with ECL luminescence reagent (Perkin-Elmer Inc., Waltham, USA). GAPDH was used as an internal normalization.
Expression determined by polymerase chain reaction (PCR)-based analysis
Total RNA was extracted from transfected and/or treated cells Trizol reagent (Invitrogen) and treated with DNase I (Invitrogen) following the protocols. Next, oligo (dT) 20 and Superscript II reverse transcriptase (Invitrogen) were used for the synthesis of the first strand (cDNA). Finally, the expression of mRNA and miRNA was detected using an SYBR green PCR Master Mix (Qiagen) taking GAPDH (for mRNA expression) or RNU6B (for miRNA expression) as an endogenous control. The relative expression levels were calculated using a 2−ΔΔCT method.
Cell viability determined by cell counting kit-8 (CCK-8) analysis
Transfected and/or treated cells (1 × 104 cells/ml) were seeded on 96-well cell culture plates for 24 h and then added with 10 μl CCK-8 agent (03285; Merck, St. Louis, MI, USA) in each well followed by another 2 h incubation at 37 °C. Next, the optical density (value) was determined at the wavelength of 450 nm in the microplate reader (Bio-rad, USA).
miR-184 binding to Tp63 3′-UTR validated by luciferase reporter assay
The Tp63 3′-UTR was amplified by PCR, cloned into the downstream of the Renilla psiCHECK2 vector (Promega, Madison, WI, USA), and named wt-Tp63 3′-UTR. The seed region of the Tp63 3′-UTR containing the predicted miR-184 binding site was mutated to construct the mutant-type vector (mut-Tp63 3′-UTR). These reporter vectors were then co-transfected into 293T cells with miR-184 mimic or miR-184 inhibitor and the luciferase activity was determined 48 h after transfection using the Dual Luciferase Reporter Assay System (Promega). Renilla luciferase activity was normalized to Firefly luciferase activity for each transfected well.
In vivo pulmonary fibrosis model in mice
Male C57BL/6 mice (6 weeks of age; 18–20 g) were obtained from SLAC China (Changsha, China) and administered with bleomycin (BLM) (Sigma, USA) intratracheally at a dose of 5 mg/kg dissolved in a total of 100 μl sterile saline. The control groups were treated with 100 μl of sterile saline using the same method. For miR-184 overexpression, mice were intravenously injected with 15 nmol agomir-184 (RiboBio, China) twice a week for 4 weeks. Each experimental group contained six mice. The mice were sacrificed on day 28. The lung tissues were collected for H&E, Masson’s, and Immunohistochemistry (IHC) staining. All animal procedures were conducted in accordance with humane animal care standards approved by the Hunan Provincial People’s Hospital Medicine Ethics Committee.
IHC staining
The tissue sections were undergoing heat-induced epitope retrieval and then blocked in 3% H2O2 in methanol for 15 min at room temperature. After washed with distilled H2O and PBS, tissue sections were incubated with anti-Tp63 antibody (bs-0723R, BIOSS) at 4 °C overnight and incubated with HRP-polymer-conjugated secondary antibody at 37 °C for 1 h. The nuclei were counterstained with hematoxylin. Sections were observed and imaged under a microscope.
Data processing and statistical analysis
Data processed and analyses were performed using GraphPad software and data from three independent experiments were presented as the mean ± SD. Differences between two groups or among more groups were compared using a Student t test or one-way ANOVA followed Turkey’s test. P values of less than 0.05 are considered as statistically significant.
Results
Tp63 expression within IPF and normal control tissues according to online and experimental data
As we have mentioned, Tp63 is an early key molecule for epithelial abnormalities in IPF [10, 21]. According to online microarray expression profiles reporting the differentially expressed genes in normal and IPF lung, including GSE32537, GSE21369, and GSE2052, the expression of Tp63 was significantly upregulated in IPF lung than that within healthy normal lung (Fig. 1a). Herein, firstly, normal control tissues and IPF tissues were collected and applied for pathophysiological examinations by H&E staining and Masson staining. The distortion of the lung could be observed in both H&E and Masson’s trichrome-stained lung sections while the lung sections in control group did not show any lung damage, and normal histology with good alveolar structure was observed (Fig. 1b, c). Fibrillar collagen deposition as an indicator of lung fibrosis was determined by Masson staining; in IPF lung sections, a greater proportion of area was stained blue, indicating the increases in collagen accumulation and deposition (Fig. 1c). These observations were further confirmed by the increased α-SMA, as revealed by IF staining (Fig. 1d) and Immunoblotting (Fig. 1f). In the meantime, the protein content and the mRNA expression of TP63 were also examined. Consistent with online data, Tp63 protein levels (Fig. 1d, f) and mRNA expression (Fig. 1e) were both significantly upregulated in IPF tissue samples, compared to normal control tissues. TP63 gene encodes two major protein isoforms TAp63 and ΔNp63, which appear opposite effect on carcinogenesis [22]. In the present study, the protein levels of Tp63 isoforms TAp63 and ΔNp63 were both increased in IPF tissue samples, compared to normal control tissues (Fig. S1A). These data further suggest that the abnormal upregulation of Tp63 might affect the IPF pathogenesis.

a The expression of Tp63 in normal control tissues and IPF tissues according to GSE32537, GSE21369, and GSE2052. b The histopathology of collected samples examined by Hematoxylin & eosin (H&E) staining. c The collagen deposition status in collected samples examined by Masson staining. d The protein content and distribution of α-SMA and Tp63 were examined by immunofluorescence (IF) staining. e The expression of Tp63 was determined in 20 normal control tissues and 20 IPF tissues by real-time PCR. f The protein levels of α-SMA and Tp63 were examined in normal control tissues and IPF tissues by Immunoblotting. **P < 0.01.
Specific role of Tp63 in TGF-β1-treated human bronchial epithelial cells and epithelioid lung adenocarcinoma cancer cells
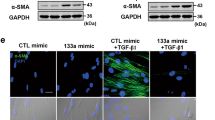
After confirmed T63 abnormal upregulation in IPF, the study further generated fibrotic cell models in human bronchial epithelial cells (BEAS-2B) and human epithelioid lung adenocarcinoma cancer cells (A549) by treating them with 0.5 ng/ml TGF-β1 for 48 h to investigate the specific effects of Tp63 on these cells. The fibrotic cell models were validated by α-SMA protein level and distribution, as examined by IF staining and Immunoblotting; the fluorescence intensity representing α-SMA (Fig. 2a) and the protein levels of α-SMA (Fig. 2b) were both dramatically upregulated under TGF-β1 stimulation. In the meantime, the protein levels of Tp63 were also remarkably enhanced in response to TGF-β1 stimulation (Fig. 2b). Similarly, the TAp63 and ΔNp63 protein levels were both increased in response to TGF-β1 stimulation (Fig. S1B).

A human bronchial epithelial cell line (BEAS-2B) and a human epithelioid lung adenocarcinoma cancer cell line (A549) was treated with 0.5 ng/ml TGF-β1 for 48 h to generate fibrotic cell models. To validate the cell models, a the protein content and distribution of α-SMA were examined by IF staining and b the protein levels of α-SMA and Tp63 were examined by Immunoblotting. c TP63 knockdown was generated in BEAS-2B and A549 cells by the transfection of si-Tp63 1/2/3 and the transfection efficiency was confirmed by real-time PCR. Si-Tp63 1 was selected for further experiments because of better transfection efficiency. Next, BEAS-2B and A549 cells were transfected with si-Tp63 in the presence or absence of 0.5 ng/ml TGF-β1 treatment and examined for (d) cell viability by CCK-8 assay and (e) the protein levels of Tp63, α-SMA, vimentin, and EpCAM by Immunoblotting. *P < 0.05; **P < 0.01, compared to the control group; ##P < 0.01, compared to si-NC (negative control) + TGF-β1 group.
To investigate the cellular effects of Tp63 on fibrotic cells, we transfected si-Tp63 1/2/3 to generate Tp63 knockdown in BEAS-2B and A549 cell lines, and performed real-time PCR to verify the transfection efficiency; we selected si-Tp63 1 for further experiments because of better transfection efficiency (Fig. 2c). Next, we transfected BEAS-2B and A549 cells with si-Tp63 with or without 0.5 ng/ml TGF-β1 treatment and examined for cell viability the protein levels of Tp63 and fibrosis markers, including α-SMA [23], vimentin [24], and EpCAM [25]. As revealed by CCK-8 assay, TGF-β1 treatment significantly promoted, while Tp63 knockdown inhibited the cell viability of both cell lines, Tp63 knockdown significantly reversed the roles of TGF-β1 treatment in cell viability (Fig. 2d). As for the fibrotic changes in cells, TGF-β1 treatment increased Tp63, α-SMA, and vimentin protein contents while decreased EpCAM protein content (Fig. 2e); on the contrary, Tp63 knockdown decreased Tp63, α-SMA, and vimentin protein contents while increased EpCAM protein content (Fig. 2e). Tp63 knockdown significantly attenuated the effects of TGF-β1 treatment upon above mentioned proteins (Fig. 2e). These data indicate that knocking down Tp63 could attenuate TGF-β1-induced fibrotic changes within BEAS-2B and A549 cell lines.
miR-184 suppresses the expression of Tp63 via targeting its 3′-UTR
Based on the essential role of miRNAs in IPF, next, the present study performed bioinformatics analysis attempting to find miRNAs related to Tp63 functions in IPF. We predicted miRNAs targeting Tp63 by two online tools, starBase V3 and Targetscan 7, and found 11 miRNAs in total (miR-140–5p, miR-216b-5p, miR-184, miR-143-3p, miR-223-3p, miR-216a-5p, miR-139-5p, miR-128-3p, miR-129-5p, miR-217, and miR-22-3p). Multiple sets of GEO chips (GSE13316, GSE32538, GSE27430, and GSE24323) were downloaded and analyzed for differentially expressed miRNAs in IPF. According to these four sets of expression profiles, 5 of the 11 miRNAs were found to be downregulated or upregulated in IPF (Table S1); among them, miR-184 was the only one miRNA that downregulated in IPF according to the four GEO chips (Figs. 3a and S2).

a A schematic diagram showing the process of miRNA selection. Two online tools, starBase V3 and Targetscan 7, were used to predict miRNAs that might target Tp63 and a total of 11 miRNAs were found. Multiple sets of GEO chips (GSE13316, GSE32538, GSE27430, and GSE24323) were downloaded and analyzed for differentially expressed miRNAs in IPF. Among significantly downregulated miRNAs in IPF according to all these 4 expression profiles, miR-184 was 1 of the 11 miRNAs that could target Tp63 and downregulated in IPF. b The expression of miR-184 was determined in normal control tissues and IPF tissues by FISH. c The expression of miR-184 was determined in 20 normal control tissues and 20 IPF tissues by real-time PCR. d The correlation of miR-184 and Tp63 expression in tissue samples was analyzed by Pearson’s correlation analysis. e BEAS-2B and A549 cells were treated with 0.5 ng/ml TGF-β1 for 48 h and examined for miR-184 expression by real-time PCR. f miR-184 overexpression or inhibition was generated in BEAS-2B and A549 cells by the transfection of miR-184 mimic or miR-184 inhibitor, as confirmed by real-time PCR. g BEAS-2B and A549 cells were transfected with miR-184 mimic or miR-184 inhibitor and examined for the protein levels of Tp63 by Immunoblotting. h Luciferase reporter assay was performed to validate the predicted binding between miR-184 and Tp63. **P < 0.01; #P < 0.05; ##P < 0.01, compared to the control group.
Before evaluating the specific effect of miR-184 on IPF, the study first examined the expression of miR-184 in tissue samples. FISH and RT-PCR results shown that the expression of miR-184 showed to be indeed remarkably downregulated within IPF tissue samples than that in normal tissue samples (Fig. 3b, c). Within tissues, there was a negative correlation between the expression of miR-184 and Tp63 (Fig. 3d). In contrary to Tp63, the expression of miR-184 showed to be considerably downregulated within BEAS-2B and A549 cell lines under TGF-β1 treatment (Fig. 3e).
Next, we transfected miR-184 mimic/inhibitor to generate miR-184 overexpression or inhibition in BEAS-2B and A549 cell lines, and performed real-time PCR to verify the transfection efficiency (Fig. 3f). As predicted, miR-184 overexpression significantly inhibited, while miR-184 inhibition promoted Tp63 protein contents within BEAS-2B and A549 cell lines (Fig. 3g). To validate the predicted miR-184 binding to Tp63, based on the “Materials and methods” section, we constructed two different types of Tp63 3′-UTR reporter vectors, wild type and mutant type. Next, we co-transfected these vectors in tool cell line 293T with miR-184 mimic or miR-184 inhibitor, and examined for the luciferase activity (Fig. 3h). As expected, wild-type Tp63 3′-UTR luciferase activity showed to be dramatically downregulated via the overexpression of miR-184 while upregulated via the inhibition of miR-184; mutating the putative miR-184 binding site could eliminate the alterations within the luciferase activity (Fig. 3h). In summary, miR-184 suppresses the expression of Tp63 via direct binding to its 3′-UTR.
Effects of miR-184 on cellular fibrotic changes under TGF-β1 treatment
After confirming miR-184 binding to Tp63 3′-UTR, the study transfected BEAS-2B and A549 cells with miR-184 mimic with or without TGF-β1 treatment and examined for the specific effects of miR-184 overexpression upon TGF-β1-induced fibrotic changes. Similar to Tp63 knockdown, miR-184 overexpression significantly inhibited TGF-β1-induced cell viability of both BEAS-2B and A549 cells (Fig. 4a). As for the fibrotic changes in cells, likewise, TGF-β1 treatment enhanced Tp63, α-SMA, and vimentin protein contents and inhibited EpCAM protein content (Fig. 4b), while miR-184 overexpression exerted an opposing effect (Fig. 4b). The overexpression of miR-184 significantly attenuated the effects of TGF-β1 treatment upon above mentioned proteins (Fig. 4b). In summary, miR-184 overexpression exerts a similar effect on TGF-β1-induced fibrotic cell lines. Since miR-184 directly binds to Tp63, miR-184 might exert its functions through targeting Tp63.

BEAS-2B and A549 cells were transfected with miR-184 mimic in the presence or absence of TGF-β1 and examined for (a) the cell viability by CCK-8 assay and (b) the protein levels of Tp63, α-SMA, vimentin, and EpCAM by Immunoblotting. *P < 0.05; **P < 0.01, compared to the control group. #P < 0.05; ##P < 0.01, compared to the NC (negative control) mimic + TGF-β1 group.
Dynamic effects of miR-184 on cellular fibrotic changes under TGF-β1 treatment
To validate the above-described speculation, the study co-transfected BEAS-2B and A549 cells with miR-184 mimic and Tp63-overexpressing vector under TGF-β1 stimulation and examined for related indexes. In contrast to miR-184 overexpression, Tp63 overexpression aggravated TGF-β1-induced fibrotic changes in both BEAS-2B and A549 cell lines by promoting cell viability, increasing Tp63, α-SMA, and vimentin protein levels, and decreasing EpCAM protein levels (Fig. 5a, b). More importantly, the overexpression of Tp63 partially reversed the roles of miR-184 overexpression, indicating that miR-184 contributes to the inducible effects of TGF-β1 treatment on fibrotic alterations within both BEAS-2B and A549 cell lines through targeting Tp63.

BEAS-2B and A549 cells were co-transfected with miR-184 mimic and Tp63-overexpressing vector and examined for (a) the cell viability by CCK-8 assay and (b) the protein levels of Tp63, α-SMA, vimentin, and EpCAM by Immunoblotting. *P < 0.05; **P < 0.01, compared to the control group. #P < 0.05; ##P < 0.01, compared to the NC (negative control) mimic + Tp63 group.
Effects of miR-184 on pulmonary fibrosis model in mice
To further validation the function of miR-184/TP63 axis in vivo, we established BLM-induced pulmonary fibrosis model. As shown by HE and Masson’s staining, a typical fibrosis changes were observed in the BLM-treated groups (Fig. 6a, b, and d). Overexpression of miR-184 by agomir-184 injection significantly attenuated the degree of pulmonary fibrosis (Fig. 6a, b, and d). Moreover, the protein levels of Tp63 were upregulated by BLM stimulation, which could be reduced by miR-184 overexpression (Fig. 6c, e). The level of miR-184 in lung tissues was decreased by BLM which could be reversed by miR-184 overexpression (Fig. 6f). These results suggested that miR-184 also plays antifibrosis function in vivo.

Mice were divided into four groups: control, bleomycin, bleomycin + agomir-NC and bleomycin + agomir-184 groups. Mice in each group were treated as described in the “Materials and methods” section. The histological changes of lung tissues were observed by (a) HE staining and (b) Masson’s staining (200×). c the protein levels of Tp63 in lung tissues were determined by IHC staining (400×). d The quantitative analysis for Masson’ staining. e The quantitative analysis for IHC staining. f the level of miR-184 in lung tissues was determined by RT-PCR. N = 6 for each experimental group. **P < 0.01, compared to the control group. ##P < 0.01, compared to the bleomycin + agomir-184 group.
Discussion
Herein, we showed Tp63 abnormal upregulation and miR-184 downregulation in IPF. TGF-β1 stimulation on BEAS-2B and A549 cell lines significantly increased the protein contents of Tp63, α-SMA, and vimentin while decreased EpCAM protein level, and promoted the cell viability of both BEAS-2B and A549 cell lines. TP63 knockdown in BEAS-2B and A549 cell lines significantly attenuated above-described TGF-β1-induced fibrotic changes. miR-184 targeted Tp63 3′-UTR to inhibit Tp63 expression. miR-184 overexpression also attenuated TGF-β1-induced fibrotic changes in vitro and BLM-induced pulmonary fibrosis in vivo. Moreover, TP63 overexpression aggravated TGF-β1-stimulated fibrotic alterations within BEAS-2B and A549 cells and significantly reversed the effects of miR-184 overexpression, indicating miR-184 relieves TGF-β1-stimulated fibrotic alterations within BEAS-2B and A549 cells through targeting Tp63, while Tp63 overexpression could reverse miR-184 cellular functions.
Clearly, interactions between cells or cell and ECM (extracellular matrix) play a crucial role in PF pathogenic mechanism. The expression of CAMs (cell adhesion molecules) is regarded as one of the basic mechanisms for the interactions between cells and microenvironment [25]. During EMT within PF, EpCAM expression is decreased, cytokeratin cytoskeletal transforms into vimentin-based cytoskeleton, and the epithelial cells acquire the mesenchymal morphology [7, 8]. As we have mentioned, a large number of factors could initiate and/or aggravate EMT to promote PF; inflammatory factors like TGF-β1 stimulation could elicit p63 expression, which is considered as one of the early molecular events of IPF epithelial cell abnormalities [10]. TP63 is known to be expressed in many epithelial stem cells [26, 27], which has several isoforms. ΔNp63 is the predominant isoform of p63 in epithelial cells, which promote EMT resulting in lung fibrosis [10]. While full-length forms of p63 (TAp63) usually exerted opposite effects to ΔNp63 on carcinogenesis [22]. In the present study, based on both online microarray expression profiles and experimental results, the mRNA expression and protein levels of Tp63 (including TAp63 and ΔNp63) showed to be dramatically upregulated within IPF tissues; in the meantime, the expression levels of fibrosis marker α-SMA was also significantly increased in IPF tissues, suggesting the potential role of TP63 in IPF. However, the specific role of Tp63 isoform during IPF process needed further investigation.
To address this speculation, the present study used two cell models, TGF-β1-stimulated human bronchial epithelial cells (BEAS-2B) and TGF-β1-stimulated human epithelioid lung adenocarcinoma cancer cells (A549), for further functional experiments. Consistent with previous studies, TGF-β1 stimulation remarkably upregulated the protein contents of fibrosis marker α-SMA in these two cell lines, indicating the fibrotic changes in both cell lines [28, 29]. As expected, after knocking down TP63 in any of the two cell lines, TGF-β1-indcued fibrotic changes, including enhanced cell proliferation, increased expression of mesenchymal markers α-SMA [23] and vimentin [24], and decreased expression of epithelial marker EpCAM [25], were all attenuated. EMT has long been thought to be a process leading to fibrosis during which epithelial cells lose their differentiated characteristics and acquire mesenchymal features, including destruction of epithelial cell morphology, overexpression of α-SMA and vimentin, loss of cell–cell adhesions through downregulation of E-cadherin, basement membrane disruption, and elevated capacity of cells to migrate and to invade [7, 30]. Thus, the suppressive effects of TP63 knockdown on TGF-β1-indcued fibrotic changes indicate the critical role of TP63 in epithelial cell EMT and PF.
IPF could be regulated by multiple factors, including miRNAs, a series of short noncoding RNAs. The expression of about 10% of the miRNAs is obviously different in lungs with IPF. Among the significantly downregulated miRNAs are members of let-7, miR-29, and miR-30 families as well as miR-17∼92 cluster among the upregulated miR-155 and miR-21 [12]. The decrease in the members of let-7 family results in the alterations coincident with EMT (epithelial–mesenchymal transition) within pulmonary epithelium both in vitro and in vivo, while mir-21 inhibition regulates fibrosis within PF BLM model [12]. Mechanically, the partial or perfect complementarity between miRNAs and target mRNAs causes repression of translation or mRNA degradation [19]. Based on the observations that TP63 knockdown attenuates TGF-β1-indcued fibrotic changes in two epithelial cell lines, herein, we detected the expression pattern of online microarray profiles for miRNAs which could be related to IPF and might target TP63, and miR-184 was selected. Further experimental results confirmed that miR-184 binds to TP63 to suppress its expression.
The effect of miR-184 on TGF-β-induced fibrotic changes has been reported. In TGF-β1-stimulated human lens epithelial cells, lentivirus-miRNA-184 infection could maintain the expression of epithelial marker gene CDH1 while reduce the expression of mesenchymal stem cell marker genes VIM and α-SMA [31]. Herein, we also observed miR-184 overexpression attenuated BLM-induced pulmonary fibrosis in mice. Moreover, forced miR-184 overexpression within two epithelial cells significantly inhibited TGF-β1-induced cell proliferation, reduced TGF-β1-induced vimentin and α-SMA protein levels, and increased TGF-β1-suppressed EpCAM protein level, indicating that miR-184 overexpression also attenuates TGF-β1-induced fibrotic changes in two epithelial cell lines. Interestingly, miR-184 overexpression inhibited TGF-β1-induced upregulation of TP63. We already confirmed miR-184 targets Tp63 to inhibit its expression; next, we further investigated whether miR-184 attenuates TGF-β1-induced fibrotic changes through targeting TP63. Under TGF-β1 stimulation, TP63 overexpression and miR-184 overexpression exerted opposite effects upon TGF-β1-stimulated fibrotic alterations within both epithelial cell lines; more importantly, the roles of miR-184 overexpression showed to be obviously reversed by the overexpression of its downstream target TP63. In summary, the suppressive roles of miR-184 overexpression in TGF-β1-stimulated fibrotic alterations are mediated via TP63.
In conclusion, the miR-184/TP63 axis modulates the TGF-β1 -induced fibrotic alterations in epithelial cell lines and BLM-induced pulmonary fibrosis in mice. Therefore, these results confirmed that miR-184/TP63 axis is involved in IPF progression.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
References
Bradley B, Branley HM, Egan JJ, Greaves MS, Hansell DM, Harrison NK, et al. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63:v1–58.
American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161:646–64.
Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377–82.
Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–5.
Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc. 2006;3:364–72.
Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–32.
Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Investig. 2003;112:1776–84.
O’Connor JW, Gomez EW. Biomechanics of TGFbeta-induced epithelial-mesenchymal transition: implications for fibrosis and cancer. Clin Transl Med. 2014;3:23.
Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–11.
Murata K, Ota S, Niki T, Goto A, Li CP, Ruriko UM, et al. p63—Key molecule in the early phase of epithelial abnormality in idiopathic pulmonary fibrosis. Exp Mol Pathol. 2007;83:367–76.
Chilosi M, Poletti V, Murer B, Lestani M, Cancellieri A, Montagna L, et al. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: the role of deltaN-p63. Lab Investig. 2002;82:1335–45.
Pandit KV, Milosevic J, Kaminski N. MicroRNAs in idiopathic pulmonary fibrosis. Transl Res. 2011;157:191–9.
Li P, Li J, Chen T, Wang H, Chu H, Chang J, et al. Expression analysis of serum microRNAs in idiopathic pulmonary fibrosis. Int J Mol Med. 2014;33:1554–62.
Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2015;165:48–60.
Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–9.
Hilmarsdottir B, Briem E, Bergthorsson JT, Magnusson MK, Gudjonsson T. Functional role of the microRNA-200 family in breast morphogenesis and neoplasia. Genes. 2014;5:804–20.
Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–4.
Liang H, Gu Y, Li T, Zhang Y, Huangfu L, Hu M, et al. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis. 2014;5:e1238.
Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–6.
Lynch JP 3rd, Fishbein MC, Saggar R, Zisman DA, Belperio JA. Idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2007;1:377–89.
Jonsdottir HR, Arason AJ, Palsson R, Franzdottir SR, Gudbjartsson T, Isaksson HJ, et al. Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab Investig. 2015;95:1418–28.
Petitjean A, Hainaut P, Caron de Fromentel C. TP63 gene in stress response and carcinogenesis: a broader role than expected. Bull Cancer. 2006;93:E126–35.
Maher TM, Wells AU, Laurent GJ. Idiopathic pulmonary fibrosis: multiple causes and multiple mechanisms? Eur Respir J. 2007;30:835–9.
Morbini P, Inghilleri S, Campo I, Oggionni T, Zorzetto M, Luisetti M. Incomplete expression of epithelial-mesenchymal transition markers in idiopathic pulmonary fibrosis. Pathol Res Pract. 2011;207:559–67.
Paine R 3rd, Ward PA. Cell adhesion molecules and pulmonary fibrosis. The Am J Med. 1999;107:268–79.
Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18:8–23.
Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. 2010;3:545–56.
Qin XJ, Zhang GS, Zhang X, Qiu ZW, Wang PL, Li YW, et al. Protein tyrosine phosphatase SHP2 regulates TGF-beta1 production in airway epithelia and asthmatic airway remodeling in mice. Allergy. 2012;67:1547–56.
Weng J, Chen H, Wu H, Tu M, Wang Z, Chen D, et al. Amiodarone induces epithelial-mesenchymal transition in A549 cells via activation of TGF-beta1. Drug Chem Toxicol. 2020;43:415–22.
Hills CE, Squires PE. The role of TGF-beta and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011;22:131–9.
Xu Q, Li Z, Zhang H. The influence of lentivirus-miRNA-184 on epithelial-mesenchcymal transition of human lens epithelial cells in vitro. Zhonghua Yan Ke Za Zhi. 2015;51:295–300.
Funding
This work was supported by the grants from the Changsha Science and Technology Program, Science and Technology Support Foundation for Social Development in 2015 (k1508037-31) and Project of Scientific Research Plan of Health and Family Planning Commission of Hunan Province (B2016013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of Hunan Province People’s Hospital and with the 1964 Helsinki declaration.
Informed consent
Informed consent to participate in the study has been obtained from participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, J., Pan, C., Tang, C. et al. miR-184 targets TP63 to block idiopathic pulmonary fibrosis by inhibiting proliferation and epithelial–mesenchymal transition of airway epithelial cells. Lab Invest 101, 142–154 (2021). https://doi.org/10.1038/s41374-020-00487-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-020-00487-0
This article is cited by
-
circGRHPR inhibits aberrant epithelial-mesenchymal transformation progression of lung epithelial cells associated with idiopathic pulmonary fibrosis
Cell Biology and Toxicology (2024)
-
microRNA-184 in the landscape of human malignancies: a review to roles and clinical significance
Cell Death Discovery (2023)

{kind=link}