
Abstract
Glucagon-like peptide-1 (GLP-1), which is well known for regulating glucose homeostasis, exhibits multiple actions in cardiovascular disorders and renal injury. However, little is known about the effect of GLP-1 receptor (GLP-1R) activation on acute lung injury (ALI). In this study, we investigated the effect of GLP-1R on ALI and the potential underlying mechanisms with the selective agonist liraglutide. Our results show that GLP-1 levels decreased in serum, though they increased in bronchoalveolar lavage fluid (BALF) and lung tissue in a mouse model of lipopolysaccharide (LPS)-induced ALI. Liraglutide prevented LPS-induced polymorphonuclear neutrophil (PMN) extravasation, lung injury, and alveolar-capillary barrier dysfunction. In cultured human pulmonary microvascular endothelial cells (HPMECs), liraglutide protected against LPS-induced endothelial barrier injury by restoring intercellular tight junctions and adherens junctions. Moreover, liraglutide prevented PMN–endothelial adhesion by inhibiting the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), and thereafter suppressed PMN transendothelial migration. Furthermore, liraglutide suppressed LPS-induced activation of Rho/NF-κB signaling in HPMECs. In conclusion, our results show that GLP-1R activation protects mice from LPS-induced ALI by maintaining functional endothelial barrier and inhibiting PMN extravasation. These results also suggest that GLP-1R may be a potential therapeutic target for the treatment of ALI.
Similar content being viewed by others


Introduction
Acute lung injury (ALI) is a fatal clinical syndrome with high morbidity and mortality and is induced by various infectious (such as pneumonia and sepsis) and noninfectious insults (such as trauma, transfusions, mechanical ventilation, or toxic smoke inhalation) [1, 2]. In patients with ALI, the disruption of alveolar capillary barrier results in the accumulation of protein-rich pulmonary edema fluid and polymorphonuclear neutrophil (PMN) entrapment/activation in the airspaces [1, 3, 4]. These pathological abnormalities sequentially lead to deficient gas exchange, arterial hypoxemia, and eventually respiratory failure.
Pulmonary endothelial cells (ECs), the major components lining in the intima of pulmonary vessels, maintain the balance of alveolar capillary barrier. On the one hand, quiescent ECs have predominantly anti-inflammation effect in rodents. On the other hand, the pro-inflammatory phenotype of ECs can initiate lung parenchymal inflammation once activated by biological, chemical, or mechanical stimuli [5]. The pro-inflammatory phenotype can produce substantial chemokines (e.g., chemokine [C-X-C motif] ligand-1 [CXCL1], CXCL2, and CXCL8) and adhesion molecules (e.g. intercellular adhesion molecule-1 [ICAM-1] and vascular cell adhesion molecule-1 [VCAM-1]) which in turn promote inflammatory cell infiltration and alveolar-capillary barrier damage in ALI [5].
Glucagon-like peptide-1 (GLP-1), a well-known molecule released from gut enteroendocrine cells, can regulate meal-related glycemic excursions by augmenting insulin production and inhibiting glucagon secretion [6]. Additionally, mounting evidence has shown that GLP-1 analogs/GLP-1 receptor (GLP-1R) agonists reduce obesity-induced chronic kidney injury via restoration of renal metabolism homeostasis [7], ameliorate renal injury resulting from diabetic kidney disease [8], attenuate neuronal death in early brain injury after subarachnoid hemorrhage [9], relieve myocardial damage by promoting autophagy [10], alleviate reperfusion injury-induced acute myocardial infarction [11, 12], and protect liver from ischemia-reperfusion injury [13]. Furthermore, GLP-1 analogs/GLP-1R agonists modulate tumor necrosis factor-α (TNF-α)-mediated endothelial activation [14], inhibit high-glucose-induced oxidative injury of vascular ECs [15], protect cardiac microvascular ECs against hypoxia/reoxygenation injury [16], and strengthen the barrier integrity in ECs [17]. In addition, GLP-1 analogs/GLP-1R agonists also attenuate lipopolysaccharide (LPS)-stimulated eosinophil activation [18], modulate macrophage cell fate towards MΦ2 pro-resolving macrophages [19], and decrease neutrophil activation [20]. Taken together, these findings indicate GLP-1 analogs or GLP-1R agonists protect multiple organs from injury, prevent ECs from dysfunction/apoptosis, and modulate inflammatory response.
Considering the anti-inflammatory and endothelial protective effects of GLP-1R, activating GLP-1R may be beneficial for ALI. Therefore, in the present study, we evaluated the effect of a GLP-1R agonist (liraglutide) on LPS-induced ALI in mice, and explored the underlying mechanism via a co-culture system of PMN and human pulmonary microvascular endothelial cells (HPMECs).
Materials and methods
Experimental animals
Male mice (between 18 and 22 g), obtained from the Animal Core Facility of Nanjing Medical University (Nanjing, China), were housed under standard laboratory conditions. The relative humidity ranged from 55% to 65%, the temperature was maintained at 22 ± 2 °C, free access to food and water was provided, and a 12 h light–dark cycle was maintained. After 5 days, eight of the mice were randomly picked up for an intra-tracheal instillation of 0.9% saline (40 μl) with a subcutaneous injection of 0.9% saline (0.1 ml), the remaining mice received an intra-tracheal instillation of LPS (15 mg/kg, O55:B5, Sigma-Aldrich, St. Loius, MO, USA) with/without an subcutaneous injection of liraglutide (NovoNordiskA/S, Denmark) at 0.2 mg/kg (LPS + Li-0.2) and 0.8 mg/kg (LPS + Li-0.8). Twenty-four hours later, mice were sacrificed for sample collection. Bronchoalveolar lavage fluid (BALF) was collected to detect the total protein concentration and the number of PMN, as well as macrophages. The concentration of GLP-1 in BALF and in blood was determined using mice GLP-1 ELISA Kit (CUSABIO Life science, Wuhan, China). Lung tissue were harvested for examination.
Measurement of lung inflammation and injury
Mouse lung tissue was fixed with 4% polyformaldehyde and embedded in paraffin, sliced into 5-µm sections, stained with hematoxylin and eosin (H&E) and subjected to immunohistochemical staining with a primary antibody against Ly6G (Abcam). The lung injury score was calculated using H&E staining according to a formula described in an American Thoracic Society report [21]. Permeability of the alveolar capillary membranes were assessed using an Evans blue dye (EB) extravasation technique [22]. Briefly, mice were given an intraperitoneal injection with EB dye (50 ml/kg) 2 h before sacrifice. At the end of exposure, the pulmonary circulation was flushed. The EB dye extracted from lung tissue were then determined at 620 and 750 nm using a microplate reader (Thermo Scientific, CA, USA). Cellular apoptosis in lung tissue from different groups was determined using a TUNEL assay kit (In Situ Cell Death Detection Kit, POD, Roche Company, Germany) described previously [23]. The TUNEL-positive cells were stained green, and the number of TUNEL-positive cells in eight different random fields from each section was counted using a Leica 2500 microscope (Leica Microsystems, Wetzlar, Germany) at ×200 magnification.
Cell culture
HPMECs, purchased from ScienCell (CA, USA), were cultured in an endothelial growth medium (ScienCell) containing 10 μg/mL streptomycin, 10 U/mL penicillin, EC growth supplement, and 5% heat-inactivated fetal bovine serum (FBS) (ScienCell). The cells were incubated at 37 °C in a 5% CO2 humidified incubator. HPMECs, between passage 4 and 6, were used for the following experiments. After starvation for 6 h, HPMECs were exposed to LPS (1 μg/ml, Sigma-Aldrich, MO, USA) for 30 min or 24 h with/without liraglutide (Selleck Chemicals, Houston, TX, USA) pretreatment for 30 min at different doses (200, 400, and 800 nM). Thereafter, cells were harvested for the following experiments.
PMN–endothelial adhesion assay
Human PMNs, obtained from the peripheral blood of healthy volunteers, were collected using Ficoll-Paque Plus (GE Healthcare, Pittsburgh, USA), then suspended in RPMI 1640 medium containing 10% FBS as described previously [24].
For the PMN–endothelial adhesion assay, the isolated PMN (106 cells/ml) were stained with 5 ng/ml calcein-AM (BD Biosciences) for 30 min. HPMECs were exposed to LPS (1 μg/ml) for 24 h with/without liraglutide pretreatment. The labeled PMN, resuspended at the concentration of 2 × 105 cells/ml, were co-incubated with HPMECs at 37 °C for 1 h. Non-adherent PMN were washed out by PBS, and the adherent cells were fixed with 4% paraformaldehyde. The number of adherent neutrophils (labeled in green) in 10 random areas from each group was counted under a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
PMN transendothelial migration assay
PMN transendothelial migration assay were performed as described previously [24]. Briefly, 200 μl HPMECs in suspension (106 cells/ml) were seeded on bovine plasma fibronectin (ScienCell)-coated Corning transwell inserts (6.5 mm diameter, 8 μm pore size). After grown for 3 days to confluency, HPMECs on the inserts were treated with LPS (1 μg/ml) for 24 h with or without liraglutide pretreatment (800 nM). Additional HPMECs, cultured in a 25 cm2 culture-flask, were challenged with LPS, after 24 h, the cellular supernatant (conditioned medium, or CM), was harvested for the following exploration. Then, after the inserts were washed with PBS and transferred into a fresh 24-well plate, the PMN suspension (105 cells in 100 μl) was added into the upper chamber of the inserts and the bottom of the well was supplemented with 600 μl CM or basic culture medium (BCM). The transendothelial migration assay system was maintained at 37 °C. Two hours later, PMN that migrated into the bottom of the well were collected and counted under a microscope.
Immunofluorescence
Confluent HPMECs monolayers grown on coverslips were subjected to immunofluorescence staining to detect the cellular junctions using the primary antibodies against vascular endothelial cadherin (VE-cadherin) (Abcam, MA, USA) and zonula occludens-1 (ZO-1) (Proteintech, Rosemont, IL). Briefly, cells, cultured on coverslips, were given the aforementioned treatment for 24 h, fixed with 4% paraformaldehyde solution for 30 min, blocked using 3% goat serum at room temperature for 30 min, incubated with primary antibodies at 4 °C overnight, and labeled with Alexa Fluor 555-conjuated anti-rabbit IgG at room temperature for 1 h. Thereafter, cells were counterstained with DAPI and obtained under a fluorescence microscope (Leica Microsystems).
RhoGTPase activation assay
RhoGTPase activation was determined by the use of Rho Activation Assay Kit (Millipore, Billerica, MA). Cleared lysates from HPMECs, using Mg2+ lysate containing 1 mM phenylmethylsulfonyl fluoride and a cocktail of protease inhibitors, were incubated with 50 µg GST-fusion protein of the Rhotekin-binding domain (RBD) along with glutathione agarose resin to pull down GTP-bound Rho. Western blot analysis was used to determine active Rho (Rho-GTP) with primary antibody against Rho.
Western blot and RT-qPCR
Total protein was extracted from mice lung tissue and HPMECs using RIPA lysis and extraction buffer (Thermo Fisher Scientific, Middletown, VA, USA) containing 0.4 mM phenylmethylsulfonyl fluoride and a protease inhibitor cocktail (Roche Company, Germany) as described previously [25]. The protein concentration was measured using a BCA Protein Assay Kit (Beyotime Biotechnology, Shanghai, China). Thereafter, 30 μg of total protein was separated via 10% SDS–PAGE, electroblotted to a PVDF membrane, and incubated with primary antibodies against GLP-1 (Abcam), toll like receptor-4 (TLR-4, CST), VE-cadherin (Abcam), ZO-1 (Proteintech), occludin (Proteintech), caspase 3 (CST, Danvers, MA), intercellular cell adhesion molecule-1 (ICAM-1, CST), vascular cell adhesion molecule 1 (VCAM-1, CST), myosin phosphatase target subunit 1 (MYPT1, CST), phosphorylated MYPT1 (p-MYPT1, CST), p65 (CST), and phosphorylated p65 (CST) following by incubation with HRP-conjugated second antibody. At last, chemiluminescence (ECL, Cell Signaling Technology) were used to label protein band. Band intensities were analyzed using NIH Image 1.46 software.
Total RNA was isolated from HPMECs with Trizol reagent (Invitrogen), and quantified (1000 ng). Reverse transcription was conducted with SYBR Premix Ex Taq™ kit (TaKaRa Bio Inc., Dalian, China), following by quantitative RCR using an ABI Prism 7500 FAST apparatus (Applied Biosystems, Foster City, CA, USA). PCR primers are listed in Table 1, among them, β-actin was selected as housekeeping gene.
Statistical analysis
The results were presented as mean ± S.E.M. Differences among groups were analyzed using one-way ANOVA followed by least significant difference (LSD) post hoc test. P < 0.05 was considered statistically significant. All the experiments for in vitro studies were performed at least three times.
Results
GLP-1 alterations in LPS-induced ALI
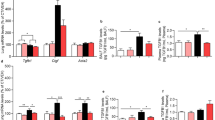
As presented in Fig. 1a, LPS inhalation induced a significant decrease in GLP-1 concentration in mice serum. However, the effect could be reversed by treatment with liraglutide (a GLP-1R agonist) at 0.8 mg/kg. On the contrary, a marked increase in GLP-1 in BALF was noted in LPS-challenged mice (Fig. 1b), but was significantly suppressed by liraglutide administration (0.8 mg/kg). As seen in BALF, elevated GLP-1 protein was also noted in lung tissue from LPS-challenged mice, and was inhibited by liraglutide treatment (Fig. 1c). Similarly, upregulation of GLP-1R mRNA caused by LPS-challenge in lung tissue was abolished by liraglutide at 0.2 and 0.8 mg/kg (Fig. 1d). Additionally, liraglutide significantly reversed the LPS-induced increase in TLR-4 expression at the mRNA and protein levels (Fig. 1e, f).

Glucagon-like peptide-1 (GLP-1) content change in lipopolysaccharide (LPS, 15 mg/kg)-induced acute lung injury (ALI). a Liraglutide restored LPS-induced decrease of GLP-1 in serum, b reduced LPS-induced increase of GLP-1 in bronchoalveolar lavage fluid (BALF), and c inhibited LPS-induced up-regulation of GLP-1 in lung tissue at 0.8 mg/kg. d-f Up-regulation of GLP-1R and TLR4 mRNA and protein expression resulting from LPS-challenge were suppressed by liraglutide treatment. Data are presented as the mean ± S.E.M. *P < 0.05
Effect of liraglutide on lung injury and inflammation
H&E staining (Fig. 2a) revealed diffuse alveolar damage, characterized by PMN accumulation in the interstitial and alveolar spaces, interstitial thickening, and fusion of pulmonary alveoli in lung tissue from LPS-challenged mice. These pathological changes were alleviated by treatment with liraglutide. The quantitative lung injury score was calculated using a formula defined in an American Thoracic Society report. The results show that liraglutide at both low dosage and high dosage significantly inhibited LPS-induced lung injury in mice (Fig. 2b). Using immunohistochemical staining with a primary antibody against Ly6G (a glycosylphosphatidylinositol-linked differentiation antigen expressed by myeloid-derived cells) (Fig. 2a), liraglutide effectively attenuated LPS-induced Ly6G-positive PMN infiltration (Fig. 2c). Liraglutide also successfully inhibited the extravasation of PMN into BALF in alveolar spaces (Fig. 2d). In addition, extensive apoptosis contributes to the irreversible alveolar damage during ALI. TUNEL analysis showed that the increase in apoptotic cells in lungs resulting from LPS challenge was significantly reduced by liraglutide treatment (Fig. 2a, e). Consistent with PMN, liraglutide also suppressed LPS-induced accumulation of macrophages in BALF. Liraglutide also inhibited the expression of macrophage-derived cytokines, including macrophage migration inhibitory factor (MIF), interleukin-6 (IL-6), chemokines [the chemokine (C-X-C motif) ligand 1/2, CXCL1, CXCL2] in lung tissue from LPS-challenged mice.

Liraglutide inhibited LPS-induced lung injury and inflammatory response. a Representative photomicrographs from lung tissue with HE staining, Ly6G staining, and TUNEL staining. b Liraglutide (0.2 and 0.8 mg/kg) alleviated LPS-induced lung injury (diffuse alveolar damage characterized by inflammatory cells accumulation in the interstitial and alveolar spaces, interstitial thickening, and fusion of pulmonary alveoli) as shown by H&E staining, c reduced Ly6G-positive PMN infiltration, d attenuated PMN extravasation into BALF, and e decreased TUNEL-positive cells (undergoing apoptosis) in lung tissue. Liraglutide also suppressed LPS-induced accumulation of macrophages in BALF (f), and inhibited the expression of macrophages-derived cytokines, including macrophage migration inhibitory factor (MIF), interleukin-6 (IL-6), and chemokines [the chemokine (C-X-C motif) ligand 1/2, CXCL1, CXCL2] (g). Data are presented as the mean ± S.E.M. *P < 0.05
Effect of liraglutide on LPS-induced alveolar capillary barrier dysfunction
Alveolar-capillary barrier dysfunction is another fundamental pathological profile of ALI, which leads to capillary hyperpermeability and leakage of protein-rich fluid to the interstitial and alveolar space. EB extravasation analysis (Fig. 3a) showed that the alveolar capillary hyperpermeability challenged by LPS was significantly inhibited by the treatment of liraglutide at 0.8 mg/kg. Similarly, LPS-induced accumulation of protein in BALF was also decreased by liraglutide administration (Fig. 3b). It is well-recognized that loss of intercellular tight junctions (which contain ZO-1 and occludin) and adherens junctions (which contain VE-cadherin) contributes the pulmonary microvascular leakage in ALI. As shown in Fig. 3c, the protein levels of VE-cadherin, ZO-1, and occludin in lung were markedly decreased in response to LPS challenge, and they were restored after the administration of liraglutide (0.8 mg/kg).

Liraglutide alleviated LPS-induced alveolar capillary barrier dysfunction. a Liraglutide suppressed the extravasation of Evans blue (EB) dye from the vascular to the interstitial and alveolar space (0.8 mg/kg), b decreased the protein content in BALF (0.2 and 0.8 mg/kg), and c reversed the LPS-induced down-regulation of proteins in intercellular tight junctions (occludin), adherens junctions (vascular endothelial cadherin, VE-cadherin), and adaptor molecule zonula occludens-1 (ZO-1) (at 0.8 mg/kg). Data are presented as the mean ± S.E.M. *P < 0.05
Effect of liraglutide on LPS-induced disruption of HPMEC barrier
A functional pulmonary endothelial barrier plays a key role in inhibiting PMN extravasation. In cultured HPMECs, LPS significantly downregulated the mRNA transcription of intercellular junctions (ZO-1 and occludin), but it was restored by liraglutide in a dose-dependent manner (Fig. 4a). Western blot analysis also confirmed that LPS-induced downregulation of intercellular tight junction (ZO-1) protein and intercellular AJ (VE-cadherin) protein were reversed by the treatment with liraglutide (Fig. 4b). Moreover, immunofluorescence staining indicated that liraglutide also prevented LPS-induced loss of VE-cadherin and ZO-1 between neighboring cells (Fig. 4c).

Effect of liraglutide on LPS-induced disruption of human pulmonary microvascular endothelial cell (HPMEC) barrier. a Liraglutide dose-dependently (200, 400, and 800 nM) reversed intercellular junctions (ZO-1, occludin) in HPMECs using RT-qPCR. b Liraglutide (800 nM) reversed the protein expression of intercellular junctions (VE-cadherin and ZO-1), and c restored the junctions between HPMECs indicated by VE-cadherin and ZO-1 immunofluorescence staining (VE-cadherin and ZO-1, red; all cell nuclei labeled by DAPI, blue). Data are presented as the mean ± S.E.M. *P < 0.05
Effect of liraglutide on PMN–endothelial adhesion and PMN transendothelial migration
PMN extravasation during ALI is a dynamic process, consisting of PMN–endothelial adhesion and sequential transendothelial migration. Thus, mRNA adhesion molecule expression in HPMECs was determined using RT-qPCR. In response to LPS challenge, adhesion molecule mRNA levels, including ICAM-1 and VCAM-1, were up-regulated markedly in cultured HPMECs (Fig. 5a), and were reduced by liraglutide in a dose-dependent manner. Consistent with mRNA expression patterns, both ICAM-1 and VCAM-1 protein levels increased significantly in LPS-challenged HPMECs compared with the control group, and were effectively reduced by the treatment of liraglutide (800 nM) (Fig. 5b). PMN and HPMECs were then co-cultured to evaluate the effect of liraglutide on LPS-induced PMN–endothelial adhesion. As expected, the number of adherent PMNs (green) in LPS-challenged HPMECs was larger than that for control HPMECs, but was markedly reduced by liraglutide (Fig. 5c). Thereafter, PMN transendothelial migration was measured to evaluate whether a GLP-1R agonist would reduce PMN extravasation. Consistent with the results of the in vivo study, liraglutide effectively attenuated the PMN transendothelial migration (Fig. 6a).

Effect of liraglutide on PMN–endothelial adhesion. a Liraglutide inhibited LPS-induced evaluated mRNA expression related to adhesion molecules (ICAM-1 and VCAM-1) in a dose-dependent manner (200, 400, and 800 nM), b suppressed LPS-induced increased ICAM-1 and VCAM-1 protein expression (800 nM), and c inhibited PMN–endothelial adhesion (PMN stained by calcein-AM, green; all cell nuclei labeled by DAPI, blue). Data are presented as the mean ± S.E.M. *P < 0.05

Effect of liraglutide on PMN transendothelial migration and LPS-induced Rho/NF-κB signaling. a The PMN transendothelial migration system, b Liraglutide (800 nM) attenuated PMN transendothelial migration (fewer PMN in the lower chamber), c reduced active Rho (Rho-GTP), d suppressed phosphorylation of MYPT1, and e inhibited phosphorylation of p65. Data are presented as the mean ± S.E.M. *P < 0.05. CM conditioned medium (cellular supernatant collected from HPMECs treated with LPS for 24 h); BCM basic culture medium
Effect of liraglutide on LPS-induced Rho/NF-κB signaling
Rho/NF-κB signaling is involved in the regulation of pulmonary vascular barrier function [26] and endothelial inflammatory response [27]. Notably, mice with LPS-induced ALI benefited from Rho/NF-κB signaling inhibition [24]. Therefore, Rho/NF-κB signaling in HPMECs was determined by western blotting. As shown in Fig. 6b, liraglutide effectively reduced LPS-induced evaluated active Rho (Rho-GTP), suppressed phosphorylation of MYPT1(Fig. 6c), and inhibited the downstream phosphorylation of p65 (Fig. 6d).
Discussion
ALI is a fatal complex clinical syndrome characterized by intense and diffuse alveolar damage secondary to various injurious stimuli, including infectious and noninfectious insults [2]. An intense inflammatory response with PMN recruitment, infiltration, and activation contributes to the pathology of ALI. Endothelial injury and endothelial inflammation are believed to mediate the alveolar-capillary barrier dysfunction and PMN extravasation. In the present study, we demonstrate that a GLP-1R agonist alleviated PMN infiltration, reduced injury of lung tissue, and restored the functional alveolar-capillary barrier in mice with LPS-induced ALI. Our in vitro studies indicate that a GLP-1R agonist restored barrier function of endothelial monolayers via restoration of intercellular junctions in HPMECs, inhibited LPS-induced PMN–endothelial adhesion through reducing the expression of adhesion molecules, and suppressed PMN transendothelial migration. The protective role of the GLP-1R agonist in LPS-challenged HPMECs may be attributed to Rho/NF-κB signaling inhibition.
Damage to the endothelial barrier not only induces hyperpermeability of the alveolar capillary membrane, but also promotes PMN transendothelial migration. Under physiological conditions, the microvascular endothelial barrier is mainly regulated by the intercellular junction between neighboring ECs. These intercellular junctions are composed of tight junctions (containing occludin) and adherens junctions (containing VE-cadherin), which maintain the endothelium integrity. The adaptor molecule ZO-1, connecting tight junctions to the cellular actin cytoskeleton, is also important for endothelial barrier stability. Under pathological conditions, biological, chemical, or mechanical stimuli disrupt inter-endothelial junction and decrease the endothelial integrity, ultimately leading to alveolar capillary barrier dysfunction. In cultured ECs, our data indicate LPS directly induces the down-regulation of VE-cadherin and ZO-1 and the loss of intercellular junction. In LPS-challenged mice, markedly down-regulation of intercellular junctions (occludin, VE-cadherin, and ZO-1) in the lung suggests that LPS induces pulmonary capillary barrier dysfunction, which is confirmed by increased protein concentrations in BALF or EB dye extravasation in the lung. Our study reveals that the alveolar capillary barrier dysfunction, cell apoptosis, and PMN extravasation were alleviated by the treatment of liraglutide, which means mice with LPS-induced ALI may benefit from treatment with a GLP-1R agonist. Consistent with the present study, a GLP-1R agonist was reported to protect the blood-retinal barrier and reduce retinal vascular permeability [28, 29], attenuated LPS-induced increase in gut permeability [30], and restored blood–brain barrier [17]. Taken together, these results indicate that the GLP-1R agonist can help maintain the endothelial barrier in various organs.
Apart from hyperpermeability of the alveolar capillary membrane, inflammatory infiltrate is another key feature of LPS-induced ALI. PMN extravasation during ALI is a dynamic process including PMN–endothelial adhesion and PMN transendothelial migration [31]. In the present study, we found that treatment with a GLP-1R agonist inhibits PMN–endothelial adhesion via reduction in the expression of adhesion molecules in HPMECs. As previously reported, the GLP-1R agonist inhibited TNFα/LPS-induced THP-1 monocyte-endothelial adhesion via reduction of the protein expression of adhesion molecules VCAM-1 [32]. Similarly, GLP-1 analog also modulated tumor–endothelial cell interactions through reducing production of adhesion molecules (ICAM-1 and VCAM-1) [33]. Additionally, the GLP-1 analog suppressed the expression of cell adhesion molecules including ICAM-1 and VCAM-1 in diabetic mice [34]. Taken together, these results indicate that a GLP-1 analog/GLP-1R agonist can regulate cell–cell adhesion via modulating the expression of adhesion molecules.
We previously demonstrated that mice with LPS-induced ALI benefited from Rho inhibition [24]. In addition, Rho GTPases have also been reported to be key regulator in regulating endothelial barrier function in response to a variety of environmental factors and signaling agents [26]. In addition, Rho GTPases play an important role in cell adhesion and migration including leukocyte extravasation [27]. In the present study, LPS-induced Rho/NF-κB signaling was successfully inhibited by the GLP-1R agonist. As reported, the GLP-1R agonist also protected against cardiac microvascular injury via reduction of Rho expression [35], stimulated insulin secretion by inhibiting RhoA/ROCK signaling [36], and restored endothelial function through suppression of Rho/NF-κB signaling [37]. Therefore, the protective effect of a GLP-1R agonist on LPS-induced ALI may be at least partially attributed to Rho/NF-κB signaling inhibition.
GLP-1 is mainly secreted by intestinal L-cells in response to feeding [38]. Our recent study demonstrated that GLP-1 was also expressed in kidney and was upregulated in response to a challenge by monocrotaline [39]. In the present study, GLP-1 was also upregulated in lung tissue when mice were exposed to LPS. Thus, GLP-1 can be produced by organs other than intestinal L-cells under pathological conditions. GLP-1 levels were lower in peripheral blood from LPS-challenged mice compared to controls, but much higher in BALF and lung tissue from LPS-challenged mice. Based on our results, activation of GLP-1R by the GLP-1R agonist liraglutide exerts a potent protective effect on LPS-induced ALI in mice. Thus, decrease of GLP-1 in serum and increase of GLP-1 in lung suggest a redistribution of GLP-1, which could be a self-protective mechanism for the lung to resist the injury from LPS. However, this observation may also be due to depletion of GLP-1 in serum and the release of GLP-1 from pulmonary cells in response to the LPS challenge. These hypotheses are the subjects of future studies. It will be interesting to learn whether the ratio of GLP-1 in serum to BALF is a potential parameter for evaluating the severity of ALI.
In conclusion, the present study indicates that GLP-1 may be involved in ALI. GLP-1R activation protected mice from LPS-induced ALI through restoration of endothelial barrier function and reduction of PMN extravasation, which may result from the inhibitory role of a GLP-1R agonist in Rho/NF-κB signaling. These findings suggest that GLP-1 may be an important target conferring endothelial protection under inflammatory stress, and GLP-1R agonists may be potential therapeutic drugs for ALI.
References
Fanelli V, Ranieri VM. Mechanisms and clinical consequences of acute lung injury. Ann Am Thorac Soc. 2015;12(Suppl. 1):S3–8.
Petty TL, Ashbaugh DG. The adult respiratory distress syndrome. Clinical features, factors influencing prognosis and principles of management. Chest. 1971;60:233–9.
Muller-Redetzky HC, Suttorp N, Witzenrath M. Dynamics of pulmonary endothelial barrier function in acute inflammation: mechanisms and therapeutic perspectives. Cell Tissue Res. 2014;355:657–73.
Herold S, Gabrielli NM, Vadasz I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2013;305:L665–81.
Oliveira SDS, Castellon M, Chen J, et al. Inflammation-induced caveolin-1 and BMPRII depletion promotes endothelial dysfunction and TGF-beta-driven pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2017;312:L760–71.
Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 2018;27:740–56.
Wang C, Li L, Liu S, et al. GLP-1 receptor agonist ameliorates obesity-induced chronic kidney injury via restoring renal metabolism homeostasis. PLoS ONE. 2018;13:e0193473.
Chen P, Shi X, Xu X, et al. Liraglutide ameliorates early renal injury by the activation of renal FoxO1 in a type 2 diabetic kidney disease rat model. Diabetes Res Clin Pract. 2018;137:173–82.
Xie Z, Enkhjargal B, Wu L, et al. Exendin-4 attenuates neuronal death via GLP-1R/PI3K/Akt pathway in early brain injury after subarachnoid hemorrhage in rats. Neuropharmacology. 2018;128:142–51.
Zhang Y, Ling Y, Yang L, et al. Liraglutide relieves myocardial damage by promoting autophagy via AMPK-mTOR signaling pathway in zucker diabetic fatty rat. Mol Cell Endocrinol. 2017;448:98–107.
Kim S, Jeong J, Jung HS, et al. Anti-inflammatory effect of glucagon like peptide-1 receptor agonist, exendin-4, through modulation of IB1/JIP1 expression and JNK signaling in stroke. Exp Neurobiol. 2017;26:227–39.
Hu SY, Zhang Y, Zhu PJ, et al. Liraglutide directly protects cardiomyocytes against reperfusion injury possibly via modulation of intracellular calcium homeostasis. J Geriatr Cardiol. 2017;14:57–66.
Abdelsameea AA, Abbas NA, Abdel Raouf SM. Liraglutide attenuates partial warm ischemia-reperfusion injury in rat livers. Naunyn Schmiede Arch Pharmacol. 2017;390:311–9.
Garczorz W, Gallego-Colon E, Kosowska A, et al. Exenatide exhibits anti-inflammatory properties and modulates endothelial response to tumor necrosis factor alpha-mediated activation. Cardiovasc Ther. 2018;36:12317
Li Q, Lin Y, Wang S, et al. GLP-1 inhibits high-glucose-induced oxidative injury of vascular endothelial cells. Sci Rep. 2017;7:8008.
Zhang Y, Zhou H, Wu W, et al. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2+)-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic Biol Med. 2016;95:278–92.
Fukuda S, Nakagawa S, Tatsumi R, et al. Glucagon-like peptide-1 strengthens the barrier integrity in primary cultures of rat brain endothelial cells under basal and hyperglycemia conditions. J Mol Neurosci. 2016;59:211–9.
Mitchell PD, Salter BM, Oliveria JP, et al. Glucagon-like peptide-1 receptor expression on human eosinophils and its regulation of eosinophil activation. Clin Exp Allergy. 2017;47:331–8.
Bruen R, Curley S, Kajani S, et al. Liraglutide dictates macrophage phenotype in apolipoprotein E null mice during early atherosclerosis. Cardiovasc Diabetol. 2017;16:143.
Dokken BB, La Bonte LR, Davis-Gorman G, et al. Glucagon-like peptide-1 (GLP-1), immediately prior to reperfusion, decreases neutrophil activation and reduces myocardial infarct size in rodents. Horm Metab Res. 2011;43:300–5.
Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–38.
Moitra J, Sammani S, Garcia JG. Re-evaluation of Evans Blue dye as a marker of albumin clearance in murine models of acute lung injury. Transl Res. 2007;150:253–65.
Wang JJ, Zuo XR, Xu J, et al. Evaluation and treatment of endoplasmic reticulum (ER) stress in right ventricular dysfunction during monocrotaline-induced rat pulmonary arterial hypertension. Cardiovasc Drugs Ther. 2016;30:587–98.
Wang J, Xu J, Zhao X, et al. Fasudil inhibits neutrophil-endothelial cell interactions by regulating the expressions of GRP78 and BMPR2. Exp Cell Res. 2018;365:97–105.
Xu J, Wang J, He M, et al. Dipeptidyl peptidase IV (DPP-4) inhibition alleviates pulmonary arterial remodeling in experimental pulmonary hypertension. Lab Invest. 2018;98:1333–46.
Duluc L, Wojciak-Stothard B. Rho GTPases in the regulation of pulmonary vascular barrier function. Cell Tissue Res. 2014;355:675–85.
Cernuda-Morollon E, Ridley AJ. Rho GTPases and leukocyte adhesion receptor expression and function in endothelial cells. Circ Res. 2006;98:757–67.
Fan Y, Liu K, Wang Q, et al. Exendin-4 alleviates retinal vascular leakage by protecting the blood-retinal barrier and reducing retinal vascular permeability in diabetic Goto-Kakizaki rats. Exp Eye Res. 2014;127:104–16.
Goncalves A, Lin CM, Muthusamy A, et al. Protective effect of a GLP-1 analog on ischemia-reperfusion induced blood-retinal barrier breakdown and inflammation. Invest Ophthalmol Vis Sci. 2016;57:2584–92.
Nozu T, Miyagishi S, Kumei S, et al. Glucagon-like peptide-1 analog, liraglutide, improves visceral sensation and gut permeability in rats. J Gastroenterol Hepatol. 2018;33:232–9.
Kreuger J, Phillipson M. Targeting vascular and leukocyte communication in angiogenesis, inflammation and fibrosis. Nat Rev Drug Discov. 2016;15:125–42.
Krasner NM, Ido Y, Ruderman NB, et al. Glucagon-like peptide-1 (GLP-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS ONE. 2014;9:e97554.
Kosowska A, Gallego-Colon E, Garczorz W, et al. Exenatide modulates tumor-endothelial cell interactions in human ovarian cancer cells. Endocr Connect. 2017;6:856–65.
Gumuslu E, Cine N, Ertan Gokbayrak M, et al. Exenatide alters gene expression of neural cell adhesion molecule (NCAM), intercellular cell adhesion molecule (ICAM), and vascular cell adhesion molecule (VCAM) in the hippocampus of type 2 diabetic model mice. Med Sci Monit. 2016;22:2664–9.
Wang D, Luo P, Wang Y, et al. Glucagon-like peptide-1 protects against cardiac microvascular injury in diabetes via a cAMP/PKA/Rho-dependent mechanism. Diabetes. 2013;62:1697–708.
Kong X, Yan D, Sun J, et al. Glucagon-like peptide 1 stimulates insulin secretion via inhibiting RhoA/ROCK signaling and disassembling glucotoxicity-induced stress fibers. Endocrinology. 2014;155:4676–85.
Tang ST, Zhang Q, Tang HQ, et al. Effects of glucagon-like peptide-1 on advanced glycation endproduct-induced aortic endothelial dysfunction in streptozotocin-induced diabetic rats: possible roles of Rho kinase- and AMP kinase-mediated nuclear factor kappaB signaling pathways. Endocrine. 2016;53:107–16.
Cheang JY, Moyle PM. Glucagon-like peptide-1 (GLP-1)-based therapeutics: current status and future opportunities beyond type 2 diabetes. ChemMedChem. 2018;13:662–71.
Xu J, Wang J, Cheng Y, et al. Glucagon-like peptide-1 mediates the protective effect of the dipeptidyl peptidase iv inhibitor on renal fibrosis via reducing the phenotypic conversion of renal microvascular cells in monocrotaline-treated rats. Biomed Res Int. 2018;2018:1864107.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81273571, No. 81870054), the Jiangsu Clinical Research Center for Respiratory Diseases Project under Grant BL2012012, the Jiangsu Province Scientific Research Innovation Project of University graduate students (JX22013361), and by a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) (JX10231802).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, J., Wei, G., Wang, J. et al. Glucagon-like peptide-1 receptor activation alleviates lipopolysaccharide-induced acute lung injury in mice via maintenance of endothelial barrier function. Lab Invest 99, 577–587 (2019). https://doi.org/10.1038/s41374-018-0170-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0170-0
This article is cited by
-
Repurposing existing drugs for COVID-19: an endocrinology perspective
BMC Endocrine Disorders (2020)
-
Challenging Issues in the Management of Cardiovascular Risk Factors in Diabetes During the COVID-19 Pandemic: A Review of Current Literature
Advances in Therapy (2020)
