
Abstract
Cell-to-cell transmission of proteopathic alpha-synuclein (α-syn) seeds is increasingly thought to underlie the progression of neurodegenerative diseases including Parkinson’s disease, dementia with Lewy bodies, multiple system atrophy, and related synucleinopathies. As such, it is important to understand the chemical and biological relationships between cells and pathological aggregates of α-syn. This brief review updates our understanding of the templated spread of α-syn pathology in neurodegenerative disease from the perspective of proteopathic α-syn seeds, including how these seeds are processed by cells as well as their effects on cellular function. Recent advances in understanding the conformations of α-syn seeds are highlighted, and the possible structural basis for the observed heterogeneity of synucleinopathies is discussed. Finally, we propose the possibility that some known risk factors for synucleinopathies may in fact potentiate the cell-to-cell transmission of α-syn pathology via imbalances in interrelated cell biological processes.
Similar content being viewed by others
Introduction to α-syn pathology and transmission
The movement disorder that eventually came to be known as Parkinson’s disease (PD) was originally reported by James Parkinson in 1817 [1], and although significant progress has been made in elucidating the molecular basis for this neurodegenerative disease, debate still exists today around the myriad proposed genotypic and phenotypic underpinnings of this devastating aging-related disease. α-Synuclein (α-syn) had been discovered as a synaptic and nuclear protein of unknown function in the 1980s [2], and was later found as a non-amyloid β component of amyloid plaques in AD patients [3]. It was not until 1997 that a PD related familial point mutation in the gene encoding α-syn, and subsequent studies showing α-syn as the major component of intracellular amyloid inclusions known as Lewy bodies (LBs), that we had our first key clue into the underlying biochemistry of PD and related synucleinopathies [4, 5]. These α-syn inclusions in PD are known as LBs and Lewy neurites (LN) and they have come to characterize PD, Parkinson’s disease dementia, and Dementia with LBs (DLB) depending on the spatial and temporal development [6]. α-Syn pathology also appears frequently in brains of Alzheimer’s disease (AD) patients both as mature LBs and LNs, wherein the later are a minority component of Aβ amyloid plaques [3, 7]. Insoluble α-syn is also found in distinct neuronal cytoplasmic inclusions and glial cytoplasmic inclusions in the aggressive neurodegenerative synucleinopathy known as multiple system atrophy (MSA) [8, 9]. Of the major breakthroughs in understanding the etiology and pathogenesis of PD and related synucleinopathies, what remains the most enduring and significant observation is the centrality of α-syn pathology and its link to cell death.
It is now well understood that α-syn pathology spreads through stereotypic patterns unique to phenotypically distinct neurodegenerative synucleinopathies [10,11,12]. Early hypotheses for direct cell-to-cell transfer of α-syn pathology originated from postmortem analysis of cohorts of Swedish patients who received fetal dopamine neuron grafts into their putamen or both the caudate nucleus and putamen. While many patients derived therapeutic benefit from the resulting compensatory dopaminergic innervation, and it was found at autopsy that these relatively young grafts could develop LB pathology after about 10 years post-implantation, suggesting that the environment of the diseased brain may support the development of LBs even in exogenous, ~10–20 year old neurons lacking naturally occurring connectivity [13,14,15,16,17]. It has since been shown that aggregated, insoluble α-syn pre-formed fibrils (PFFs) generated from recombinant α-syn or pathological α-syn isolated from human diseased brains can induce synucleinopathy-like pathological processes in numerous model systems. Inoculation of α-syn PFFs into the brains of rodents is sufficient to induce LB-like pathology characterized by phosphorylated, insoluble, intracellular α-syn inclusions [18,19,20,21,22]. Further, transduction of immortalized cells overexpressing α-syn or primary neurons with α-syn PFF suspensions results in the spontaneous development of similar LB-like aggregates [23,24,25].
To develop a complete mechanistic model for disease transmission, it is critically important to understand how cells respond to α-syn proteopathic seeds on a sub-cellular level, why certain cells and brain regions are impacted in different neurodegenerative synucleinopathies, why α-syn pathology so often is comorbid with other amyloidogenic proteins, and how α-syn inclusions may affect cell biology leading to impairment of normal biological processes, cell death, and ultimate manifestation of clinical symptoms. This review examines recent advances in the understanding of synucleinopathy transmission from the perspective of synucleinopathy brain-derived insoluble α-syn or synthetic α-syn PFF seeds and the effects they exert on neural cells. Emerging structural data for α-syn seeds will be discussed, along with hypotheses relating to selective vulnerability to α-syn pathology and modulating factors of α-syn transmission.
Life cycle of α-syn seeds in the cell
Uptake of pathogenic α-syn species into cells
Over the last several years, numerous uptake pathways and cellular receptors for α-syn fibrils have been identified. (Fig. 1a) The first specific mediator of uptake to be found was a class of glycosylated extracellular matrix proteins known as heparan sulfate proteoglycans (HSPGs) [26]. It was previously known that infectious prion protein binds to HSPGs [27, 28], and Holmes et al. [25] identified that α-syn and tau PFFs but not huntingtin PFFs bound to cell-surface HSPGs and were taken up via macropinocytosis. In a followup study testing effects of specific sulfation moieties on binding and uptake of fibrils, it was found that overall sulfation of HSPGs contributed to this process, although no specific individual sulfation moiety was required for binding and uptake of α-syn fibrils [29], suggesting a relatively non-specific nature of α-syn pff binding to HSPGs. The immune receptor Lag3 was recently described as a receptor for α-syn PFFs in neurons, although the overall contribution of Lag3 to cellular binding and uptake of fibrils appears low and the same initial binding assay identified amyloid-β precursor-like protein 1 (APLP1) and neurexin 1β as additional potential receptors for α-syn PFF internalization [30]. The proposed pathway for Lag3-dependent uptake is clathrin-mediated endocytosis (CME). Some evidence suggests that CME may play a role in α-syn monomer uptake, although the role in fibril uptake remains unclear [31]. It has also been found that the normal prion protein (PrPc) can partially mediate internalization of α-syn fibrils [32]. Indeed, the in vitro data from this study revealed that the presence of PrPC fosters the higher uptake of α-Syn amyloid fibrils, which was also confirmed in vivo in wild type (Prnp+/+) compared to PrP knock-out (Prnp−/−) mice. Remarkably, α-syn amyloids blocked the replication of the diseased version of PrPc, known as scrapie prions (PrPSc) in vitro and ex vivo. Thus, although PrPC may mediate the internalization of α-syn amyloids, PrPSc is not able to replicate in their presence, and it is notable that several case studies report that the accumulation of α-Syn amyloid deposits in Creutzfeldt-Jakob disease patients is accompanied by a longer disease course [32].

Proposed life cycle of α-syn seeds in the cell. a Multiple mechanisms have been shown to mediate α-syn fibril uptake into cells, including receptor-mediated endocytosis and fluid-phase endocytosis. Additionally, it has been demonstrated in model systems that various extracellular vesicles or tunneling nanotubes can mediate α-syn entry. b After endocytosis, fibrils are trafficked via the endocytic pathway to lysosomes where many remain sequestered (See J). If α-syn PFF uptake bypasses endocytosis, as in the case of some microvesicle, endosome, or tunneling nanotube-mediated transmission, these α-syn PFFs can likely reach the cytosol where they can immediately recruit monomeric α-syn into pathological inclusions. c If lysosomes are impaired due to genetic anomalies, age, or if the α-syn fibrils themselves disrupt and cross the lysosomal membrane, fibrils can escape to the cytosol where they d interact with and recruit monomeric α-syn into mature intracellular inclusions. e Release of pathogenic α-syn seeds is thought to be possible, although specific mechanisms are poorly understood. f α-Syn inclusions can stress the cells by various means, leading to loss of specific cellular functions and eventually cell death. This may be mediated in some contexts by (h) impaired mitochondrial function. i Cells can mount an autophagic response to inclusions, possibly disrupting mitophagy and inhibiting autophagic flux. j Internalized fibrils can remain sequestered in intact lysosomes. Also, it is possible that some aggregated α-syn species can become degraded in lysosomes via autophagy, k resulting in the production of α-syn degradation products which may be less or more pathogenic than full length α-syn PFFs. Biochemically-distinct α-syn strains (see discussion of strains below) likely exhibit different behaviors at each or any of these steps, potentially resulting in different disease manifestations
Still other mechanisms have been identified as potential pathways of synucleinopathy transmission. Extracellular vesicles derived from cells or patient CSF have been described to contain α-syn [33, 34]. When applied to neurons, these extracellular vesicles may merge with the plasma membrane and deposit their contents directly into the cytosol or are taken up by endocytosis and trafficked throughout the cell. It has been shown that DLB patient-derived extracellular vesicles originating from CSF can generate pathological inclusions in mouse brain upon intracerebral injection [33], and that exosomes isolated from DLB patient brain tissue can induce α-syn seeding in cultured cells and the mouse brain following intracerebral injection [35]. Tunneling nanotubes represent another distinct mechanism by which seeds or lysosomes containing seeds may directly transfer from cell to cell [36]. While these structures are known to form in cultured cells, their presence in tissue remains controversial and uncharacterized.
Development of a therapeutic approach aimed at reducing α-syn seed uptake in cells for the treatment of synucleinopathies remains a tantalizing prospect. Some success in reducing the development of pathology from α-syn seeds has been achieved in proof-of-concept or preclinical studies with monoclonal antibodies targeting extracellular aggregates [37, 38]. If the transmission hypothesis is true, reduction of cell-to-cell spread of pathological species could slow the progression of disease even if pathological inclusions are already formed in certain regions of the brain. Given the many distinct receptors and endocytic mechanisms that have been identified for α-syn in various model systems, it remains a considerable challenge to develop such a therapy targeted at reducing uptake alone.
Trafficking of internalized α-syn aggregates
What happens to α-syn seeds after endocytic uptake? It has been demonstrated in multiple studies that seeds are trafficked through the endocytic pathway (Fig. 1b). Cell-based α-syn PFF transduction experiments have demonstrated at least partial colocalization of internalized seeds with components of the endocytic pathway [39, 40]. A caveat of these studies is the inability to readily distinguish internalized from extracellular α-syn seeds, which may provide significant background signal. In primary neurons transduced with recombinant α-syn PFFs labeled with environmentally-responsive fluorophores, it appears that the vast majority of PFFs are acidified along the endocytic pathway and remain there for a week or more [41].
Intercellular trafficking of internalized α-syn seeds in primary neurons has been characterized by fluorescence microscopy. Freundt, et al. grew neurons in microfluidic chamber devices, effectively separating neuronal somata from processes, and microgrooves between the chambers allowed for neuronal processes to project into adjacent chambers [42]. After transduction of cells in a single chamber with α-syn PFFs against a gradient of hydrostatic pressure, internalized α-syn seeds were observed moving along axons with kinetics consistent with axoplasmic transport, specifically slow component b of axonal transport. Interestingly, seeds were observed in cells not directly exposed to α-syn PFFs, suggesting release of internalized seeds is possible. This was further characterized in a recent study comparing the behavior of α-syn PFFs to Aβ and HTTExon1, where both anterograde and retrograde motion of internalized α-syn species were detected, although the relative efficiencies of transport differed among protein aggregates [43]. Secretion of internalized α-syn was detected from intact, healthy neurons, suggesting that seeds may be transferred from cell to cell through neuronal processes. Taken together, these results strongly suggest that fibrillar α-syn in the endocytic pathway can be shuttled back to the cell surface for release. This does not, however, address how mature cytosolic pathology is released.
Endocytic escape of α-Syn species and recruitment
How pathological α-syn species initiate recruitment in the cell remains a crucial unknown. A prevailing hypothesis has been that escape of internalized seeds from the endocytic pathway is required in order to allow contact with the soluble pool of monomeric α-syn (Figs. 1c, d). Galectin-3 is a carbohydrate-binding lectin usually expressed throughout the cytosol that binds galactosides on the luminal side of the lysosomal membrane when the membrane is perturbed. Gal3 redistribution to lysosomes has been observed in cells after treatment with a high concentration of α-syn PFFs, suggesting that fibrils themselves are capable of disrupting the lysosomal membrane [44, 45]. It has also been shown in a similar system that phosphorylation of ser-129 on PFF seeds increases the degree of PFF-induced lysosomal membrane disruption [46]. Chemical impairment or disruption of lysosomes with the lipophilic weak base chloroquine increases the rate of aggregate growth in neurons, suggesting that decreased α-syn seed degradation, increased escape from damaged lysosomes, or both can potentiate recruitment of α-syn into aggregates [41, 47]. While proteolytic degradation of α-syn PFFs in the lysosome has been shown to protect cells from α-syn seed pathogenicity [47], certain truncated species of proteopathic α-syn display higher seeding propensity [48], suggesting the possibility that tuning of lysosomal protease activity under different conditions may modulate pathogenicity of the α-syn seeds contained within. After treatment of cultured primary neurons with α-syn PFFs, a small proportion of α-syn seeds have been found to colocalize with recruited endogenous α-syn, suggesting direct conversion and recruitment of monomeric synuclein by a small number of seeds within the cell is sufficient to initiate robust α-syn pathology [23, 41]. It remains unclear exactly how seeds escape from the lysosome and whether this is entirely necessary for the initial recruitment of soluble α-syn to form aggregates. Regardless, the relationship between α-syn seeds and lysosomes is emerging as an important area of study in the understanding of synucleinopathy transmission.
Dysfunction observed in α-syn pff-treated neurons
How do α-syn seeds and the resulting recruited pathological α-syn inclusions affect neuronal chemistry and biology? It has been demonstrated that recombinant α-syn PFFs are not inherently toxic to cultured neurons, as neurons lacking α-syn (α-syn KO) display no toxicity in response to transduction with the same concentration of α-syn PFFs that would generate robust p-syn inclusions and eventual cell death in WT neurons [23]. Conversely, mature, recruited intracellular α-syn aggregates resulting from α-syn PFF transduction are known have several effects on neurons. (Figs. 1f–h) Aggregates in cultured neurons can selectively impair intracellular vesicle trafficking, including the trafficking of autophagosomes, Rab5, and the receptor tyrosine kinase (RTK) TrkB which functions as the cell-surface receptor for the growth factor BDNF [49]. The potential implications of impaired RTK trafficking are not just on cell survival, but on neuronal health, synaptic dynamics and plasticity, and paracrine signaling with possible effects on the function of the brain regions affected. This could, in part, underlie synaptic dysfunction which is known to predate complete cell loss and degeneration. Any cell-specific effects or lack thereof on RTK trafficking could play a role in selective vulnerability of different neuronal populations to pathological load. It has relatedly been shown that pathological burden can dysregulate synaptic signaling in cultured primary neurons [23]. Indeed, electrophysiological changes and a decrease in postsynaptic density have been observed to result from treatment of hippocampal mouse neurons with PFFs [50]. The effects of seeded intracellular aggregates on neuronal function in cultured cells could provide insight into the mechanisms of dysfunction which precede cell loss in neurodegenerative disease.
LB-like and LN-like α-syn inclusions have been shown to interact with the autophagic-lysosomal pathway. (Figs. 1i–k) Mature intracellular α-syn inclusions resulting from α-syn PFF transduction are positive for P62 and LC3, and have been shown to inhibit autophagy in immortalized cells and primary neurons [24]. It is possible that this inhibition interferes with mitochondrial quality control via inhibition of mitophagy; interestingly both of the synucleinopathy risk factors parkin and PINK1 are known to regulate mitophagy [51]. Using a novel antibody against phosphorylated α-syn (p-Ser129), it has been suggested that partially-degraded aggregates can branch from growing p-syn inclusions and directly interact with mitochondria, raising the possibility that yet-uncharacterized α-syn species may exhibit toxic effects on mitochondria [52].
The ultimate mechanism of toxicity resulting from cytosolic α-syn inclusions remains obscure, as do exact mechanisms of selective vulnerability to this pathology. It has been observed that susceptibility to seeded aggregation resulting from α-syn PFF treatment correlates with total α-syn levels in cultured cells, and eventual toxicity correlates with the extent of the burden of α-syn pathology within cells [23, 53, 54]. Selective vulnerability of brain regions and specific cells affected in synucleinopathies likely involves a complex interplay between the α-syn seeds and the unique biology of the affected cell population.
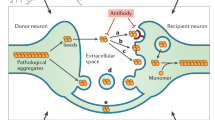
Release of proteopathic seeds
Although it is now understood that fibrils can be relayed back to the extracellular space via endocytic recycling after uptake, questions remain as to how aggregated endogenous α-syn is released from cells as a key component of the transmission paradigm. (Fig. 1e) Aggregated α-syn could be released through non-specific means including cell death, or through specific, tightly regulated cellular pathways. Folding state-dependent release of α-syn has been demonstrated under various stress conditions [55] and upon dopamine treatment [56], and cell-to-cell transfer has been observed under conditions of oxidative stress [57]. It is important to note in these studies that either the amyloid character of these species has not been definitively determined, or the released species were found to be non-amyloid oligomers. Autophagic impairment has been shown in a donor-recipient co-culture cell model to increase cell to cell transfer of α-syn, although it is not clear the extent to which impaired autophagy in donor cells leads to increased release, or if impairment of autophagy in recipient cells leads to increased deposition and retention of transferred species [58]. Unconventional secretion through an ER-dependent process chaperoned by USP19, an ER-associated deubiquitylase, has been identified for α-syn but not tau [59]. It has also been found that the HSP70 co-chaperone DNAJC5 can play a key role in secretion of aggregated α-syn species via misfolding-associated protein secretion (MAPS) [60]. Misfolded aggregates in the cytosol were observed to be shuttled to the cell surface in a DNAJC5-dependent manner, taken up by endocytosis, and degraded in the lysosome. While these experiments were conducted in immortalized cells, if the conclusions extend to neurons, this may represent a microcosm of the transmission hypothesis.
Impairment of autophagy has been demonstrated to increase secretion of α-syn species by numerous pathways. Exosomal secretion of α-syn aggregates has been observed in response to knockdown of macroautophagy component ATG5 [61]. Inhibition of lysosomes with bafilomycin leads to an upregulation of release of autophagosome-like extracellular vesicles from primary neurons as well as an increase in α-syn association with these extracellular vesicles, and PD or DLB patient-derived extracellular vesicles can induce α-syn pathology upon intracerebral injection into mice [33]. It was further demonstrated that extracellular vesicles containing α-syn are released by primary neurons under stress induced by the lipid peroxidation product 4-hydroxynonenal [34]. These extracellular vesicles were observed to be taken up by secondary neurons and trafficked within axons. These results suggest that α-syn transfer from cell-to-cell via extracellular vesicles may be at least partially involved in the spread of synucleinopathies throughout the brain.
One general challenge in interpreting many of the above experiments is the relatively low level of α-syn that cultured cells are capable of releasing. It is thus difficult to distinguish specific release of α-syn species under various conditions from non-specific release resulting from toxicity or cell death, even at low levels. Transmission of α-syn species in the brain may rely on multiple forms of release including non-specific release from cell death, these mechanisms may be present in a spectrum in any particular synucleinopathy, or the mechanism of release may possibly define the progression of individual synucleinopathies.
The varied structures of α-Syn
monomeric α-Syn
Under physiological conditions, monomeric α-syn is natively unfolded with an amphipathic n-terminus, a hydrophobic core necessary and sufficient for fibrillization, and an acidic c-terminus [62, 63]. When bound to curved anionic lipids such as synaptic vesicles, the n-terminus has been shown to adopt an α-helical conformation and interact directly albeit weakly with the lipid membrane and as such, α-syn is highly localized to synapses [2, 62, 64, 65]. The function of α-syn has been somewhat mysterious, although emerging work suggests it plays a role in synaptic vesicle trafficking, recycling, and dynamics of the exocytotic fusion pore [63, 66,67,68]. Although controversial, it has been demonstrated that α-syn can exist in solution as a stable tetrameric unit and that this association could protect cells from aggregation of α-syn into pathological species via sequestration of monomer [69]. It has further been shown through crosslinking experiments that genetic mutations of α-syn associated with PD can perturb the equilibrium between tetrameric and monomeric α-syn, and this equilibrium may underlie the propensity of α-syn aggregates to form in the cell [70]. It should be noted that there is no consensus in the field regarding the presence or physiological relevance of tetrameric α-syn species, and it is possible that synaptic vesicles or other membranes could act as a similar reservoir for α-syn monomer. The existence of α-syn tetramers notwithstanding, exposure of α-syn PFFs to neurons in vitro and in vivo leads to the rapid formation of PD-like LBs and LNs with the subsequent death of affected neurons containing α-syn aggregates.
The initial pathogenesis of synucleinopathy is still a poorly understood process thought to involve the conversion of soluble α-syn species into insoluble aggregates via spontaneous nucleation. This generation of pathological species could be caused by countless environmental or genetic factors, and is quite likely the result of interaction among multiple factors. Although these processes could differ for distinct synucleinopathies, detailed discussion is outside the scope of this review.
Conformations of pathological α-syn aggregates
Regardless of the factors driving initial nucleation and early recruitment of pathological α-syn in the body, it is known that α-syn can misfold in disease into insoluble, β-sheet rich fibrillar structures that associate together as intracellular inclusions. It is entirely possible that biochemical differences among distinct fibrillar species can underlie the different behaviors of α-syn observed across the spectra of proteopathic neurodegenerative diseases, as has been observed for pathological prion protein [71].
Progress has been made recently toward elucidation of the tertiary and quaternary structure of fibrillary α-syn generated from recombinant monomer. A seminal solid-state NMR study recently described the structure of α-syn fibrils in high resolution. The structure of the fibrillization core of α-syn PFFs generated in phosphate buffer was determined to be an in-register Greek key β-sheet motif containing several stabilizing features including a glutamine latter, an intermolecular salt bridge, several steric zippers, and hydrophobic-core interactions [72]. Using a similar technique, the same group determined that α-syn fibrils prepared in a tris buffer system displayed a remarkably similar structure, with small differences between T44 and V55 as well as the primarily unstructured n-terminus, suggesting this motif as a possible general structure for fibrils [73].
In addition, recent structural studies employing the powerful technique of cryo-EM have shown a similar, though not identical, Greek key motif within the fibril core [74,75,76]. These studies have the added benefit of observing whole α-syn fibrils, and can more closely interrogate intermolecular contacts. As such, the back-to-back paired filament structure of the α-syn fibrillization core has been observed. The cryo-EM structure of fibrils consisting of α-syn αα residues 1–121 was reported first, showing a similar Greek key motif as determined by NMR [74]. A similar study of fibrils generated from full length α-syn identified two distinct fibrillar species that originate from intertwined protofilaments [75]. Each species displays approximate 21 screw axis symmetry with right handed helical twist, but the species differ in fold and contacts between protofilaments. One fibril species defined as the “rod” is straight, displaying a similar Greek key motif as other structures recently determined. The second observed structure, a “twister,” appears twisted in projection views and consists of an ordered β-arch motif. Shortly after these reports, a third group described a similar cryo-EM analysis of full length recombinant α-syn fibrils, showing yet again a similar Greek key fold and intermolecular contacts among 2 identical protofibrils with approximate 21 screw axis symmetry, although these fibrils displayed left-handed helicity [76]. For each of the high-resolution structures summarized here, many of the familial α-syn mutations known to be risk factors for disease are found within the fibrillization core. We speculate that these different structures may be linked to the properties of biologically distinct α-syn strains.
The observation by high-resolution structural techniques of numerous similar but distinct fibrillar species originating from recombinant monomeric α-syn suggests that distinct conformations with diverse biological activities in cells are likely to be found across the spectrum of synucleinopathies. These structural differences likely give rise to distinct biochemical properties and may in fact define the behavior of α-syn strains in disease. An emerging push to deduce the structure of fibrillar α-syn isolated from different synucleinopathies will likely soon provide insight into the relationships between pathological α-syn conformation and disease.
A structural basis for seed behavior?
Could biochemically-distinct strains explain or cause divergent pathologies, either in terms of spatiotemporal spread or the presence of co-pathologies? (Fig. 2a) A study systematically mutating human α-syn (hα-syn) to mouse α-syn (mα-syn) has identified an array of behaviors of chemically distinct α-syn species, and that different species of α-syn monomer can largely inherit the intrinsic properties of seeds including conformation and pathogenicity [77]. This suggests that properties imparted to α-syn monomer by seeds may play a role in the formation of distinct strains of α-syn. Further, evidence of numerous α-syn strains with different biological properties in vitro and in vivo has recently emerged. It has been shown that serial passaging of α-syn PFFs generated with recombinant α-syn monomer leads to the generation of different strains of α-syn PFFs [78]. De novo (Strain A) α-syn PFFs recruit α-syn PD-like LBs and LNs in both primary neurons and upon intracerebral injection into mouse brain, while PFFs from later passages (Strain B) are capable of recruiting tau into neurofibrillary tangles similar to AD, alongside α-syn PD-like LBs and LNs suggesting the possibility that frequently-observed comorbid proteinopathies may result directly from the cross-seeding of tau by pathological α-syn seeds under certain conditions, and that distinct strains may define specific co-morbidities. It has also been demonstrated that the cellular environment in which α-syn seeds develop can dictate the structure and pathogenicity of these α-syn seeds [79]. Distinct properties can be imparted on α-syn fibrils by the environment in which they grow [80,81,82]. MSA brain-derived α-syn seeds have been shown to be considerably more potent than PD brain derived seeds, consistent with the aggressive nature of MSA [79, 83], and have also been characterized as remarkably stable compared to other fibrillar α-syn species [84]. Additionally, it appears that certain PD-related α-syn mutations, most notably E46K, are resistant to recruitment by MSA seeds [85].

Distinct α-syn strains may underlie the phenotypic diversity of synucleinopathies. a General schematic of α-syn strain formation. Monomeric α-syn can aggregate into conformationally-distinct pathological α-syn species as a result of spontaneous nucleation, through seeded aggregation by distinct or maturing strains, or through conversion within a specific cellular milieu. b It is possible that the observed heterogeneity of synucleinopathies results, in part, from differential effects of α-syn strains on distinct neuronal populations (Neuron A and Neuron B) and other cell types within the central nervous system such as glia. Indicated are two putative strains of α-syn seeds (blue and gray objects) differentially affecting distinct cell types (neurons and glia)
An important outstanding question is how α-syn pathology spreads to different cell populations in the brain in distinct synucleinopathies. (Fig. 2b) Differential degradation of α-syn PFFs has been characterized in different cell culture systems, where it was observed that astrocytes can degrade exogenous α-syn PFFs more efficiently than neurons under similar conditions [86]. More work is necessary to understand the basis for this differential cell-type specific processing of pathological α-syn and its subsequent spread in cells, particularly in the case of MSA GCIs.
Complex relationships exist between fibril conformation and the biological processes that the α-syn fibrils are subjected to once interacting with a cell. Given the observed difference in seeding potency among distinct α-syn species in neurons, it is entirely possible that the biochemical properties of the seeds could regulate uptake, endolysosomal degradation or escape, recruitment of soluble α-syn, and toxicity; distinct strain properties of seeds may in fact underlie the heterogeneity of synucleinopathies by responding differentially to cell populations and brain regions selectively vulnerable or selectively resilient to the pathogenic properties of different seeds. A wealth of information should be forthcoming over the next decade as these structural techniques are applied to the elucidation of tertiary and quaternary structure of pathology isolated from distinct diseases.
Possible modulators of pathological α-syn transmission: a brief overview
While it is outside the scope of this review to thoroughly discuss the vast array of genetic or environmental risk factors for development of synucleinopathies, we can hypothesize that a handful may be involved in modulating disease spread through transmission. Many of the known genetic risk factor alleles or genetic mutations that are pathogenic for synucleinopathies can be grouped into 3 categories. The first to consider are abnormalities in the α-syn gene (SNCA), the second are genes involved in lysosomal/autophagic processes, and the third are genes involved in the health of mitochondria. How do these systems interrelate, and how may α-syn or transmission of α-syn form a link among them to disease? It is known that aggregate-promoting SNCA mutations or increased expression of α-syn due to multiplication of copies of the SNCA gene can promote disease [4, 87] (Fig. 3a). Once formed, these aggregates interact with and likely disrupt autophagic processes in affected cells through substrate inhibition [24] (Fig. 3b). While it is possible that the resulting proteostatic inhibition could affect general protein quality control by impaired clearance of amyloidogenic proteins and lead to an increase in seeding events, it is also possible that resulting cellular stress could increase the release of proteopathic seeds with subsequent templated propagation of pathological α-syn [55, 58].

Mediators of α-syn pathology. a α-Syn aggregation is promoted by various genetic mutations (i.e., the A53T SNCA mutation) or increased cytosolic concentration via whole-gene SNCA duplication and triplication. b Aggregated α-syn species, as well as deficient activity of a number of lysosomal synucleinopathy risk factors (i.e., variants in the ATP13A2, GBA or CTSD genes), can interfere with autophagic/lysosomal function resulting in increased aggregation of α-syn. c Lysosomal impairment can directly or indirectly affect mitochondrial health and function, which may ultimately mediate toxicity in synucleinopathies; release of α-syn seeds upon cell death or as a regulated function of cell stress may initiate the transmission of individual α-syn seeds. Cell-to-cell transmission of pathological α-syn in diverse synucleinopathies could be mediated via imbalances in any of these processes
Of particular relevance to the transmission hypothesis are genes regulating endosomal trafficking, lysosomal function, and lysosomal integrity. Genetic anomalies in the endolysosomal pathway could accelerate the generation of pathology resulting from endocytic flux of α-syn seeds. Variants in the genes encoding lysosomal hydrolases including cathepsin D (CTSD) and glucocerebrosidase A (GBA) are known genetic risk factors risk factors for synucleinopathies, and deficiency in the production of the corresponding proteins has shown decreased lysosomal activity or integrity [88, 89]. A loss of function mutation of the gene encoding the lysosomal protein probable cation-transporting ATPase 13A2 (ATP13A2) is associated with Kufor-Rakeb syndrome, which is characterized in part by Parkinsonism [90]. Cellular studies have demonstrated that impairment of ATP13A2 leads to lysosomal failure and accumulation of α-syn [91] and this has been shown to regulate α-syn release [92]. Another interesting possible lysosomal risk factor for synucleinopathies is the lysosomal K + channel TMEM175, which has been shown to modulate lysosomal activity and integrity [93]. Importantly, variants in TMEM175 have been proposed as PD risk factors, and knockout of TMEM175 was found to perturb lysosomal function and potentiate α-syn PFF-seeded aggregation of monomeric α-syn in neurons [94]. Further, mitochondrial impairment was observed in this model, supporting a link between lysosomal function and mitochondrial health. Vacuolar protein sorting-associated protein 35 (VPS35) is known to regulate endosome maturation via retromer trafficking from late endosomes to the trans-Golgi network, which is closely tied to lysosomal function, and missense mutation of VPS35 is also a risk factor for familial PD [95]. VPS35 likely modulates autophagic turnover of α-syn by regulating the lysosomal level or activity of hydrolases such as cathepsin D [96]. Additionally, although the neuronal expression of the PD related gene LRRK2 is low, mutations in this gene are known risk factors for synucleinopathies and aberrantly regulate endolysosomal trafficking [97,98,99]. Impairments in lysosomal function caused by any of these faulty processes could have direct effects on mitochondria resulting from impaired mitophagy or the generation of toxic ROS, or even the generation of species interacting directly with mitochondria [51, 52, 94] (Fig. 3b). Indirect effects on mitochondrial health are also possible from lack of availability of critical metabolites.
The convergence of the evidence that endocytosed fibrillar α-syn species are trafficked to lysosomes along with a growing understanding of endolysosomal risk factors for synucleinopathy highlights the possibility of a direct relationship between lysosomal function and the spread of synucleinopathies. Decreased lysosomal activity or integrity resulting from age or genetic mutations may in fact potentiate the transmission of fibrillar or oligomeric α-syn pathology once it is formed in the nervous system.
Concluding remarks
Amazingly, the last decade has seen an explosion in our understanding of many of the chemical and biological processes underlying the onset and progression of human synucleinopathies, and transmission of proteopathic α-syn seeds is increasingly thought to play a role in the gradual progression of these diseases throughout the brain. Cell and animal models have provided us with relatively facile or tractable systems to interrogate the complex dynamic relationships between α-syn seeds and the cell. We are learning about the uptake and intracellular trafficking of α-syn seeds, the recruitment of monomeric α-syn into mature α-syn pathology, and the processing and release of proteopathic α-syn species as the transmission process continues in an autocatalytic manner. However, a complete understanding how different neural cells respond to proteopathic α-syn seeds of likely divergent conformations or biochemical properties is still elusive, considering the potential number of protective factors, vulnerability factors, and conformational effects of fibrillar α-syn seeds. With emerging structural and biological assays and an improving understanding of factors mediating α-synuclein transmission, the future will likely see the development of targeted therapies to suppress or remedy any of the pathological processes discussed here, and almost certainly some that have yet to be discovered.
References
Parkinson J. An essay on the shaking palsy (Reprinted). J Neuropsychiatry Clin Neurosci. 2002;14:223–36.
Maroteaux L, Campanelli JT, Scheller RH. Synuclein—a neuron-specific protein localized to the nucleus and presynaptic nerve-terminal. J Neurosci. 1988;8:2804–15.
Ueda K, Fukushima H, Masliah E, et al. Molecular-cloning of Cdna-encoding an unrecognized component of amyloid in Alzheimer-disease. Proc Natl Acad Sci USA. 1993;90:11282–6.
Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7.
Spillantini MG, Schmidt ML, Lee VMY, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40.
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–73.
Joachim CL, Duffy LK, Morris JH, Selkoe DJ. Protein chemical and immunocytochemical studies of meningovascular beta-amyloid protein in Alzheimer’s disease and normal aging. Brain Res. 1988;474:100–11.
Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998;249:180–2.
Tu PH, Galvin JE, Baba M, et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. 1998;44:415–22.
Marui W, Iseki E, Nakai T, et al. Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2002;195:153–9.
Braak H, Del Tredici K, Bratzke H, et al. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol. 2002;249(Suppl 3):III/1–5.
Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211.
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–6.
Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord. 2008;23:2303–6.
Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–3.
Li JY, Englund E, Widner H, et al. Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov Disord. 2010;25:1091–6.
Kurowska Z, Englund E, Widner H, et al. Signs of degeneration in 12-22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson’s disease. J Park Dis. 2011;1:83–92.
Luk KC, Kehm V, Carroll J, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53.
Luk KC, Kehm VM, Zhang B, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–86.
Paumier KL, Luk KC, Manfredsson FP, et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015;82:185–99.
Osterberg VR, Spinelli KJ, Weston LJ, et al. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015;10:1252–60.
Recasens A, Dehay B, Bove J, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–62.
Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71.
Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem. 2013;288:15194–210.
Luk KC, Song C, O’Brien P, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci USA. 2009;106:20051–6.
Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA. 2013;110:E3138–47.
Schonberger O, Horonchik L, Gabizon R, et al. Novel heparan mimetics potently inhibit the scrapie prion protein and its endocytosis. Biochem Biophys Res Commun. 2003;312:473–9.
Horonchik L, Tzaban S, Ben-Zaken O, et al. Heparan sulfate is a cellular receptor for purified infectious prions. J Biol Chem. 2005;280:17062–7.
Stopschinski BE, Holmes BB, Miller GM, et al. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and beta-amyloid aggregates. J Biol Chem. 2018;293:10826–40.
Mao X, Ou MT, Karuppagounder SS, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353:aah3374.
Konno M, Hasegawa T, Baba T, et al. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol Neurodegener. 2012;7:38.
Aulic S, Masperone L, Narkiewicz J, et al. Alpha-synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci Rep. 2017;7:10050.
Minakaki G, Menges S, Kittel A, et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. 2018;14:98–119.
Zhang S, Eitan E, Wu TY, Mattson MP. Intercellular transfer of pathogenic alpha-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol Aging. 2018;61:52–65.
Ngolab J, Trinh I, Rockenstein E, et al. Brain-derived exosomes from dementia with Lewy bodies propagate alpha-synuclein pathology. Acta Neuropathol Commun. 2017;5:46.
Abounit S, Bousset L, Loria F, et al. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016;35:2120–38.
Tran HT, Chung CH, Iba M, et al. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep. 2014;7:2054–65.
Bae EJ, Lee HJ, Rockenstein E, et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454–69.
Apetri MM, Harkes R, Subramaniam V, et al. Direct observation of alpha-synuclein amyloid aggregates in endocytic vesicles of neuroblastoma cells. PLoS ONE. 2016;11:e0153020.
Masaracchia C, Hnida M, Gerhardt E, et al. Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta Neuropathol Commun. 2018;6:79.
Karpowicz RJ Jr., Haney CM, Mihaila TS, et al. Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem. 2017;292:13482–97.
Freundt EC, Maynard N, Clancy EK, et al. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol. 2012;72:517–24.
Brahic M, Bousset L, Bieri G, Melki R, Gitler AD. Axonal transport and secretion of fibrillar forms of alpha-synuclein, Abeta42 peptide and HTTExon 1. Acta Neuropathol. 2016;131:539–48.
Freeman D, Cedillos R, Choyke S, et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE. 2013;8:e62143.
Jiang P, Gan M, Yen SH, McLean PJ, Dickson DW. Impaired endo-lysosomal membrane integrity accelerates the seeding progression of alpha-synuclein aggregates. Sci Rep. 2017;7:7690.
Samuel F, Flavin WP, Iqbal S, et al. Effects of serine 129 phosphorylation on alpha-synuclein aggregation, membrane association, and internalization. J Biol Chem. 2016;291:4374–85.
Sacino AN, Brooks MM, Chakrabarty P, et al. Proteolysis of alpha-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J Neurochem. 2017;140:662–78.
Sorrentino ZA, Vijayaraghavan N, Gorion KM, et al. Physiological carboxy-truncation of alpha-synuclein potentiates the prion-like formation of pathological inclusions. J Biol Chem. 2018;293:18914–32.
Volpicelli-Daley LA, Gamble KL, Schultheiss CE, et al. Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell. 2014;25:4010–23.
Froula JM, Henderson BW, Gonzalez JC, et al. alpha-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol Commun. 2018;6:35.
Plotegher N, Duchen MR. Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front Cell Dev Biol. 2017;5:110.
Grassi D, Howard S, Zhou M, et al. Identification of a highly neurotoxic alpha-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc Natl Acad Sci USA. 2018;115:E2634–E43.
Luna E, Decker SC, Riddle DM, et al. Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol. 2018;135:855–75.
Mahul-Mellier AL, Vercruysse F, Maco B, et al. Fibril growth and seeding capacity play key roles in alpha-synuclein-mediated apoptotic cell death. Cell Death Differ. 2015;22:2107–22.
Jang A, Lee HJ, Suk JE, et al. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113:1263–74.
Lee HJ, Baek SM, Ho DH, et al. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp Mol Med. 2011;43:216–22.
Bae EJ, Ho DH, Park E, et al. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of alpha-synuclein. Antioxid Redox Signal. 2013;18:770–83.
Lee HJ, Cho ED, Lee KW, et al. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med. 2013;45:e22.
Lee JG, Takahama S, Zhang G, Tomarev SI, Ye Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol. 2016;18:765–76.
Xu Y, Cui L, Dibello A, et al. DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov. 2018;4:11.
Fussi N, Hollerhage M, Chakroun T, et al. Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018;9:757.
Eliezer D, Kutluay E, Bussell R Jr., Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–73.
Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron. 2013;79:1044–66.
Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9.
Iwai A, Masliah E, Yoshimoto M, et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–75.
Logan T, Bendor J, Toupin C, Thorn K, Edwards RH. Alpha-synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci. 2017;20:681–9.
Burre J, Sharma M, Tsetsenis T, et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–7.
Diao J, Burre J, Vivona S, et al. Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife. 2013;2:e00592.
Bartels T, Choi JG, Selkoe DJ. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–10.
Dettmer U, Newman AJ, Soldner F, et al. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. 2015;6:7314.
Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336:1511–3.
Tuttle MD, Comellas G, Nieuwkoop AJ, et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 2016;23:409–15.
Barclay AM, Dhavale DD, Courtney JM, Kotzbauer PT, Rienstra CM. Resonance assignments of an alpha-synuclein fibril prepared in Tris buffer at moderate ionic strength. Biomol NMR Assign. 2018;12:195–9.
Guerrero-Ferreira R, Taylor NM, Mona D, et al. Cryo-EM structure of alpha-synuclein fibrils. eLife. 2018;7:e36402.
Li B, Ge P, Murray KA, et al. Cryo-EM of full-length alpha-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun. 2018;9:3609.
Li Y, Zhao C, Luo F, et al. Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res. 2018;28:897–903.
Luk KC, Covell DJ, Kehm VM, et al. Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity. Cell Rep. 2016;16:3373–87.
Guo JL, Covell DJ, Daniels JP, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–17.
Peng C, Gathagan RJ, Covell DJ, et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature. 2018;557:558–63.
Peelaerts W, Bousset L, Van der Perren A, et al. Alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–4.
Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575.
Kim C, Lv G, Lee JS, et al. Exposure to bacterial endotoxin generates a distinct strain of alpha-synuclein fibril. Sci Rep. 2016;6:30891.
Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA. 2015;112:E5308–17.
Woerman AL, Kazmi SA, Patel S, et al. MSA prions exhibit remarkable stability and resistance to inactivation. Acta Neuropathol. 2018;135:49–63.
Woerman AL, Kazmi SA, Patel S, et al. Familial Parkinson’s point mutation abolishes multiple system atrophy prion replication. Proc Natl Acad Sci USA. 2018;115:409–14.
Loria F, Vargas JY, Bousset L, et al. Alpha-synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017;134:789–808.
Singleton AB, Farrer M, Johnson J, et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841.
Bae EJ, Yang NY, Lee C, et al. Haploinsufficiency of cathepsin D leads to lysosomal dysfunction and promotes cell-to-cell transmission of alpha-synuclein aggregates. Cell Death Dis. 2015;6:e1901.
Bae EJ, Yang NY, Lee C, et al. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and alpha-synuclein aggregation. Exp Mol Med. 2015;47:e153.
Ramirez A, Heimbach A, Grundemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–91.
Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240–6.
Tsunemi T, Hamada K, Krainc D. ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J Neurosci. 2014;34:15281–7.
Cang C, Aranda K, Seo YJ, Gasnier B, Ren D. TMEM175 Is an Organelle K(+) Channel Regulating Lysosomal Function. Cell. 2015;162:1101–12.
Jinn S, Drolet RE, Cramer PE, et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc Natl Acad Sci USA. 2017;114:2389–94.
Zimprich A, Benet-Pages A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–75.
Miura E, Hasegawa T, Konno M, et al. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol Dis. 2014;71:1–13.
Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600.
Khan NL, Jain S, Lynch JM, et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128:2786–96.
Steger M, Tonelli F, Ito G, et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016;5:e12813.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Karpowicz, R.J., Trojanowski, J.Q. & Lee, V.MY. Transmission of α-synuclein seeds in neurodegenerative disease: recent developments. Lab Invest 99, 971–981 (2019). https://doi.org/10.1038/s41374-019-0195-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0195-z
This article is cited by
-
In vivo effects of the alpha-synuclein misfolding inhibitor minzasolmin supports clinical development in Parkinson’s disease
npj Parkinson's Disease (2023)
-
Effects of acupuncture and moxibustion on PINK1/Parkin signaling pathway in substantia nigra of Thy1-αSyn transgenic mice with Parkinson disease
Journal of Acupuncture and Tuina Science (2023)
-
Mechanisms of enhanced aggregation and fibril formation of Parkinson’s disease-related variants of α-synuclein
Scientific Reports (2022)
-
Glycoconjugate journal special issue on: the glycobiology of Parkinson’s disease
Glycoconjugate Journal (2022)
-
Binding Stability of Antibody—α-Synuclein Complexes Predicts the Protective Efficacy of Anti-α-synuclein Antibodies
Molecular Neurobiology (2022)
