
Abstract
The widespread use of genome-wide diagnostic screening methods has greatly increased the frequency with which incidental (but possibly pathogenic) copy number changes affecting single genes are detected. These findings require validation to allow appropriate clinical management. Deletion variants can usually be readily validated using a range of short-read next-generation sequencing (NGS) strategies, but the characterization of duplication variants at nucleotide resolution remains challenging. This presents diagnostic problems, since pathogenicity cannot generally be assessed without knowing the structure of the variant. We have used a novel Cas9 enrichment strategy, in combination with long-read single-molecule nanopore sequencing, to address this need. We describe the nucleotide-level resolution of two problematic cases, both of whom presented with neurodevelopmental problems and were initially investigated by array CGH. In the first case, an incidental 1.7-kb imbalance involving a partial duplication of VHL exon 3 was detected. This variant was inherited from the patient’s father, who had a history of renal cancer at 38 years. In the second case, an incidental ~200-kb de novo duplication that included DMD exons 30–44 was resolved. In both cases, the long-read data yielded sufficient information to enable Sanger sequencing to define the rearrangement breakpoints, and creation of breakpoint-spanning PCR assays suitable for testing of relatives. Our Cas9 enrichment and nanopore sequencing approach can be readily adopted by molecular diagnostic laboratories for cost-effective and rapid characterization of challenging duplication-containing alleles. We also anticipate that in future this method may prove useful for characterizing acquired translocations in tumor cells, and for precisely identifying transgene integration sites in mouse models.
Similar content being viewed by others


Introduction
The ubiquitous adoption of short-read NGS platforms has transformed the availability of diagnostic tests for the analysis of Mendelian disease genes. Laboratory workflows typically rely on hybridization enrichment reagents that target the coding regions of genes of interest and can be incorporated into an automated common laboratory process that also includes production of instrument-specific sequencing libraries. Assay designs are typically constrained by sequencer capacity; instruments producing a higher yield can accommodate assays targeting a larger genomic footprint and/or sequence more patient libraries in a single run. “Population-scale” sequencers, capable of performing cheap whole genome sequencing (WGS) have been used extensively by large research programs, but they are now also being deployed in diagnostic practice [1]. Despite this shift towards WGS, which simplifies laboratory workflows by eliminating the need for target enrichment, a number of clinically relevant genomic regions are known to be intractable to analysis by short-read-sequencing [2].
Two manufacturers, Pacific Biosciences and Oxford Nanopore Technologies (ONT), currently lead in the production of long-read “third generation” sequencers. “Real-time” single-molecule sequencing is a defining characteristic of these new technologies. In the Pacific Biosciences single molecule real-time (SMRT) workflow, library preparation involves ligation of hairpin adapters to both ends of the target molecule, creating a circular ‘SMRTbellTM’ molecule. In combination with a sequencing primer and polymerase, library fragments diffuse into a nanoscale observation chamber where the incorporation of fluorescently labeled nucleotides is recorded. High per-base consensus accuracy is achieved following multiple polymerase passes around the SMRTbellTM, with shorter fragment inserts enabling a greater number of circuits. Regardless of accuracy, maximum read lengths are determined by polymerase processivity. By contrast, nanopore sequencing is defined by changes in ionic current across an isolated membrane, as DNA molecules are ratcheted through a protein pore [3]. Typically, “1D”-sequencing is performed, which results in a single pass of the DNA strand. While this restricts per-base accuracy, a read length is limited only by the size of the DNA molecule (under optimal conditions sequencing reads of up to 2 Mb have been reported).
Exploiting the long-read capabilities of third generation sequencers presents a number of technical challenges. To undertake targeted enrichment of specific genomic regions, while also maintaining large fragment lengths, various PCR-free approaches have been developed. Several of these methods are based on components of the bacterial clustered regularly interspaced short palindromic repeats (CRISPR) system. A ribonucleoprotein complex comprising a CRISPR-associated protein (such as Streptococcus pyogenes Cas9) together with two RNAs (a generic tracrRNA and a unique crRNA) is sufficient to generate target-specific double-strand breaks. After such cleavage, various selection methods have been described, ranging from pulse-field gel electrophoresis [4] to hybridization and magnetic bead capture [5]. Genomic regions with tandem repeat arrays are particularly attractive targets for PCR-free workflows, and analyses of a number of clinically relevant loci have been reported [6, 7]. One UK hospital has obtained ISO15189 diagnostic accreditation to characterise the HTT CAG repeat using nanopore sequencing, albeit using a PCR-based enrichment approach [8].
Here we describe an approach that enables targeted clinical validation of duplication sequence variants. We used locus-specific guide RNAs and Cas9 endonuclease to linearize bulk genomic DNA prior to library preparation and long-read sequencing on the ONT MinION. Our enrichment strategy exploits the sequencer’s ability to process DNA fragments bound by only a single adapter molecule, thus eliminating any requirement to perform PCR amplification. We exemplify this approach using two cases for which the duplication integration site, originally identified by array comparative genomic hybridization (aCGH), was resolved at nucleotide resolution. In each case, our long-read findings were confirmed by Sanger sequencing, allowing a facile diagnostic assay specific for the duplication breakpoint to be deployed in the extended family.
Materials and methods
DNA was isolated from peripheral blood lymphocytes using a Chemagic 360 DNA (Perkin Elmer, Waltham, MA, USA). Ethical approval for this study was given by the Leeds East Research Ethics Committee (07/H1306/113).
Diagnostic aCGH analysis for Case 1 was performed using an Illumina ISCA 8 × 60 K OligoArray (v.2.0) and analyzed using BlueFuse Multi (v.4.1) (Illumina, Inc., San Diego, CA, USA). This array provides a median resolution of 50 kb (backbone resolution is 64 kb, increasing to 3.5 kb in genic regions). Array CGH analysis performed on Case 1’s father and Case 2 was undertaken using a CytoSure Constitutional v.3 (8 × 60 K) oligo-array and was analyzed using CytoSure Interpret (v.4.6.85) (Oxford Gene Technology, Begbroke, UK).
Multiplex ligation-dependent probe amplification (MLPA) was performed using probe-set P016-C2 (targeted to the VHL locus) in addition to P034-B1 and P035-B1 (targeting the DMD locus) following the manufacturer’s protocol throughout (MRC Holland, Amsterdam, Netherlands). Three normal controls were included in the comparator group, and data analysis was undertaken using Coffalyser v.140721.1958 (www.mlpa.com).
For WGS of Case 1, 1 μg of genomic DNA was first sheared using a Covaris S2 (Covaris, Inc., MA, USA). An Illumina sequencer-compatible library was prepared using NEBNext® UltraTM reagents (New England Biolabs, Ipswich, MA, USA) following manufacturer’s protocols. The final library was sequenced on an Illumina NextSeq500 using paired-end 151-bp reads (Illumina, Inc, San Diego, CA, USA). Raw data were converted from BCL to fastq.gz format using bcl2fastq v.2.17.1.14. These data were processed using Cutadapt (v.1.9.1) [9] (for adapter trimming and read quality filtering) prior to alignment to the human reference genome (build hg19) using BWA mem (v.0.7.13) [10]. File manipulation was performed using samtools (v.1.8) [11] and Picard (v.2.8.3) (http://broadinstitute.github.io/picard).
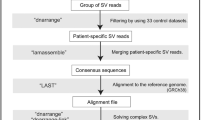
Cas9 target enrichment was performed for both Cases 1 and 2, prior to library preparation and long-read sequencing. The methodology is outlined in Fig. 1. For each locus, PAM sites for two custom Alt-R® CRISPR-Cas9 guide RNAs, designed using an online tool (https://eu.idtdna.com/), were targeted to linearize the genomic DNA on either the “+” or “−” strand (design IDs and sequences are listed in Supplementary Table 1). A crRNA-tracrRNA duplex was first created by incubating 1 μl of 100 μM crRNA, 1 μl of 100 μM Alt-R® CRISPR-Cas9 tracrRNA and 8 μl of duplex buffer (Integrated DNA Technologies, Inc., Skokie, IL, USA) at 95 °C for 5 min. For each guide RNA, ribonucleoprotein complexes (RNPs) were created by combining 6 μl of 10 × CutSmart® buffer (New England Biolabs, Ipswich, MA, USA), 48.8 μl nuclease-free H2O, 4.8 μl of the annealed crRNA-tracrRNA duplex (10 μM) and 0.4 μl of Alt-R® S.p. HiFi Cas9 V3 (62 μM) (Integrated DNA Technologies, Inc., Skokie, IL, USA), which was incubated at room temperature for 30 min. A dephosphorylation reaction comprising 3 μl of 10 × CutSmart® buffer, 3 μl nuclease-free H2O, 21 μl genomic DNA (357 ng/μl) and 3 μl Quick CIP (New England Biolabs, Ipswich, MA, USA) was incubated at 37 °C for 10 min then 80 °C for 2 min. The dephosphorylated DNA sample was halved before being linearized in separate (+) or (−) strand-specific cleavage and dA-tailing reactions that comprised 5 μl of Cas9 RNPs, 0.5 μl 10 mM dATP (New England Biolabs, Ipswich, MA, USA) and 0.5 μl Taq DNA polymerase (5,000 U/ml) (New England Biolabs, Ipswich, MA, USA). The reaction was incubated at 37 °C for 15 min and then 72 °C for 5 min. Directional bias is created at the cut site, due to the retention of bound RNPs on cleaved DNA fragments that are distal to the PAM site. Cleaved genomic samples were combined, and adapter ligation was performed in a reaction comprising 20 μl LNB (Oxford Nanopore Technologies, Oxford, UK), 10 μl T4 DNA Ligase (New England Biolabs, Ipswich, MA, USA), 3 μl nuclease-free H2O and 5 μl AMX (Oxford Nanopore Technologies, Oxford, UK), which was incubated for 10 min at room temperature. A 0.3× Agencourt AMPure XP bead cleanup (Beckman Coulter, Indianapolis, IN, USA) was performed to allow buffer exchange of the sequencer-ready library and remove excess unligated adapters and short DNA fragments. The beads were washed twice with 250 μl long fragment buffer and 12 μl of buffer EB eluted library was recovered following a 10-min incubation. A MinION FLO-MIN106 flowcell was primed for loading using SpotON priming mix (30 μl FLT and 1170 μl FLB); 800 μl was initially loaded via the priming port and incubated for 5 min. A further 200 μl was subsequently loaded prior to dropwise addition of the sequencing mix (which consisted of 12 μl of the eluted library, 25 μl of SQB, and 13 μl of LB) via the SpotON port. A 48-h sequencing run was then initiated using MinKNOW software (Oxford Nanopore Technologies, Oxford, UK).

A schematic overview of the Cas9 enrichment workflow showing the wet laboratory (a) and informatics steps (b). Cleavage reactions for (+) and (−) strand guide RNAs are performed separately to prevent interference
Offline basecalling was performed using Guppy (v.2.1.3) to convert raw data from fast5 to fastq format (https://nanoporetech.com/). Adapter sequences were trimmed from the resulting reads using Porechop (v.0.2.3) (https://github.com/rrwick/Porechop) prior to read quality filtering (-q 4) using NanoFilt (v2.2.0) [12], alignment to the human reference sequence (build hg19) using minimap2 (v.2.10) [13] and file manipulation using samtools (v.1.7) [11]. NanoStat (v.1.1.0) [12] was used to obtain read metrics. To collate the genomic information and visualize read mapping positions, a combination of the Integrative Genome Browser (v.2.4.10) [14] and UCSC genome browser (http://genome.ucsc.edu/) [15] was used. The UCSC BLAT server (https://genome.ucsc.edu/cgi-bin/hgBlat), with default parameters, was used to determine alignment coordinates for the 5′ and 3′ ends of informative duplication-spanning reads. Genome-wide read distribution was assessed by partitioning aligned reads into 500-kb windows using bedtools (v.2.28.0) [16].
For each case, a PCR assay was then developed to amplify across the integration site identified by nanopore sequencing. The primers used to amplify the duplication-containing allele for Case 1 were dTCCAGTTTTCCTTCTACTCCGA (forward) and dCTTGACTAGGCTCCGGACAA (reverse) and those for Case 2 were dTGGTTTACGGGAGGTCTGAA (forward) and dTCAGGCTGGGTTTCTTGGAA (reverse). Reagents and reaction volumes are recorded in Supplementary Table 2 and thermocycling conditions in Supplementary Table 3. Amplification products were resolved on a 1.5% Tris-borate-EDTA agarose gel. For Case 1, the predominant band was gel-slice extracted and processed using a QIAquick purification column (Qiagen, Venlo, Netherlands). An internal primer (dGGTGCGATCTCTGCTCACTA) was then used to perform Sanger sequencing on an ABI 3730 according to manufacturer’s protocols (Life Technologies Ltd, Paisley, UK). For Case 2, the PCR amplification products were sequenced directly. Sequence chromatograms were visualized using 4Peaks v.1.8 (http://www.nucleobytes.com).
Results
To allow the nucleotide-resolution analysis of genomic duplication events that are refractory to analysis by short-read sequencing, we developed a simplified method for target enrichment and long-read sequencing on the ONT MinION sequencer. Our goals were to obtain a workflow suitable for rapid deployment in a diagnostic setting. In addressing this, we aimed both to eliminate the need for laborious purification of size-selected fragments, and to avoid the use of PCR. The principle of the approach is that targeted cleavage of the duplicated region is followed by adapter ligation to the cleaved ends; background from random DNA fragmentation is reduced by a dephosphorylation reaction prior to cleavage. In our implementation, a “+” and a “−” strand-specific guide RNA, positioned within the duplicated sequence, were used to perform independent strand-specific cleavage reactions. Below, we demonstrate the utility of this method with reference to two exemplar cases, in each of which an incidental duplication variant had been identified using routine aCGH.
Case selection and initial molecular analyses
A 13-year-old boy was referred with suspected autistic spectrum disorder and learning and social communication difficulties. Array CGH was undertaken, and a small copy number gain was identified at 3p25.3, which included VHL exon 3. The estimated minimum size of the duplicated region was 1.7 kb (chr3:10,191,757-10,193,407) and the maximum size was 12.1 kb (chr3:10,191,474-10,203,584). The aCGH log2 profile is displayed in Supplementary Figure 1A. The duplication was confirmed by MLPA analysis, in which there were two VHL exon 3 probes whose dosage quotient values (1.43 and 1.57) were indicative of one extra copy of the target. Their genomic locations (01161-L00717 at chr3:10,191,523/4 and 01162-L00718 at chr3: 10,191,592/3) were upstream of the aCGH-defined minimum boundary, enabling the duplicated interval to be refined by 234 bp.
The duplication variant was subsequently shown to have been paternally inherited, with minimum and maximum sizes of 2.48 kb (chr3:10,191,660-10,194,136) and 14.92 kb (chr3:10,188,692-10,203,613). (The difference in the reported genomic interval, between the proband and his father, is due to the two different aCGH platforms; Supplementary Figure 1B.) Given the involvement of VHL, the father’s past history of renal cancer at 38 years of age was of particular concern. The histology of this had been of a mixed clear cell and papillary type. Magnetic resonance imaging of the spine showed a number of L3/L4 vertebral hemangiomas, but computerized tomography imaging of the left kidney was normal, and no retinal angiomas were detected. No other notable family history was reported.
To more closely delineate the variant identified at the VHL locus, whole genome short-read sequencing was performed in the proband. Summary sequencing metrics for these data are displayed in Supplementary Table 4. Six aligned read pairs were identified with a discordant (outward-facing) read orientation and a larger than expected library insert (>3.7 kb) (Table 1). Having mapped these data to the genome browser, we concluded that the duplication boundaries lay outside the aCGH maximum interval originally identified in the proband. We also identified only a single region of discontinuity with the reference sequence, suggesting the presence of only one integration site.
The second case, a 3-year-old girl, was referred with speech and communication delay in addition to learning difficulties. Behavioral abnormalities, including a lack of play skills, poor spatial awareness, and repetitive movements, were also noted. On aCGH analysis, a copy number gain was identified at Xp21.1, including DMD exons 30–44. The aCGH log2 profile displaying the 195-kb minimum extent (chrX:32,234,746-32,430,152) and 211-kb maximum extent (chrX:32,226,980-32,437,515) of the duplicated interval is presented in Supplementary Figure 1C. MLPA analysis confirmed that the duplication was a single copy gain and was absent from parental lymphocyte-derived DNA, consistent with it having arisen de novo.
Variant characterization by long-read sequencing
We sought to characterize the exact structure of each duplication-containing allele using the locus-specific Cas9-mediated target enrichment approach and long-read nanopore sequencing. The methodology is outlined in Fig. 1 and the performance metrics from the resulting MinION long-read sequencing runs are displayed in Table 2. We assessed the number of reads mapping to the neighborhood of each Cas9 cleavage site and estimated an approximate 500-fold enrichment above the expected background read coverage.
Reads that mapped to more than one location within the target gene were identified, and manually curated according to their likely Cas9 cleavage site (Data File 1). This analysis yielded 28 reads for Case 1 and 51 reads for Case 2. The remaining reads were not informative, either because they were derived from the normal allele or because they were too short to span the duplication integration site.
For both patients, BLAT-determined 5′ and 3′ mapping positions confirmed that the duplicated sequence contained only a single integration site and that the duplication was arranged in tandem (without inversion). For Case 1, the duplicated sequence begins within the exon 3 untranslated region and incorporates both the distal end of intron 2 and the proximal end of exon 3 (Fig. 2). Reads originating from duplication-containing alleles are mostly restricted in length, because the genomic DNA fragments are truncated due to the presence of two copies of the cleavage site. However, four VHL-aligned reads from Case 1 were identified for which there was incomplete Cas9 cleavage. The longest of these was a 53,757-bp read which mapped to the (+) strand and extended into intron 2 of the adjoining IRAK2 gene (NM_001570.3) (Fig. 2a). The longest incompletely digested (−) strand read was 11,497 bp and extended as far as VHL intron 1 (Fig. 2b).

Long-read analysis of the VHL locus (Case 1). a Reads originating from guide RNA CD.Cas9.XWFV6878.AC and b reads originating from guide RNA CD.Cas9.XWFV6878.AA. Each read alignment was split into its 5′ and 3′ component; these data can be reconciled using the displayed read ID. MinION reads mapping to the (+) strand are colored gray, and those mapping to the (−) strand are green. For each Illumina read pair, the read 1 alignment is colored purple and the read 2 alignment is turquoise. The hat symbol denotes a read’s start site. Asterisk denotes a read’s end position. Genomic coordinates refer to human reference sequence build hg19
For Case 2, the duplication begins within DMD intron 29 and extends to DMD intron 44 (Fig. 3); no incompletely digested reads were identified, which was probably due to the large genomic distance (~200 kb) between recurring Cas9 probe sites.

Long-read analysis of the DMD locus (Case 2). a Reads originating from guide RNA CD.Cas9.MGPL4222.AA and b reads originating from guide RNA CD.Cas9.MFWR5781.AA. Each read alignment was split into its 5′ and 3′ component; these data can be reconciled using the displayed read ID. MinION reads mapping to the (+) strand are colored gray, and those mapping to the (−) strand are green. The hat symbol denotes a read’s start site. Asterisk denotes a read’s end position. Genomic coordinates refer to human reference sequence build hg19
Genome-wide read distributions were assessed by determining read count across 500-kb windows. Maximum values, for both cases, were in the window containing the target locus (see Data File 2). Read counts exceeded the baseline for a number of other windows (especially those in centromeric regions) however manual inspection of the read alignments within these windows revealed low-quality mapping scores.
Validation of long-read sequencing data
The presence of RepeatMasker-identified elements near the integration sites of both variants made specific amplification of the region challenging. Despite this, we generated amplification products, which when sequenced, were consistent with the long-read data. For Case 1, we confirmed that the variant intersects two SINE family elements (AluSc8 and AluYa5). A 26-bp region of identity at either side of the integration site, which may have led to the formation of the variant, was identified (Fig. 4a). The variant integration site was also confirmed to be identical in the proband and his father. For Case 2, the intron 44 breakpoint intersects an L1MEc LINE family repeat, and a 10-bp insertion was identified at the duplication junction (Fig. 4b).

Schematic representation of the normal and duplication-containing alleles for each exemplar case. a Case 1: note the partial duplication of VHL intron 2 and the exon 3 untranslated region. The panel inset displays a sequence chromatogram that shows the beginning of the duplicated intron 2 sequence (vertical dashed red line). A region of 100% sequence identity, within intron 2, is adjacent to the duplication breakpoint (see green-colored coordinates and annotated region of surrounding homology). The start and end sites of the duplicated sequence intersect SINE family repeats. b Case 2, showing the duplicated region extending from DMD intron 29 to intron 44. A 10 bp insertion was identified at the duplication junction. Introns are colored gray and exons blue, with hatching denoting 5′ and 3′ untranslated regions. Genomic coordinates are displayed according to chromosome 3 of human reference genome build hg19
Discussion
When copy number gains are detected by genomic screening methods such as aCGH or WGS, the genomic localization and orientation of the duplicated material often remain unknown. This hinders clinical interpretation, particularly when the patient phenotype does not match the known pathologies attributable to the duplicated gene. Possible questions that arise include (but are not limited to): Is the extra genomic material inserted at another genomic locus? Is the duplicated locus rearranged in another way (e.g., inverted)? Is there loss or gain of material at the breakpoints, not detected by the aCGH? A recently reported example of an exon 45–51 DMD duplication localized to chromosome 17 (and therefore not disrupting the X-linked DMD gene) highlights the importance of nucleotide-resolution variant characterization [17].
Robust and sensitive techniques for copy number assessment include MLPA and qPCR. However, these approaches can only be applied in a targeted locus-specific way. More recently, widely-used hybridization-capture-based NGS methods (such as exome sequencing) have incorporated comparative read-depth analysis to detect copy number changes. In principle, this allows a single laboratory method to be used for detecting both sequence and copy number variants (CNVs) across a large number of genes [18]. This approach has increased diagnostic yield for many rare disease genes, for which analyses of copy number were previously infeasible due to the limited scalability of lower-throughput methods. However, the sensitivity of mutation detection by this method remains incompletely understood. The size and class (deletion vs. duplication) and genomic architecture of the variant are all likely to influence the sensitivity of detection. In addition, the disproportionately high GC content of many first exons makes them harder to capture and sequence, reducing the sensitivity of mutation detection in these regions.
As an alternative to target comparative read-depth analysis, aCGH platforms and low-coverage WGS copy number detection techniques, are enabling the identification of CNVs in a genome-wide, hypothesis-free manner [19]. This has led to an increase in the detection of incidental findings, in the form of CNVs that intersect known OMIM morbid genes [20]. Making clinical predictions in the face of such incidental findings can be difficult, often requiring more detailed laboratory analysis. While there are numerous short-read NGS approaches for characterizing genomic deletions at nucleotide resolution [21], defining the integration site and orientation of a duplicated sequence is a more challenging task. That these variants typically arise in so-called “dark” regions of the genome, defined by low-confidence alignments of short-read sequences, further complicates their analysis.
The use of Cas9 that we describe, to specifically cleave genomic DNA in the neighborhood of the duplication, offers a rapid and straightforward assay for the characterization of duplication variants identified using standard molecular genetic methods (aCGH, MLPA, or comparative read-depth analysis of targeted NGS data). The enrichment methodology is particularly well suited to use with the ONT range of sequencers, since only one end of the target DNA fragment requires a ligated adapter in order for sequencing to proceed. Furthermore, there is also no requirement for a “large-fragment” DNA isolation workflow, allowing the approach to be readily adopted by diagnostic healthcare laboratories using samples for which retrospective variant characterization may be required. We note that only standard laboratory equipment is required to perform our Cas9 enrichment protocol and less than a few hours of hands-on time are required to complete the protocol.
In Case 1, we sought to refine the risk of VHL-associated cancer in a patient identified to be an incidental carrier of an exon 3 VHL duplication. While we were able to determine that the duplicated sequence is located at the VHL locus, in tandem, in this particular case the functional and therefore clinical consequence of the rearrangement remains uncertain. Despite this, molecular characterization of the locus is now complete. Our observation that the maximal extent of the duplication, in the proband, was inaccurately determined by aCGH highlights a weakness resulting from the limited number of data points examined by this technology (as well as our inability to empirically validate the performance of each aCGH probe). With the adoption of sequence-based methods to identify copy number variation, using low-coverage WGS and comparative read-depth analysis, it is anticipated that this limitation will be overcome [19].
For Case 2, we again confirmed that the variant is an intragenic duplication (with 10-bp insertion) arranged in tandem, which is predicted in this case to retain the reading frame in the mRNA if all exons are spliced. This result also confirms that the presenting neurodevelopmental phenotype is not accounted for by the duplicated sequence insertionally disrupting a disease-associated gene elsewhere in the genome.
While Cas9 enrichment is a widely applicable methodology, each new assay requires a specific guide RNA to target the region of interest. In vitro assays using PCR amplification products or synthetic gene fragments (e.g., the Integrated DNA Technologies’ gBlocks®) as the reaction substrate, can be used to assess RNP activity. Despite this, probe specificity cannot be known without empirical data from a successful cleavage and sequencing reaction on a genomic DNA sample. To improve the likelihood of cleavage, and the yield of reads at the target locus, it may be preferable to design multiple guide RNAs located close to one another. Future improvements to guide RNA design software may also increase probe specificity, enabling either sample multiplexing or use of the lower-throughput sequencing devices (e.g., the ONT “Flongle” which is available for a tenth of the price). We achieved ~500-fold enrichment in the vicinity of the cleavage site, which is comparable to a recent report by Gilpatrick et al. (2019) [22]. Our assessment of genome-wide read distribution revealed a more or less random distribution of off-target reads. Manual inspection of windows whose read count visually exceeded the baseline identified the presence of low-quality alignments and we found no evidence of off-target Cas9 cleavage hotspots.
Early short-read library preparation workflows made extensive use of focused acoustic fragmentation, for the random shearing of genomic DNA. More recently restriction enzyme-based fragmentation has become a popular alternative. This is in part due to the lack of expensive equipment needed to setup the digestion reactions, and the reduction in sample transfer steps between proprietary shearing vessels. Adjustments to enzyme cocktails, and incubation times (resulting in a partial digest), allows accurate prediction of restriction-site-associated DNA tags and has become a popular short-read method for SNP discovery and mapping, especially for model organisms [23]. As long-read workflows continue to be developed it is likely that the utility of restriction enzyme-based approaches will be further demonstrated.
The lack of PCR, both for fragment enrichment and sequencing, is a distinctive aspect of our present workflow, and is likely to be advantageous in some applications. For example, access to genomic variants in some regions can be difficult (e.g., due to the high GC-content typical around exon 1 regions). Similarly, some insertion variants can be refractory to PCR-based methods because generic thermocycling conditions fail to amplify the modified locus (e.g., when a repeat element expands or a mobile element integrates within a coding region). Against this must be set the requirement for a larger mass of starting DNA, which will not be available for all clinical specimens.
The ability of long-read “third generation” sequencers to identify single-nucleotide and small insertion/deletion variants continues to be assessed, with several encouraging reports [24]. In our approach, the small number of reads required to determine the integration site of a targeted structural variant means that manual data analysis remains feasible. As base-calling accuracy continues to improve, we envisage that read filtering, on a per-allele (normal vs. mutant) basis, could be performed using the sequence content of each read. This would reduce ambiguity between reads derived from the “normal allele” and those reads which were too short to provide informative information about the integration site.
In other settings, we anticipate that the approach could be applied to the identification of somatic gene rearrangements in cancer. That only a single cleavage reaction is required could aid the discovery and characterization of unknown translocation partners. Further studies are however needed to determine the sensitivity of the approach when the allelic fraction of the target locus is skewed. Outside diagnostics, the method may be useful for the precise characterization of transgene integration sites in established transgenic mouse models.
The accurate molecular characterization of structural variants, in cases where the imbalance is inherited, enables a less resource-intensive assay to be designed for subsequent testing of family members. In practice, the time required to iterate through the design and optimization process is highly dependent on the genomic architecture of the target locus. The two cases reported here as exemplars proved challenging due to the number of low-complexity repeats close to the integration site. While the immediate clinical utility of the Cas9-based enrichment and ONT sequencing for the characterization of other complex variants is evident, the difficulties associated with the validation of these data will require a more thorough assessment on a case-by-case basis.
References
Davies SC. Annual report of the Chief Medical Officer 2016, generation genome. London: Department of Health; 2017. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/631043/CMO_annual_report_generation_genome.pdf.
Ebbert MTW, Jensen TD, Jansen-West K, Sens JP, Reddy JS, Ridge PG, et al. Systematic analysis of dark and camouflaged genes: disease-relevant genes hiding in plain sight. Genome Biol. 2019;20:97. https://www.ncbi.nlm.nih.gov/pubmed/31104630.
van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. The third revolution in sequencing technology. Trends Genet. 2018;34:666–81.
Gabrieli T, Sharim H, Fridman D, Arbib N, Michaeli Y, Ebenstein Y. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Res. 2018;46:e87.
Lee J, Lim H, Jang H, Hwang B, Lee JH, Cho J, et al. CRISPR-Cap: multiplexed double-stranded DNA enrichment based on the CRISPR system. Nucleic Acids Res. 2019;47:e1.
Tsai YC, Greenberg D, Powell J, Höijer I, Ameur A, Strahl M., et al. Amplification-free, CRISPR-Cas9 targeted enrichment and SMRT sequencing of repeat-expansion disease causative genomic regions. 2017. https://www.biorxiv.org/content/10.1101/203919v1.
Hafford-Tear NJ, Tsai YC, Sadan AN, Sanchez-Pintado B, Zarouchlioti C, Maher GJ, et al. CRISPR/Cas9-targeted enrichment and long-read sequencing of the Fuchs endothelial corneal dystrophy-associated TCF4 triplet repeat. Genet Med. 2019. https://doi.org/10.1038/s41436-019-0453-x.
Heger M. UK Hospital Launches Nanopore Sequencing Huntington’s Dx as Reflex Test. San Francisco: GenomeWeb; 2019. https://www.genomeweb.com/sequencing/uk-hospital-launches-nanopore-sequencing-huntingtons-dx-reflex-test#.XLCmLy3MzOY.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9.
De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 2018;34:2666–9.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34:3094–100.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2.
Lauridsen MF, Koldby KM, Krogh LN, Graakjaer J, Jensen TD, Fagerberg C, et al. A non-pathogenic duplication of DMD exon 45-51, inserted in chromosome 17, in three Danish patients. Presented at the European Human Genetics Conference, Milan (2018). Poster number P10.20D. http://www.abstractsonline.com/pp8/#!/4652/presentation/2271.
Watson CM, Crinnion LA, Berry IR, Harrison SM, Lascelles C, Antanaviciute A, et al. Enhanced diagnostic yield in Meckel-Gruber and Joubert syndrome through exome sequencing supplemented with split-read mapping. BMC Med Genet. 2016;17:1.
Hayes JL, Tzika A, Thygesen H, Berri S, Wood HM, Hewitt S, et al. Diagnosis of copy number variation by Illumina next generation sequencing is comparable in performance to oligonucleotide array comparative genomic hybridisation. Genomics 2013;102:174–81.
Newman S, Hermetz KE, Weckselblatt B, Rudd MK. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet. 2015;96:208–20.
Watson CM, Crinnion LA, Tzika A, Mills A, Coates A, Pendlebury M, et al. Diagnostic whole genome sequencing and split-read mapping for nucleotide resolution breakpoint identification in CNTNAP2 deficiency syndrome. Am J Med Genet A. 2014;164A:2649–55.
Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, et al. Targeted nanopore sequencing with Cas9 for studies of methylation, structural variants and mutations. 2019. https://www.biorxiv.org/content/10.1101/604173v1.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008;3:e3376.
Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36:338–45.
Acknowledgements
We thank the staff at Oxford Nanopore Technologies, particularly James Graham, Etienne Raimondeau, and Andy Heron for insight into the use of Cas9 reagents, and provision of early access protocols.
Funding
This work was supported by a UK Medical Research Council grant awarded to DTB (MR/M009084/1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Watson, C.M., Crinnion, L.A., Hewitt, S. et al. Cas9-based enrichment and single-molecule sequencing for precise characterization of genomic duplications. Lab Invest 100, 135–146 (2020). https://doi.org/10.1038/s41374-019-0283-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0283-0
This article is cited by
-
capTEs enables locus-specific dissection of transcriptional outputs from reference and nonreference transposable elements
Communications Biology (2023)
-
Beyond assembly: the increasing flexibility of single-molecule sequencing technology
Nature Reviews Genetics (2023)
-
Multiplex structural variant detection by whole-genome mapping and nanopore sequencing
Scientific Reports (2022)
-
Assessing the utility of long-read nanopore sequencing for rapid and efficient characterization of mobile element insertions
Laboratory Investigation (2021)
-
A novel Cas9-targeted long-read assay for simultaneous detection of IDH1/2 mutations and clinically relevant MGMT methylation in fresh biopsies of diffuse glioma
Acta Neuropathologica Communications (2020)
