
Abstract
Inflammation always accompanies infection during sepsis. Mitochondrial dysfunction and the role of reactive oxygen species (ROS) produced by mitochondria have been proposed in the pathogenesis of sepsis. Maresins have protective and resolving effects in experimental models of infection. In the present study, we investigated the effects of maresin 1 (MaR1) on mitochondrial function in cecal ligation and puncture (CLP)-induced sepsis and sepsis patients to identify mechanisms underlying maresin 1-mediated stimulation of ROS in mitochondria. We found that treatment with MaR1 significantly inhibited production of cytokines, decreased bacterial load in the peritoneal lavage fluid, reduced the number of neutrophils, decreased lactic acid level and upregulated cyclic AMP (cAMP) concentration, with the outcome of decreased lung injury in CLP-induced sepsis in mice. The effects of MaR1 on downregulation nitric oxide (NOX) activity, improvement CAT and SOD activity to inhibit ROS production in mitochondria was dependent on lipoxin receptor (ALX) and cAMP. Survival rates were significantly increased after the treatment of mice with MaR1. In BMDM stimulated with LPS, MaR1 inhibited the ROS production, downregulated enzyme activity, reduced mtO2 production, increased mitochondrial membrane potential, improved adenosine triphosphate (ATP) content and mitochondrial DNA (mtDNA) copy number. Finally, the effects of MaR1 on ROS production in the blood of healthy volunteers stimulated with LPS or sepsis patients were associated with ALX and cAMP. Taken together, these data suggest that treatment with MaR1 could attenuate mitochondrial dysfunction during sepsis through regulating ROS production.
Similar content being viewed by others

Introduction
Sepsis is a complex clinical syndrome, an expression of the host in reaction to severe pathogenic invasion and is defined as an uncontrolled inflammatory response [1]. Numerous trials were conducted of agents that block the inflammatory cascade and high levels of neutrophil activation can cause tissue injury [2]. Patients who die of sepsis usually succumb to the ensuing multi-organ failure instead of the initial acute inflammation.
Mitochondria are organelles that supply energy by generating adenosine triphosphate (ATP) by oxidative phosphorylation. Their additional functions include the generation and detoxification of reactive oxygen species (ROS). Mitochondrial dysfunction is thought to play an important role in the pathogenesis of many different disease states such as organ dysfunction in sepsis [3, 4]. The terms ‘function’ and ‘dysfunction’ in relation to mitochondria are widely employed in bioenergetics and cell biology. Research data emerging over the last decade has demonstrated that ROS can influence a range of cellular events such as the damage to lipids, proteins, and DNA in a manner similar to that seen for traditional second messenger molecules [5, 6].
Serhan et al. [7, 8] found a new pathway of mediators, operative in resolution of acute inflammation, that possess potent action on polymorphonuclear neutrophils and macrophages. Identification of these new mediators, called macrophage mediators in resolving inflammation--maresins, provides evidence for autacoids produced from essential omega-3 fatty acids and biosynthesized via 12-lipoxygenase, termed specialized pro-resolving mediators (SPMs) [7, 8]. Maresin 1 (7,14-dihydroxydocosa-4Z,8Z,10,12,16Z,19Z-hexaenoic acid, MaR1) has been shown to be a potent mediator to stop polymorphonuclear infiltration and stimulate macrophages phagocytosis [9,10,11]. Spite et al. [12] found that SPM, such as resolvin D2, improved survival in cecal ligation and puncture (CLP)-initiated sepsis by playing a potent anti-inflammation and pro-resolving effect. Soon after, some other SPMs, such as lipoxin A4 [13], resolvin D1 [14] were found successively to be effective in sepsis of rodents. Unexpectedly, in addition to controlling inflammation and its resolution, the pro-resolving mediators were found to stimulate bacterial killing and clearance, rendering these molecules not to be immunosuppressive [15]. However, so far there are no studies about the effects and mechanisms underlying the actions of MaR1 on mitochondria.
In this study, we investigated the effects of MaR1 on mitochondrial function in sepsis. The study was designed to test the hypothesis that administration of MaR1 will inhibit ROS production to attenuate mitochondrial dysfunction and then increase the survival rate of the CLP mice (in vivo) and protect lipopolysaccharide (LPS)-stimulated Bone Marrow-Derived Macrophages (BMDM) (in vitro). Our secondary hypothesis was that treatment with MaR1 will inhibit ROS production associated with ROS enzyme, such as NADPH oxidase (NOX), catalases (CAT) and superoxide dismutase (SOD). Our third hypothesis was that sepsis patient would also be correlation with ROS production and MaR1 could attenuate ROS production via regulating the enzyme. Finally, to gain a better understanding of the mechanisms, we also investigated the effect of butoxycarbonyl-Phe-Leu-Phe-Leu-Ph (BOC-2 (ALX antagonist)) and SQ22536 (cAMP inhibitor) on ROS production both in vivo and in vitro, to understand how their regulation affects mitochondrial dysfunction.
Materials and methods
Materials
Maresin 1 (7,14-dihydroxydocosa-4Z,8Z,10,12,16Z,19Z-hexaenoic acid, MaR1) was obtained from Cayman Chemical Company (Ann 105Arbor, MI). Lipopolysaccharide (LPS; E.coil serotype 055:B5), cAMP inhibitor (SQ22536) and 2% sodium pentobarbital were obtained from Sigma (St. Louis, MO). BOC-2 (ALX inhibitor) was obtained from Biomol-Enzo Life Sciences (Farmingdale, NY). Myeloperoxidase (MPO), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), MIP-2, IL-10, cAMP ELISA kits were from R&D Systems (Minneapolis, MN). Anti-mouse LyG FITC, anti-mouse F4/80 APC, isotype control mouse IgG1 FITC and isotype control mouse IgG1 APC were from eBioscience (San Diego, CA). MitoSOX Red superoxide indicator, 2, 7-dichlorodihydrofluorescein diacetate (DCFH-DA) and JC-1 were from Invitrogen (Carlsbad, CA), firefly luciferase ATP assay kit was from Beyotime (Shanghai, China). NOX, SOD, and CAT activity kits were from Jiancheng (Nanjing, China). JC-1 mitochondrial membrane potential assay kit was from Beyotime Biotechnology Inc (Nantong, China). Anti-COX I and Anti-COX IV were obtained from Abcam (Cambridge, MA). BCA protein assay kits and RT-PCR kits were obtained from Thermo Scientific (Rockford, IL). SYBR Green Real-time PCR Master Mix was obtained from Toyobo (Osaka, Japan).
Animal and Preparation
8-week-old male C57B6/L mice weighing 20–25 g (Shanghai, China), were housed at four per cage and maintained in a specific pathogen-free room with controlled temperature (22–24 °C) and humidity (60–65%) under a 12 h light/dark cycle. The mice were given standard laboratory chow and water ad libitum. All animal experiments were approved by the Animal Studies Ethics Committees of the Second Affiliated Hospital of Wenzhou Medical University.
Mice were anesthetized with 2% sodium pentobarbital (80 mg/kg, intraperitoneally) and randomly assigned to nine groups (n = 8): Sham group, CLP group, slight CLP group, CLP+MaR1 group (MaR1:10 ng/mice), CLP+MaR1 group (MaR1:50 ng/mice), CLP+MaR1 group (MaR1:100 ng/mice), CLP+MaR1 post group (MaR1:100 ng/mice), CLP+MaR1+BOC-2 group (MaR1:100 ng/mice, BOC-2:600 ng/kg) and CLP+BOC-2+SQ22536 group (SQ22536:10 mg/kg).
The CLP model was produced as described previously [16]. Midline abdominal incision was made after the abdomen was disinfected and the cecum was exposed. Then, cecum was ligated below the ileocecal valve and a through and through puncture was performed with a 20-gauge needle for mid-grade or slight-grade sepsis. Finally, it was relcoated into the abdominal cavity without spreading feces from the cecum onto the abdominal wall wound margins. The sham mice underwent the same procedure but the cecum was neither ligated nor punctured. Maresin 1, BOC-2, and SQ22536 were, respectively, injected into peritoneum half an hour after operation. For the CLP+MaR1 post group, Maresin 1 were injected into peritoneum 6 h later after operation.
Survival analysis
Mice which had undergone CLP were randomized to receive various concentration of MaR1 (10, 50, and 100 ng) in the presence or absence of BOC-2, or vehicle in Sham group (intraperitoneal injection; N = 12 for each group). The survival rate of mice was monitored for 8 days after operation and recorded everyday. The control groups for measuring the survival rate of mice were the same set of animals.
Pathological studies
The right lower lung lobes were collected and fixed with 10% neutral buffered formalin for 24 h, then embedded in paraffin and stained with hematoxylin and eosin (H&E) for light microscope analysis. A semi-quantitative scoring system was adopted to evaluate the lung injury including alveolar congestion, alveolar hemorrhage, infiltration, or aggregation of neutrophils in the airspace or vessel wall, and thickness of alveolar wall/hyaline membrane formation and inflammatory cell infiltration. The grading scale to score the pathologic findings was as follows: 0 = no injury; 1 = slight injury (25%); 2 = moderate injury (50%); 3 = severe injury (75%); and 4 = very severe injury (almost 100%). The results were graded from 0 to 4 for each item, as described previously [17, 18]. The four variables were summed to represent the lung injury score (total score: 0–16).
Blood samples were collected after MaR1 treatment for 24 h, and then centrifuged at 3000×g for 10 min to collect plasma. The plasma level of IL-1β, MPO, TNF -α, MIP-2, and IL-10 was determined, respectively, using ELISA kits.
Bacterial culture
50 μl peritoneal lavage fluid was diluted with PBS 1:10–1:104, and 50 μl of each dilution was evenly cultured on agarose gel plate. All plates were incubated at 37 °C in aerobic ordinary incubator for 12–14 h, and then the number of colony-forming units (CFU)/ml was calculated.
Neutrophils and macrophages/monocytes count in peritoneal lavage fluid
3 ml peritoneal lavage fluid was taken out at 24 h after MaR1 with or without BOC-2 treatment, all cells were stained with fluorochrome-conjugated antibody to cell subset-specific surface marker (FITC)-conjugated Ly6G for neutrophils and (APC)-conjugated F4/80 for macrophages after lysing the erythrocytes [19]. Then C6 flow cytometry (FCM) was used to analyse the cells.
Measurement of inflammatory cytokines and cAMP concentration
At 24 h after MaR1 with or without BOC-2 treatment, mice were anesthetized and blood and lungs were collected. Blood samples were centrifuged at 3000×g for 10 min to collect plasma. Lung tissue samples were homogenized in 50 mM potassium phosphate buffer (PB, pH 6.0). After that, inflammatory cytokines- TNF-α, IL-1β, MPO, MIP-2, and IL-10 were measured in plasma and cAMP was measured in lung tissue homogenate using ELISA kits according to the manufacturer’s instructions.
Assessment of Lactic acid level
Blood samples were collected after MaR1 with or without BOC-2 treatment for 24 h, and blood gas analysis was used to measure the Lactic acid.
Western blotting
Cell lysate was obtained using RIPA lysis buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100,1% sodium deoxycholate, 0.1% SDS, sodium orthovanadate, sodium fluoride, EDTA, and leupeptin) and PMSF. Samples were ultrasonicated three times, for 5 s, and then spun at 12,000×g for 30 min. Protein concentrations of the supernatants were determined using a BCA protein assay kit. Proteins were separated using 10% SDS polyacrylamide gel and transferred to PVDF membranes. After blocking with 5% nonfat dried milk in TBS containing 0.05% Tween 20, the membranes were incubated with primary anti-COX I and anti-COX IV (1:1000) and β-actin (internal control, 1:500) antibodies overnight at 4 °C. After washing the excess antibody, the membranes were incubated with HRP-conjugated secondary Abs (1:1000) at room temperature for 1.5 h. The protein bands were detected by ECL and visualized by UVP Gel imaging system (Upland, CA). The band intensity was analyzed by AlphaEaseFC.
Cell culture
Bone Marrow-Derived Macrophages (BMDM) were generated from 6 to 9 weeks old C57BL/6 mice. Briefly, total BMDM cells were cultured with 5% CO2/95% humidified air in Dulbecco’s modified Eagle’s medium supplemented with 0.05 mmol/L 2-ME, 1% nonessential amino acids, 10% fetal calf serum, and 20% L929 cell-conditioned medium. After 6 days of culture, the cell population regularly contained more than 90% macrophages (CD11b high F4/80 high).
Cells were randomly assigned to four groups (n = 4): CTR group, LPS group (LPS:100ng/ml), MaR1 group (MaR1:10 nM), LPS+MaR1 group.
Fluorescence microscopy
Fluorescence microscopy was performed under a Zeiss Axiophot 2 fluorescent microscope with rhodamine/fluoresceine dual filter set and a Spot 2 digital camera and imaging system. Cells were treated with JC-I (2.5 mmol/l), DCFDA (10 μ/l), MitoSOX Red (5 μg/ml) for 30 min in a 37 °C, CO2 incubator. Cells were then washed twice with PBS and examined immediately under a 40 aqueous immersible objective. We developed images using Spot 2 Software with the camera shutter set at 0.3 s for red light and 0.5 s for green light.
Assessment of ROS production and NOX, SOD, and CAT activity
ROS production in lung tissue and BMDM cells was detected using an oxygen radical sensitive probe, DCFH-DA, according to the manufacturer’s instructions. Briefly, the supernatant of lung tissue homogenate and BMDM cells were incubated with DCFH-DA solution for 20 min at 37 °C. Then, rinsed with PBS again and detected by a BD C500 flow cytometer (BD Biosciences).
For the experiment, different concentrations of MaR1 including 2.5, 5, 10, 20 nM were treated to measure the ROS production.
NOX, SOD, and CAT activity in lung tissue and blood from healthy volunteers or sepsis patients was tested using the kits as per manufacturer’s instructions.
Quantification of mtO2 production
MitoSOX Red superoxide indicator is a fluorogenic dye that is selective for mtO2 in living cells. BMDM was incubated with MitoSOX Red (5 μg/ml) for 10 min at 37 °C. Cells were collected by trypsinization and washed in PBS supplemented with 2% FBS, and then fixed in 2% paraformaldehyde and suspended in PBS. Measurements were performed in duplicates using the BD C500 flow cytometer (BD Biosciences) MitoSOX Red was excited at 488 nm, and the data collected at 575 = 26 nm (FL2) channel. The data was presented as histograms in terms of the mean intensity of MitoSOX fluorescence normalized to those of the static controls.
Mitochondrial membrane potential (ΔΨm)
The mitochondrial membrane potential (MMP) of BMDM was monitored using JC-1, a MMP-sensitive fluorescent dye, according to the manufacturer’s instructions. Briefly, BMDM were washed twice with Hank’s balanced salt solution, and incubated in the dark with JC-1 (2.5 mmol/l) for 30 min at 37 °C). Mitochondria and cells were washed with JC-1 washing buffer, and fluorescence was detected by a BD C500 flow cytometer (BD Biosciences). The relative MMP was calculated using the ratio of J-aggregate/monomer (590/520 nm). Values are expressed as the fold-increase in J-aggregate/monomer fluorescence over control cells.
ATP content measurement
Cellular ATP levels were measured using a firefly luciferase ATP assay kit according to the manufacturer’s instructions. Briefly, the BMDM were incubated in a mixed medium for 24 h. Next, a 0.5 ml culture medium was transported to 1.5 ml EP tube and was kept on ice to precisely determine the extracellular ATP concentration. Then, the cells were schizolysised and centrifuged at 12,000×g for 5 min. In 1.5 ml EP tubes, 100 µl of each supernatant was mixed with 100 µl of ATP detection working dilution. Luminance (RLU) was measured by a GloMax 20/20 luminometer. Standard curves were generated, and the protein concentration of each treatment group was determined using the Bradford protein assay. The intracellular ATP level was normalized by protein content in each sample (unit: nmol/mg protein).
Quantitative real-time PCR and reverse transcriptase-PCR
Total DNA from cultured BMDM were extracted by DNeasy Tissue Kit. Real-time PCR was used to detect mtDNA copy number. Real-time PCR amplification was performed using the ABI 7300 Real-time PCR Detection System with the SYBR Green master mix. Cycling conditions were 95 °C for10min followed by 40 repeats of 95 °C for 15 s and 60 °C for 1 min. Relative amounts of mtDNA copy number were normalized to 18 S and calculated using the comparative cycle threshold (DDCt) method.
Statistics
Data are represented as mean ± SEM. All data were analyzed by the Student t test or by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons. Survival of the two subgroups was estimated by Kaplan–Meier survival curves; comparisons were performed by the log-rank test. Significance was determined at the p < 0.05 level. Statistical analyses were performed using Prism 5.0 software (GraphPad Software, San Diego, CA).
Results
MaR1 improves survival rate and protects lung tissues in sepsis mice
First, to investigate whether MaR1 was of benefit to CLP mice, survival rate was observed for 8 days after CLP (Fig.1a). Polymicrobial sepsis resulted in a mortality rate of 81.82 % within 8 days, which is much higher than that in the Sham group (0%; p < 0.01). Conversely, mice treated with 100ng MaR1 for 24 h after CLP showed an improved survival rate during the study period (62.63%; p < 0.01), but not with 10ng or 50ng of MaR1 (p > 0.01). Moreover, after CLP operation for 6 h, then mice were treated with 100ng MaR1 for 24 h showed an improved survival rate during the study period (52.53%; p < 0.01). However, there was no difference of survival rate between Sham group and MaR1 group (p > 0.05; data not shown).

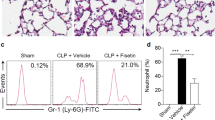
MaR1 protected lung tissues and increased the survival rate in cecal ligation and puncture (CLP)-induced Sepsis. MaR1 (100ng/mice, i.p.) was administered to C57BL/6 mice after surgery or 6 h after CLP, survival rate was observed for 8 days (a). Slight CLP model was also used to observe the effect of MaR1 (b). The lung tissues were collected 24 h after CLP and the effect of MaR1 was assessed by histology in H&E-stained sections(original magnification × 100) (c). Lung injury scores (d) were recorded from 0 (no damage) to 16 (maximum damage) according to the criteria described in Materials and Methods. Data are presented as mean ± SEM. n = 6–12. *p < 0.05, **p < 0.01 vs. SHAM group. #p < 0.05, ##p < 0.01 vs. CLP group
As human septic patients typically only have about a 20% mortality over 7 days, we next investigated the effect of MaR1 in a lower lethality model (Fig.1b). We found the survival rate was about 66% in slight CLP group and about 80% in CLP+MaR1 group.
In addition, we evaluated the effect of MaR1 on CLP-induced sepsis (Fig.1c). The Sham group showed normal pulmonary histology. In contrast, lung tissues in the CLP group were significantly damaged, with hemorrhage, thickening of the alveolar wall and infiltration of inflammatory cells into the interstitium and alveolar spaces, as evidenced by an increase in lung injury score (Fig.1d, p < 0.01). All the pathologic changes were less pronounced in the CLP+MaR1 group. MaR1 significantly attenuated CLP-induced organ damage as shown by the reduction in lung injury score (Fig.1d, p < 0.01). Incidentally, there was no significant difference between the Sham group and CLP+MaR1 group (p > 0.05).
MaR1 reduces the release of inflammatory cytokines in the plasma of sepsis mice
At 24 h post-CLP, the levels of some inflammatory cytokines were detected in the plasma of mice. The concentrations of TNF-α, IL-1β, MPO, MIP-2 and IL-10 was significantly increased in the CLP group compared with the Sham group (p < 0.01). However, the cytokines levels were greatly attenuated in the MaR1 treatment group compared to CLP group (p < 0.05; Fig. 2).

MaR1 inhibited the level of inflammatory factors in CLP-induced Sepsis. Blood samples were collected after MaR1 (100ng/mice, i.p.) treatment for 24 h, and then centrifuged at 3000 g for 10 min to collect plasma. The plasma level of IL-1β, MPO, TNF -α, MIP-2 and IL-10 was measured to quantitatively define the resolution of infiltrated cells using mouse ELISA kits. Data are presented as mean ± SEM. **p < 0.01 vs. SHAM group. #p < 0.05, ##p < 0.01 vs. CLP group
MaR1 enhances bacterial clearance and modulates immune cells in CLP-induced sepsis
Peritoneal lavage fluid samples were collected 24 h after operation and the CFU and immune cells was counted. Mice exhibited higher peritoneal bacteria content following CLP compared to the Sham group (p < 0.01). However, the bacterial colony formation in peritoneal lavage fluid markedly decreased in CLP+MaR1 group compared with CLP group (p < 0.01; Fig. 3a). Meanwhile, the amount of leukocytes in the peritoneal lavage fluid were detected by flow cytometry (Fig. 3b). The increased peritoneal Ly6G neutrophils recruitment 24 h post-CLP, whereas the total numbers of F4/80 macrophages was lower in CLP group compared with the Sham group. After treatment with MaR1, the macrophages count was increased and the neutrophils count was decreased compared to that seen with CLP group (p < 0.05; Fig. 3c,d)

MaR1 enhances bacterial clearance and modulation of immune cells in CLP model. Peritoneal lavage fluid samples were collected 24 h after operation and the CFU (a) and immune cells (b) were counted. The macrophage (c) and neutrophil count (d) was calculated by flow cytometry. Data are presented as mean ± SEM (n = 8–12). P5: Macrophages. P6: Neutrophils. *p < 0.05, **p < 0.01 vs. SHAM group. #p < 0.05, ##p < 0.01 vs. CLP group
BOC-2 abrogates the beneficial effects of MaR1 in CLP-induced sepsis
We investigated the effects of BOC-2 (600 ng/kg), an ALX antagonist, on MaR1-treated CLP-induced sepsis [20]. The mortality of CLP group is decreased by MaR1, yet coadministration of BOC-2 and MaR1 significantly blocked the effect of MaR1 (Fig. 4a). The morphologic changes of lung injury were less pronounced in the MaR1 treatment group than CLP group and the beneficial effect was abolished by BOC-2 (p < 0.01; Fig. 4b). The mortality and histology of Sham group, CLP group and CLP+MaR1 group was the same group that has been presented in Fig. 1, which means that the control groups for Fig. 1 and Fig. 4 were the same set of animals. The inhibitory effects of MaR1 on TNF-α concentration was abrogated by the treatment with BOC-2 (p < 0.01; Fig.4c). In addition, the bacterial colony formation in peritoneal lavage fluid in CLP+MaR1 group was decreased when compared with CLP+MaR1+BOC-2 group (p < 0.01; Fig. 5a).

The beneficial effects of MaR1 were abrogated by BOC-2 in CLP-induced sepsis. MaR1 (100 ng/mice, i.p.) in the presence or absence of BOC-2 (600 ng/kg, i.p.) was administered to C57BL/6 mice after surgery and survival rate was observed for 8 days. The control groups for Fig. 1 and Fig. 4 were the same set of animals. (a). The lung tissues were collected 24 h after CLP and the effect of MaR1 or BOC-2 was assessed by histology in H&E-stained sections (original magnification ×100) (b). Lung injury scores (b) were recorded from 0 (no damage) to 16 (maximum damage) according to the criteria described in Materials and Methods. Part of the right lung was homogenized from individual mice and centrifuged, then the tissue level of TNF-α in the resulting supernatants was determined using ELISA kit. (c) Data are presented as mean ± SEM. n = 6–12. **p < 0.01 vs. CLP group. ##p < 0.01 vs. CLP+MaR1 group, $$p < 0.01 vs. SHAM group

The Effect of MaR1 on bacterial clearance and leukocytes diversification was dependent on MaR1 receptor (ALX) in CLP model. MaR1 (100ng/mice, i.p.) and BOC-2 (600 ng/kg, i.p.) were co-administered to CLP mice, and peritoneal lavage fluid samples were collected 24 h after operation and the CFU (a) and immune cells (b, c, d) was calculated. Data are presented as mean ± SEM. n = 8–12. P5: Macrophages. P6: Neutrophils. *p < 0.05 vs. CLP group. #p < 0.05, ##p < 0.01 vs. CLP+MaR1 group
Administration of BOC-2 significantly decreased the amounts of macrophages (p < 0.05; Fig. 5b,c) and increased the amounts of neutrophil (p < 0.05; Fig. 5b,d).
MaR1-induced cAMP elevation in the lung tissues from CLP is dependent on ALX
To clarify the regulatory mechanism by which CLP and/or MaR1 (100 ng/mice) affect the outcome of sepsis, we tested whether MaR1 had an impact on cAMP level in lung tissue from the mice within operation. We found cAMP concentration was increased in the MaR1 treatment group compared to the CLP group (p < 0.01), and BOC-2 treatment abrogated the increased cAMP concentration when compared with the MaR1 treatment group (p < 0.05; Fig.6).

MaR1 increased the concentration of cAMP in vivo. MaR1 (100ng/mice, i.p.) was administered to CLP mice, lung tissues were collected 24 h after CLP, and cAMP concentration in the lung tissue was detected by ELISA kits. Data are presented as mean ± SEM. n = 6–8. **p < 0.01 vs. CLP group. #p < 0.05 vs. CLP+MaR1 group
MaR1 downregulated ROS production is regulated by NOX, SOD and CAT activity in CLP mice
ROS are produced by mitochondria during aerobic respiration as a consequence of electron transport chain activity. The expression of ROS in lung homogenates was measured by ELISA (Fig.7a). We found the concentration of ROS in the MaR1 (100 ng) treatment group was significantly decreased compared with the CLP group (p < 0.05). To further investigate if the effect of MaR1 on ROS production was associated with ALX or cAMP. BOC-2 (600 ng/kg) and SQ22536 (cAMP antagonist; 10 mg/kg) was used in the present study and we found that the ROS production was decreased in CLP+MaR1+BOC-2 group (p < 0.01) and CLP+MaR1+SQ22536 group (p < 0.05) when compared with MaR1 treatment group (Fig.7a).

MaR1 downregulated ROS production via regulating the enzyme NOX, SOD and CAT activity in CLP mice. MaR1 (100ng/mice) with or without BOC-2 (600 ng/kg) or SQ22536 (10 mg/kg) was co-administrated to CLP mice, lung tissues were collected 24 h after CLP, then ROS production(a), NOX(b), SOD(c) and CAT(d) activity in the lung tissue was detected. Data are presented as mean ± SEM. n = 6–8. *p < 0.05,**p < 0.01 vs. CLP group. #p < 0.05, ##p < 0.01 vs. CLP+MaR1 treatment group
Indeed, ROS production can be a consequence of NOX, SOD and CAT activities. We found that MaR1 downregulated the activation of NOX (p < 0.05; Fig.7b) and upregulated the activation of SOD and CAT when compared with CLP group (Fig.7c, d). Furthermore, BOC-2 or SD22536 reversed these effects (p < 0.05; Fig.7).
MaR1 mitigates mitochondrial protein and lactic acid level in CLP-induced sepsis
There were significant differences between the mitochondrial ultrastructure in the Sham and CLP groups’ lung cells. The mitochondria in the CLP group were swollen with disrupted or disintegrated cristae, and the osmiophilic lamellar bodies had fused or disappeared. This mitochondrial damage was slightly mitigated in the CLP+MaR1 group (Fig. 8a).

MaR1 mitigated functional proteins of the mitochondria in vivo and lactic acid level in CLP stimulated blood. MaR1 (100ng/mice) in the presence or absence of BOC-2 (600 ng/kg) or SQ22536 (10 mg/kg) was administered to CLP mice for 24 h, the lung tissues were collected 24 h after CLP and the effect of MaR1 on mitochondria was assessed by transmission electron microscopy analysis (original magnification ×15,000) (a) by the lung tissues homogenate COX I and COX IV protein expression on mitochondria (b), and by the level of lactic acid in arterial blood (c). Data are presented as mean ± SEM. n = 6–8. *p < 0.05 vs. CLP group. #p < 0.05 vs. CLP+MaR1 group
Generation of ROS has long been regarded as a by-product of oxygen consumption by mitochondria. The proteins from electron transport chain or matrix of mitochondria play a key role in a range of diseases. We previously demonstrated that MaR1 decreased ROS production, and then we investigated the respiratory chain enzyme complexes and some proteins expression in mitochondria after CLP challenge. COX I and COX IV protein expression were all lower in CLP group than Sham group. The administration of MaR1 significantly upregulated COX I and COX IV protein expression (Fig. 8b).
The level of lactic acid in plasma is one of the prognostic markers of sepsis. At 24 h after operation, we detected the amount of lactic acid in arterial blood. The findings showed that treatment with MaR1 suppressed the concentration of lactic acid when compared with CLP group, whereas BOC-2 or SD22536 eliminated this effect of MaR1 (p < 0.05) (Fig. 8c).
MaR1 downregulates LPS induced ROS production in BMDM and mitochondria
The ROS production in BMDM and mitochondria was measured by immunofluorescence and flow cytometry. LPS improved ROS level when compared with CTR group in cells (p < 0.01; Fig. 9a, b) and mitochondria (Fig. 9c, d), however, the promotive effect of LPS on ROS production was abrogated by treatment with MaR1 in BMDM and mitochondria. In addition, there was no significant difference between MaR1 only group and CTR group (p > 0.05; Fig. 9b, d).

MaR1 downregulated ROS production in BMDM and mitochondria stimulated with LPS. BMDM were treated with MaR1 (10 nM) in the presence or absence of LPS (100ng/ml) for 6 h. After incubation, cells were collected and sonicated. Cellular Reactive Oxygen Species Detection Assay Kit (DCFDA) was used to detect the ROS production by immunofluorescence (a) and flow cytometry (b) in BMDM stimulated with LPS. MitoSOX Red superoxide indicator was used to detect the ROS production by immunofluorescence (c) and flow cytometry (d) in mitochondria stimulated with LPS. Data are presented as mean ± SEM. n = 6. *p < 0.05 vs. CLP group. #p < 0.05 vs. LPS+MaR1 group
MaR1 decreases mitochondrial membrane potential in BMDM stimulated with LPS
Loss of mitochondrial membrane potential is a sign of injury in cells. To further confirm mitochondrial damage, we stained BMDM with the cationic lipophilic dye JC-1. This dye accumulates in normal mitochondria as aggregates with red fluorescence, while in depolarized or damaged mitochondria, JC-1 transforms into monomers with green fluorescence. Immunofluorescence and flow cytometry were used to detect the mitochondrial membrane potential. LPS treatment of BMDM resulted in an increase in the percentage of green-fluorescent-positive cells, but red-fluorescent-positive cells were much more in the MaR1 treatment group (Fig. 10a). We showed—in the right (R) quadrant of flow cytometry histogram- the mitochondrial membrane potential in BMDM was reduced substantially (drastic increase in JC-1 monomer) when treated with MaR1 (Fig. 10b).

MaR1 decreased mitochondrial membrane potential in BMDM stimulated with LPS. BMDM were treated with MaR1 (10 nM) in the presence or absence of LPS (100 ng/ml) for 6 h, then stained with 2.5 mmol/l of JC-1, a membrane potential-sensitive fluorescent dye. The dye was loaded, and after 30 min incubation fluorescent microscopic images were taken using both the red and green channels (a). Flow cytometry (b) was also used to measure the mitochondrial membrane potential in BMDM stimulated with LPS using JC-1 dye
MaR1 improves mitochondrial DNA (mtDNA) copy number and ATP content in BMDM treated with LPS
To monitor the effect of MaR1 on the mtDNA copy number and ATP content in BMDM, we used RT-PCR to measure the mtDNA copy number and firefly luciferase ATP assay kit to measure the ATP content. The inhibitory effect of LPS on mtDNA copy number (Fig. 11a) and ATP content (Fig. 11b) was abrogated by MaR1.

MaR1 improved mitochondrial DNA (mtDNA) copy number and ATP content in BMDM stimulated with LPS. BMDM were treated with MaR1 (10 nM) with or without of LPS (100 ng/ml) for 6 h. After incubation, the cells were collected and sonicated. RT-PCR was used to detect mtDNA copy number (a). Firefly luciferase ATP assay kit was used to measure ATP content (b). Data are presented as mean ± SEM. n = 6. *p < 0.05, **p < 0.01 vs. CLP group. #p < 0.05 vs. LPS group. &p < 0.05 vs. LPS+MaR1 group
MaR1-induced decrease of ROS is regulated by enzymes in healthy volunteers and sepsis patients
Finally, we investigated the therapeutic potential of MaR1 in humans. Peripheral venous blood (PVB, 10 ml) was collected from healthy adult volunteers or sepsis patients (at admission to the ICU and before therapeutic intervention) and treated with LPS in the presences or absence of MaR1 for 6 h. The ROS serum concentrations was measured in LPS group and in MaR1 treatment groups (Fig. 12a, f). We found that administration of 2.5 nM MaR1 for 6 h had no influence on decreasing the ROS production in the blood from patients as well as in the group stimulated with LPS (p > 0.05). However, 5 nM MaR1 decreased the ROS level induced by LPS (p < 0.05). The maximal effect was observed at 10 nM MaR1 and the effect of MaR1 was similar at 10 and 20 nM. Hence 10 nM MaR1 was chosen in our experiments. To monitor the mechanism of MaR1 on the ROS production, BOC-2 and SD22536 was used in the study, we found that BOC-2 and SD22536 abolished the effect of MaR1 (Fig. 12b, g).

MaR1-induced decrease of ROS concentration was regulated by enzymes activity in healthy volunteers and sepsis patient. Peripheral venous blood (PVB, 10 ml) was collected from healthy adult volunteers, stimulated with various concentration of MaR1 (2.5, 5, 10, and 20 nM) with or without LPS (100 ng/ml), or BOC-2 (10 μM/ml) or SQ22536 (20 μM/ml) for 6 h. Then the ROS production, NOX, SOD and CAT activity was measured (a-e). Data are presented as mean ± SEM. N = 10.*p < 0.05, **p < 0.01 vs. Control group. #p < 0.05, ##p < 0.01 vs. LPS group. &p < 0.05, &&p < 0.01 vs. LPS+MaR1 group. Moreover, the ROS production, NOX, SOD and CAT activity in sepsis patient was also measured (f–j). Data are presented as mean ± SEM. N = 10.*p < 0.05,**p < 0.01 vs. sepsis group. #p < 0.05, ##p < 0.01 vs. Sepsis+MaR1 group
Furthermore, the role of MaR1 on the NOX, SOD and CAT activity in peripheral blood of healthy volunteers treated with LPS or from sepsis patient was assessed. The outcomes of LPS-treated model and patients were consistent with the findings observed in CLP mice. MaR1 treatment downregulated the activity of NOX (p < 0.0; Fig.12c, h) and upregulated the activation of SOD (p < 0.05; Fig.12d, i) when compared with CTR/Sepsis group. MaR1 treatment improved the CAT activity when compared with sepsis patient (p < 0.01; Fig. 12j). However, there was no significant changes in activity of CAT after treatment with MaR1 in LPS-treated model (p > 0.05; Fig.12e). In addition, BOC-2 and SD22536 abolished the protective effect of MaR1 in attenuating LPS/sepsis-mediated changes of enzymes. Interestingly, in LPS model, the activity of CAT still increased in the LPS+BOC-2 and SD22536 group (Fig.12e).
Discussion
Our data clearly demonstrate that MaR1 which was administered intraperitoneally improves the survival rate of slight-grade or mid-grade CLP mice at 100 ng. Our study also shows that treatment with MaR1 even after 6 h of CLP, the survival rate in the CLP+MaR1 (post) group was higher than the CLP group, suggesting the potential of MaR1 as an effective therapeutic agent. Our data also clearly demonstrate that treatment with MaR1 inhibits the plasma inflammatory cytokine, such as TNF-α, IL-1β, MPO, MIP-2, IL-10. Decreased plasma TNF-α and IL-10 levels in MaR1-treated mice are consistent with the paradigm that MaR1 reduces inflammatory response after CLP, in addition to being associated with severity of disease [21]. IL-10 is an anti-inflammatory cytokine that has been implicated in the immunosuppression of sepsis and inability to clear pathogen [22]. Previous studies showed that CLP significantly enhanced IL-10 expression at 12, 24, 48, and 72 h when compared with the sham group [23,24,25]. LPS increased the production of inflammatory cytokine--IL-10 in isolated splenocytes [26] and IL-10 was significantly higher in severe acute pancreatitis on days 7, 9, 11, and 13 [27]. Several studies show that SPMs are important in the production of IL-10 in a sterile model of ischemia-reperfusion injury [28]. In our studies, CLP improved the IL-10 level, but after treatment with MaR1 for 24 h, the plasma levels of IL-10 were significantly decreased. Previous study also showed that plasma IL-10 levels were reduced in SPM (LXA4)-treated rats compared with CLP rats [29]. The reasons maybe the expression of IL-10 in experimental sepsis may be associated with the sepsis induction model. In addition, MaR1 intervention time and the concentration of MaR1 also affects the IL-10 level. In various stages of sepsis, pro-inflammation, anti-inflammation and immunosuppression may cause IL-10 and MaR1 to maintain balance state.
In the present study, we also found that MaR1 inhibited the growth of bacteria. Previous studies have proved that MaR1 could enhance macrophage phagocytosis [8]. In addition, treatment with MaR1 improved the macrophages and decreased the neutrophils to protect the lung tissues in CLP mice, suggesting that MaR1 may play a role in the resolution of inflammation. It has been generally believed that the SPMs exert their actions by interacting with GPCRs with high affinity and stereospecificity [30]. ALX (Lipoxin A4 Receptor) is a G-protein-coupled receptor (GPCR) with cell-type-specific signaling pathways [31]. In this study, the beneficial effects of MaR1 were abrogated by potential lack of selectivity of the BOC-2 in CLP mice, indicating that ALX works as the receptor of MaR1 and the MaR1 response is ALX dependent. Recent studies by Colas et al. [32] indicate that MaR1 antagonizes the LTB4 receptor. It is likely that LTB4 plays a role in CLP in vivo. More importantly, MaR1 has not been shown to act directly at the ALX, the lipoxin A4 receptor. Earlier studies from Levy and colleagues [33] showed that resolvin E1 stimulates endogenous lipoxin A4 levels in the lung. Thus, it is very likely that in the CLP-induced sepsis, MaR1 stimulates endogenous lipoxin A4. Our previous study also showed that MaR1 stimulates alveolar fluid clearance through the alveolar epithelial sodium channel Na, K-ATPase via the ALX/PI3K/Nedd4-2 pathway [34]. Therefore, the mechanism of MaR1 on the ALX maybe as follows: on the one hand, MaR1 may act directly at the ALX receptor. On the other hand, MaR1 may stimulate endogenous lipoxin A4 production making lipoxin A4 to act on its receptor, ALX.
cAMP is an important second messenger by which cells transduce extracellular signals into intracellular responses. ALX was the first receptor cloned and identified as a GPCR for lipoxin and resolvin with demonstrated cell-type–specific signaling pathways [31, 35]. Extracellular signals interact with GPCRs to activate the adenylate cyclase (AC) and increase the intracellular cAMP and cGMP levels. cAMP and cGMP are important second messengers by which cells transduce extracellular signals into intracellular responses [36]. There are many reports pointed out that the oxidative stress originates from an increase in mitochondrial ROS production due to the loss of cAMP dependent regulation rather than the loss of cGMP [37]. Our results are consistent with previous studies which illustrated the decrease in lung cAMP level after CLP stimulation, and which is abrogated by MaR1 in the CLP group in vivo. Furthermore, our studies also show that that the MaR1-induced increases in the levels of cAMP are blocked by BOC-2, suggesting that the MaR1 response is associated with cAMP.
ROS have been traditionally regarded as toxic by-products of oxygen consumption and cellular metabolism with the potential to cause damage to lipids, proteins, and DNA [5, 6]. What is more, they can also regulate the mitochondrial function. Indeed, ROS are produced by mitochondria during aerobic respiration as a consequence of electron transport chain activity [38]. Specific ROS generation can also be achieved enzymatically in response to various stimuli: the ROS-generating NOX, ROS-destroying CAT and SOD [39]. In this study, the increased production of ROS and NOX in CLP mice was abrogated after treatment with MaR1, while the basic effect of MaR1 was inhibited by BOC-2 and SQ22536. In addition, the decreased production of SOD and CAT in CLP mice was abolished after treatment with MaR1, while the beneficial effect of MaR1 was inhibited by BOC-2 and SQ22536. The reasons maybe as follows: Firstly, the results of earlier studies have shown that resolvins, such as resolvin E1, enhance bacterial killing and clearance by increasing intracellular ROS production in leukocytes while it reduces extracellular release of ROS that could be tissue damaging. Similarly, MaR1 maybe blocking extracellular ROS production while stimulating intracellular ROS production for bacterial killing. Secondly, it is now widely recognized that ROS are necessary in order to eradicate bacteria, but excessive production of ROS may cause tissue damage. MaR1 could moderately regulate the production of ROS, inhibiting the excessive ROS production to protect the lung injury. Finally, previous studies showed that MaR1 decreased ROS levels during lung I/R Injury [40]. The expression of ROS increased in the presence of high glucose group, but it decreased after the treatment with MaR1 at different concentrations [41]. Another study showed that ROS levels were decreased by treatment of MaR1 in Carbon Tetrachloride-Induced Liver Injury in Mice [42]. Taken together, these results, indicate that MaR1 may be regulating the NOX, SOD and CAT activity to reduce ROS production in CLP mice, and these results are dependent on ALX/cAMP.
Mitochondria play a critical role in multiple cellular functions. Dysfunction of mitochondria and the role of ROS produced by mitochondria have already been proposed in the pathogenesis of sepsis [43]. Most laboratory studies, particularly those using long-term sepsis models (>16 h), have reported a decrease in mitochondrial function, often with ultrastructural changes seen on electron microscopy. Recent studies have repeated this finding [44,45,46,47,48]. In this study, we found that LPS caused mitochondrial damage as indicated by mitochondrial swelling and the loss of mitochondrial membrane potential after LPS treatment. The loss of mitochondrial membrane potential increased the mitochondrial permeability. Furthermore, we found that MaR1 could restore the morphology of mitochondria. In addition, the decreased production of the mitochondrial respiratory chain enzyme complexes—COX I and COX IV in CLP mice was abrogated after treatment with MaR1. However, all the beneficial effects of MaR1 on mitochondria proteins were reversed by BOC-2 in vivo. In vitro, the increased ROS production induced by LPS in mitochondria was abolished by the treatment with MaR1. Taken together, these results, indicate that the MaR1 response is ALX/cAMP/ROS dependent.
To gain a better understanding of MaR1, healthy volunteers and sepsis patient’s blood was analyzed to measure the ROS and its enzymatical production. As expected, the increased ROS release and NOX activity and the decreased SOD and CAT activity in LPS-stimulated healthy volunteers’ blood was abrogated by treatment with MaR1. MaR1 also reduced ROS production and NOX activity, improved SOD and CAT activity in sepsis patient. At the same time, BOC-2 and SQ22536 abolished the beneficial effects of MaR1 in healthy volunteers and sepsis patient, indicating that MaR1 not only improve ROS generation enzyme NOX, but also reduce ROS-destroying SOD, CAT activity to inhibit ROS production to protect the patients.
In conclusion, these data demonstrate that MaR1 alleviates lung injury, reduces the release of inflammatory cytokines, and improves the survival rate partially through attenuated mitochondrial dysfunction via activation of the ALX/cAMP/ROS pathway in CLP-induced sepsis. Moreover, MaR1 also downregulated the expression of ROS both in the LPS-stimulated peripheral venous blood from healthy adult volunteers and the blood from sepsis patients’ (Fig. 13). Thus, in critically ill patients with sepsis, MaR1 has the potential to relieve serious MODS and improve outcomes. Our findings reveal a novel mechanism for attenuated mitochondrial dysfunction in sepsis and MaR1 could be exploited therapeutically for the resolution of sepsis or septic shock.

MaR1 protected CLP-induced sepsis in vivo and in vitro, and sepsis patient through ALX/cAMP/ROS pathway
References
Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54.
Nathan C. Points of control in inflammation. Nature. 2002;420:846–52.
Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50.
Vaporciyan AA, DeLisser HM, Yan HC, et al. Involvement of plateletendothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science. 1993;262:1580–2.
Ameziane-El-Hassani R, Dupuy C. Detection of Reactive Oxygen Species in Cells Undergoing Oncogene-Induced Senescence. Methods Mol Biol. 2017;1534:139–45.
Rani V, Deep G, Singh RK, et al. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016;148:183–93.
Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101.
Serhan CN, Yang R, Martinod K, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. 2009;206:15–23.
Serhan CN, Dalli J, Karamnov S, et al. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012;26:1755–65.
Dalli J, Zhu M, Vlasenko NA, et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J. 2013;27:2573–83.
Titos E, Rius B, López-Vicario C, et al. Signaling and immunoresolving actions of resolvin D1 in inflamed human visceral adipose tissue. J Immunol. 2016;197:3360–70.
Spite M, Norling LV, Summers L, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–91.
Ueda T, Fukunaga K, Seki H, et al. Combination therapy of 15-epi-lipoxin A4 with antibiotics protects mice from Escherichia coli-induced sepsis*. Crit Care Med. 2014;42:e288–295.
Chen F, Fan XH, Wu YP, et al. Resolvin D1 improves survival in experimental sepsis through reducing bacterial load and preventing excessive activation of inflammatory response. Eur J Clin Microbiol Infect Dis. 2014;33:457–64.
Chiang N, Fredman G, Bäckhed F, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–8.
Rittirsch D, Huber-Lang MS, Flierl MA, et al. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36.
Mikawa K, Nishina K, Takao Y, et al. Obara, ONO-1714, a nitric oxide synthase inhibitor, attenuates endotoxin-induced acute lung injury in rabbits. Anesth Analg. 2003;97:1751–5.
Liu D, Zeng BX, Zhang SH, et al. Rosiglitazone, a peroxisome proliferator-activated receptorgamma agonist, reduces acute lung injury in endotoxemic rats. Crit Care Med. 2005;33:2309–16.
Bouabe H, Liu Y, Moser M, et al. Novel highly sensitive IL-10-beta-lactamase reporter mouse reveals cells of the innate immune system as a substantial source of IL-10 in vivo. J Immunol. 2011;187:3165–76.
Schepetkin IA, Kirpotina LN, Khlebnikov AI, et al. Highthroughput screening for small-molecule activators of neutrophils: Identification of novel N-formyl peptide receptor agonists. Mol Pharmacol. 2007;71:1061–74.
Abraham E, Wunderink R, Silverman H, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial, TNF-alpha MAb Sepsis Study Group. JAMA. 1995;273:934–41.
Muenzer JT, Davis CG, Chang K, et al. Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun. 2010;78:1582–92.
Matsutani T, Tamura K, Kutsukake M, et al. Impact of Pioglitazone on Macrophage Dynamics in Adipose Tissues of Cecal Ligation and Puncture-Treated Mice. Biol Pharm Bull. 2017;40:638–44.
Zhang H, Cui X, Fang T, et al. Immune intervention of human umbilical cord mesenchymal stem cells on sepsis rats. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2017;29:705–10.
Zhang PP, Li ZJ, Wang DQ. Effect of Liangge powder on expression of inflammatory mediators and hepatic and renal function in rats with sepsis. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2017;35:369–72.
Tanaka H, Nishikawa Y, Fukushima T, et al. Lipopolysaccharide inhibits hepatic gluconeogenesis in rats: the role of immune cells. J Diabetes Invest. 2017. doi: 10.1111/jdi.12729. [Epub ahead of print]
Thomson JT, Brand MB, FruFonteh P. The role of IL17-A in the second hit of acute pancreatitis. S Afr J Surg. 2017;55:51.
Souza DG, Fagundes CT, Amaral FA, et al. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J Immunol. 2007;179:8533–43.
Walker J, Dichter E, Lacorte G, et al. Lipoxin a4 increases survival by decreasing systemic inflammation and bacterial load in sepsis. Shock. 2011;36:410–6.
Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. 2010;177:1576–91.
Chiang N, Serhan CN, Dahlen SE, et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–87.
Colas RA, Dalli J, Chiang N, et al. Identification and Actions of the Maresin 1 Metabolome in Infectious Inflammation. J Immunol. 2016;197:4444–445.
Haworth O, Cernadas M, Yang R, et al. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. 2008;9:873–9.
Zhang JL, Zhuo XJ, Lin J, et al. Maresin 1 stimulates alveolar fluid clearance through the alveolar epithelial sodium channel Na,K-ATPase via the ALX/PI3K/Nedd4-2 pathway. Lab Invest. 2017;97:543–54.
Wang Q, Zheng X, Cheng Y, et al. Resolvin D1 stimulates alveolar fluid clearance through alveolar epithelial sodium channel, Na,K- ATPase via ALX/cAMP/PI3K pathway in lipopolysaccharide-induced acute lung injury. J Immunol. 2014;192:3765–77.
Koyama H, Bornfeldt KE, Fukumoto S, et al. Molecular pathways of cyclic nucleotide-induced inhibition of arterial smooth muscle cell proliferation. J Cell Physiol. 2001;186:1–10.
Chevtzoff C1, Yoboue ED, Galinier A, et al. Reactive oxygen species- mediated regulation of mitochondrial biogenesis in the yeast Saccharomyces cerevisiae. J Biol Chem. 2010;285:1733–42.
Angelova PR, Abramov AY. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic Biol Med. 2016;100:81–85.
Yoshida Y, Iigusa H, Wang N, et al. Cross-talk between the cellular redox state and the circadian system in Neurospora. PLoS ONE. 2011;6:e28227.
Sun Q, Wu Y, Zhao F, et al. Maresin 1 ameliorates lung ischemia/reperfusion injury by suppressing oxidative stress via activation of the Nrf-2-mediated HO-1 signaling pathway. Oxid Med Cell Longev. 2017;2017:9634803.
Tang S, Gao C, Long Y, et al. Maresin 1 mitigates high glucose-induced mouse glomerular mesangial cell injury by inhibiting inflammation and fibrosis. Mediat Inflamm. 2017;2017:2438247.
Li R, Wang Y, Zhao E, et al. Maresin 1, a proresolving lipid mediator, mitigates carbon tetrachloride-induced liver injury in mice. Oxid Med Cell Longev. 2016;2016:9203716.
Wen X, Zhou J, Zhang D, et al. Denatonium inhibits growth and induces apoptosis of airway epithelial cells through mitochondrial signaling pathways. Respir Res. 2015;16:13.
Callahan LA, Stofan DA, Szweda LI, et al. Free radicals alter maximal diaphragmatic mitochondrial oxygen consumption in endotoxin-induced sepsis. Free Radic Biol Med. 2001;30:129–38.
Markley MA, Pierro A, Eaton S. Hepatocyte mitochondrial metabolism is inhibited in neonatal rat endotoxaemia: effects of glutamine. Clin Sci (Lond). 2002;102:337–44.
Crouser ED, Julian MW, Dorinsky PM. Ileal VO(2)-O(2) alterations induced by endotoxin correlate with severity of mitochondrial injury. Am J Respir Crit Care Med. 1999;160:1347–53.
Crouser ED, Julian MW, Weinstein DM, et al. Endotoxininduced ileal mucosal injury and nitric oxide dysregulation are temporally dissociated. Am J Respir Crit Care Med. 2000;161:1705–12.
Crouser ED, Julian MW, Blaho DV, et al. Endotoxininduced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med. 2002;30:276–84.
Acknowledgements
This work was sponsored by the grants from the National Natural Science Foundation of China (no. 81570076, no. 81571862, no. 81400059,) and supported by Natural Science Foundation of Zhejiang Provincial (LY13H150007) and Wenzhou Science and Technology Bureau Project (Y20140084). We thank Wang-Ning Shangguan, Jun Li for technical assistance. We thank Vijay D’Souza for helpful revision of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Gu, J., Luo, L., Wang, Q. et al. Maresin 1 attenuates mitochondrial dysfunction through the ALX/cAMP/ROS pathway in the cecal ligation and puncture mouse model and sepsis patients. Lab Invest 98, 715–733 (2018). https://doi.org/10.1038/s41374-018-0031-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0031-x
This article is cited by
-
Maresin1 ameliorates MSU crystal-induced inflammation by upregulating Prdx5 expression
Molecular Medicine (2023)
-
Impact of critical illness on cholesterol and fatty acids: insights into pathophysiology and therapeutic targets
Intensive Care Medicine Experimental (2023)
-
Maresin 1 protects the liver against ischemia/reperfusion injury via the ALXR/Akt signaling pathway
Molecular Medicine (2021)
-
MAR1 suppresses inflammatory response in LPS-induced RAW 264.7 macrophages and human primary peripheral blood mononuclear cells via the SIRT1/PGC-1α/PPAR-γ pathway
Journal of Inflammation (2021)
