
Abstract
Non-alcoholic fatty liver disease (NAFLD) has become the most prevalent chronic liver disease. The presence of portal hypertension has been demonstrated in NAFLD prior to development of inflammation or fibrosis, and is a result of extrahepatic and intrahepatic factors, principally driven by vascular dysfunction. An increased intrahepatic vascular resistance potentially contributes to progression of NAFLD via intralobular hypoxia. However, the exact mechanisms underlying vascular dysfunction in NAFLD remain unknown. This study investigates systemic hemodynamics and both aortic and intrahepatic vascular reactivity in a rat model of severe steatosis. Wistar rats were fed a methionine-choline-deficient diet, inducing steatosis, or control diet for 4 weeks. In vivo hemodynamic measurements, aortic contractility studies, and in situ liver perfusion experiments were performed. The mean arterial blood pressure was lower and portal blood pressure was higher in steatosis compared to controls. The maximal contraction force in aortic rings from steatotic rats was markedly reduced compared to controls. While blockade of nitric oxide (NO) production did not reveal any differences, cyclooxygenase (COX) blockade reduced aortic reactivity in both controls and steatosis, whereas effects were more pronounced in controls. Effects could be attributed to COX-2 iso-enzyme activity. In in situ liver perfusion experiments, exogenous NO donation or endogenous NO stimulation reduced the transhepatic pressure gradient (THPG), whereas NO synthase blockade increased the THPG only in steatosis, but not in controls. Alpha-1-adrenergic stimulation and endothelin-1 induced a significantly more pronounced increase in THPG in steatosis compared to controls. Our results demonstrate that severe steatosis, without inflammation or fibrosis, induces portal hypertension and signs of a hyperdynamic circulation, accompanied by extrahepatic arterial hyporeactivity and intrahepatic vascular hyperreactivity. The arterial hyporeactivity seems to be NO-independent, but appears to be mediated by specific COX-2-related mechanisms. Besides, the increased intrahepatic vascular resistance in steatosis appears not to be NO-related but rather to vasoconstrictor hyperreactivity.
Similar content being viewed by others


Introduction
Non-alcoholic fatty liver disease (NAFLD) has become the most prevalent chronic liver disease in the Western world [1]. It describes a spectrum of disease, characterized by the accumulation of fat in hepatocytes (steatosis) in the absence of secondary causes of steatosis like alcohol, viruses, or drugs. If more than 5% of the hepatocytes are affected by steatosis [1] without signs of hepatocellular damage, it is referred to as non-alcoholic fatty liver (NAFL) or simple steatosis. In about 20–30% of patients, there is associated inflammation and degenerative injury of the hepatocytes (ballooning), defining non-alcoholic steatohepatitis (NASH). NASH potentially leads to (pericellular) fibrosis ending in cirrhosis and hepatocellular carcinoma [2], and NAFLD is estimated to become the main indication for liver transplantation within a decade [3]. Moreover, NAFLD is not only closely associated with the metabolic syndrome (describing obesity, diabetes mellitus, arterial hypertension, and dyslipidemia), but adds independently to the already increased cardiovascular risk in this patient population [4,5,6].
The pathophysiology of NAFLD is not yet fully elucidated [4], but is believed to be subject to multiple ‘hits’ [7]. Several data suggest an early role for vascular alterations in this process [8] that may be one of these hits. This is supported by the fact that steatotic livers appear to be more vulnerable to ischemia–reperfusion injury [9, 10].
The changes in liver blood flow were documented both in animals [11] and in patients [12]. Previous research demonstrated the presence of portal hypertension in patients with NAFLD prior to the development of inflammation or fibrosis [13, 14]. An increased portal blood pressure can be a result of intra- and extrahepatic alterations.
Splanchnic vasodilation was demonstrated, causing an increased portal blood inflow, contributing to an increase in portal pressure (PP) [15,16,17]. Furthermore, intrahepatic vascular resistance (IHVR) has been shown to be increased, both by dynamic and structural alterations of the hepatic vasculature (e.g., as a result of swollen hepatocytes due to ballooning or fat accumulation) and an increased vascular tone [18]. In physiological circumstances, the intrahepatic vascular tone is maintained at a constant level by several regulatory mechanisms in the hepatic vasculature [19]. These appear to be impaired in chronic liver diseases, favoring intrahepatic vasoconstriction [20, 21]. Recently, endothelial dysfunction was reported in animal models of NAFL as well [18, 22,23,24]. Via local hypoxia these mechanisms possibly contribute to and promote the progression of NAFL to more severe stages of NAFLD [8, 13]. Therefore, the presence of both intra- and extrahepatic vascular alterations in NAFL, outside the setting of cirrhosis, might be important in the pathophysiology of NASH and the reported link between NAFLD and cardiovascular disease. At present, little is known about the underlying mechanisms of these alterations.
The aim of this study was to examine both hepatic and extrahepatic vascular alterations in severe steatosis, with emphasis on possible underlying mechanisms of the observed portal hypertension in NAFL, in an animal model of NAFLD.
Materials and methods
Animal model
Male Wistar rats (Charles River, France) (200–250 g) were fed a methionine-choline-deficient (MCD) diet or a standardized control diet (ICN Biomedicals SA, Asse, Belgium) for 4 weeks. The MCD diet is an established model for induction of severe steatosis after 3–4 weeks of diet, without, at this early stage, the histological presence of inflammation or fibrosis [8, 18, 25]. The animals were allowed unlimited access to their food and water, were kept in cages of up to two animals, and were treated according to the Helsinki declaration, the national guidelines for animal protection and the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health, 2011). The protocols were approved by the Antwerp University Ethical Committee on Animal Experiments (ECD 2012-40 and 2016-66).
Hemodynamic measurements
After 4 weeks of diet and after an overnight fast, animals were weighed and anesthetized (see section ‘Drugs’). In animals dedicated to organ bath experiments (n = 12), a tracheal tube (PE 240; ID 1.67 mm OD 2.42 mm, Intramedic Clay Adams Brand) was inserted by tracheostomy and a 24G catheter was inserted into the carotid artery. The abdomen was subsequently opened by median incision. The portal vein was exposed and cannulated with a 24G catheter under microscopy. The abdominal caval vein was cannulated with a 22G catheter, which was moved into the retrohepatic part of the vein. The different catheters were connected to in-house pressure monitoring equipment. Carotid artery pressure (mean arterial blood pressure, MABP), pulse rate, PP, and caudal caval vein pressure were measured, and the transhepatic pressure gradient (THPG) was calculated after subtracting the caudal caval vein pressure from the PP.
After hemodynamic measurements, the animals were sacrificed by cardiac excision. The thoracic and abdominal aorta were removed and stored in cold Krebs–Ringer solution for organ bath experiments. The liver was removed and weighed. Tissue samples of two different liver lobes were fixed in formalin aldehyde 40% for histology.
Organ bath experiments
In a first series of experiments, referred to as series 1, the abdominal and thoracic aorta of control rats and steatotic rats (n = 12 in each group) were cleaned from surrounding tissue, and 2 mm rings were mounted in an organ bath chamber and connected to a tension transducer as previously described [26]. The organ chambers were filled with Krebs–Ringer solution at a constant temperature of 37 °C and continuously oxygenated (with a gas mixture 95% O2/5% CO2). The aortic rings were stabilized under a preload tension of 2 g, which was established as the optimal preload in a preliminary set of experiments (data not shown). Once stabilization was achieved, the basal tension was set as the zero level.
The organ chamber was then flushed with 50 mM K+ Krebs–Ringer solution, causing non-receptor-dependent contraction. The absolute K+-induced contraction forces were compared between the control animals and the steatotic animals. The absolute value of tension was subsequently used as the 100% reference contraction value, to correct for differences in aortic ring length, possible surrounding tissue, and other confounding factors. The 50 mM K+ solution was subsequently removed by flushing the organ chambers at least three times with isotonic Krebs–Ringer solution. After the vessels had returned to the basal contraction level, a dose–response curve of phenylephrine (PE, an alpha-1 adrenergic receptor agonist, 3 × 10−9 to 3 × 10−5 M) was established. Contractions were expressed as the percentage of the potassium-induced pre-contraction. The Emax (maximal tension/contraction) and the pD2 (i.e., the negative logarithm of the concentration corresponding with a contraction of 50% of the maximum contraction) were established. After flushing and a return of the vessel tension to the basal level, the dose of PE corresponding to the pD2 was added. When a stable contraction was achieved, a dose–response curve to ACh (causing an endothelium-dependent smooth muscle cell relaxation, 3 × 10−9 to 10−5 M) was established. The ACh-induced relaxation served as a control for endothelial integrity. After flushing and returning to baseline, the vessels were randomly divided into two groups. In one group Nω-nitro-l-arginin methyl ester (l-NAME, a non-specific NO synthase [NOS] inhibitor) was added in a final organ bath concentration of 3 × 10−4 M, while the other group served as a time control. Afterwards, a new dose–response curve to PE was established.
To verify the reproducibility of the findings of series 1, the entire protocol was subsequently repeated in a second series of experiments referred to as series 2 (n = 12 in each group). In the last part of the protocol, the vessels were divided in three groups: a time control, a group with addition of l-NAME, and a group with addition of indomethacin (a non-specific cyclooxygenase [COX] inhibitor) in a final organ bath concentration of 10−5 M.
As indomethacin appeared to have a significant effect that differed between rats with steatosis and controls (vide infra), the entire protocol was subsequently repeated in another series of experiments referred to as series 3 (n = 6 in each group). Since indomethacin not only has COX inhibitory features, but might exert its action via direct calcium channel-mediated effects [27], we repeated the experiments with another non-specific COX inhibitor, piroxicam (Px), which lacks these direct effects on calcium channels. Px was used in an organ bath concentration of 10−5 M. Furthermore, two groups were added to differentially study two COX iso-enzymes using SC560, a selective COX-1 inhibitor, and NS398, a selective COX-2 inhibitor, both in a final organ bath concentration of 10−5 M.
Liver perfusion studies
The THPG was assessed directly by in situ ex vivo liver perfusion experiments (n = 4–11) as described previously [28, 29] (Fig. 1). Briefly, the abdomen was opened via median laparotomy and vascular structures were identified. Heparin (1400 U/kg) was injected intravenously in the caudal caval vein. Subsequently, the portal vein was cannulated with a 14G catheter. The thorax was opened and the suprahepatic caval vein was cannulated through the right atrium with a 16G catheter. The liver was perfused in single-pass mode by oxygenated Krebs–Ringer solution (37 °C) and the catheters were connected to a pressure and flow monitoring equipment. In all experiments, the portal (inflow) and caval (outflow) pressure were measured continuously and the THPG was calculated after subtracting the outflow from the inflow pressure.

Schematic representation of the in situ ex vivo liver perfusion model. The portal vein and the suprahepatic caval vein are cannulated, whereas the caudal caval vein, hepatic artery, and bile duct are ligated. A calibrated flow is applied by a pump and perfuses the liver. An bubble trap prevents air emboli. Pressure is measured in the inflow and in the outflow tract
In dose–response experiments, a syringe pump with compound was connected to the perfusion model. It was turned on after a stabilization period of 20 min at a flow of 30 mL/min. The dose was increased by 0.5 log every 5 min and the THPG was continuously measured while increasing the dose. The following compounds were tested in dose–response experiments: 10−7–3 × 10−5 M ACh (endothelial-dependent vasodilator), 10−6–3 × 10−4 M methoxamine (Mx, alpha-1-adrenoceptor agonist), 3 × 10−7–10−4 M prazosin (alpha-1-adrenoceptor antagonist, 10−5–10−2 M sodium nitroprusside (SNP, NO donor), and 10−8–3 × 10−10 M endothelin-1 (ET-1). Results are expressed in relative changes (i.e., increase or decrease in THPG compared to the starting value before adding the compound).
In flow–pressure experiments, the liver was connected to the perfusion model. After 10 min of stabilization of the THPG at a constant flow of 10 mL/min Krebs–Ringer solution, drugs were added to the perfusate, followed by another stabilization period of 10 min. Subsequently, flow was increased by 5 mL/min once every 5 min, up to a flow of 50 mL/min. The following doses were used in flow–pressure experiments: 10−6 M ACh, 10−5 M Mx, 10−3 M Nω-nitro-l-arginine (l-NNA, non-specific inhibitor of NOS), 10−3 M SNP, and 10−10 M ET-1.
Histology
After hemodynamic measurements, the liver tissue samples were fixated, sectioned, and stained with hematoxylin–eosin and Masson’s trichrome, and scored in a blinded way by one single experienced pathologist using the NASH Clinical Research Network Scoring System [26].
Drugs
Animals were anesthetized with Nembutal® (Ceva Sante Animale, Brussels, Belgium): Natrii Pentobarbitalum 60 mg/1 mL, 30 mg/kg body weight intraperitoneally. The isotonic Krebs–Ringer solution had the following composition: KCl 4.75 mM, MgSO4·7H2O 1.2 mM, KH2PO4 1.2 mM, CaCl2·2H2O 2.5 mM, CaEDTA 0.03 mM, NaCl 118.5 mM, NaHCO3 25 mM, glucose 10 mM; pH = 7.4. The 50 mM K+ Krebs–Ringer solution has the following composition: KCl 48.8 mM, MgSO4·7H2O 1.2 mM, KH2PO4 1.2 mM, CaCl2·2H2O 2.5 mM, CaEDTA 0.03 mM, NaCl 118.5 mM, NaHCO3 25 mM, glucose 10 mM; pH = 7.4. ACh was purchased from Pharmacobel SA, Laboratoires Sterop SA, Brussels, Belgium. K+, PE hydrochloride, SC560, NS398, l-NAME, l-NNA, SNP, Px, and ET-1 were purchased from Sigma-Aldrich Chemie Gmbh (Steinheim, Germany). Indomethacin was purchased from Federa SA (Brussels, Belgium). Prazosin was purchased from Alfa Aesar (Karlsruhe, Germany). Because of fast degradation, SNP was not brought to body temperature until 10 min before its administration to the liver and the reservoir was protected against light during the experiment. All drugs were first dissolved in aqua destillata before diluting them in the Krebs–Ringer solution to obtain the given concentrations. All solutions were freshly prepared on the day the experiments were performed.
Statistics
The contractions measured in organ bath experiments were analyzed with GraphPad Prism© Version 4.00. Dose–response curves were compared with nonlinear regression (curve fit). The Emax and pD2 were subsequently analyzed with a two-way ANOVA (with the absence or presence of steatosis as first factor [between] and the drug used as second factor [within]) and Bonferroni post-hoc testing if appropriate using SPSS v22.0. The other results were analyzed with Student’s t-test (continuous variables), Chi-square (categorical variables), and Mann–Whitney U test (histology scores). Flow–pressure curves were compared using the generalized estimating equation model followed by least significant difference post-hoc test when appropriate, using SPPS. p-Values <0.05 were considered significantly different.
Results
Establishing NAFL without inflammation or fibrosis
After 4 weeks of diet, all animals receiving the MCD diet developed signs of severe liver steatosis as further validated in the rats of series 1 and 2, resulting in marked hepatomegaly: the liver/body weight ratio was significantly increased in steatotic compared to control animals (4.93 ± 0.13 vs. 3.23 ± 0.08, p < 0.001). The increase in liver volume and liver/body weight ratio corresponded with histology, confirming severe steatosis (>60% of the hepatocytes affected) in all the treated rats (Table 1 and Figure S1). Besides discrete lobular inflammation (score 1) in 4/24 (17%) and 3/24 (13%) in series 1 and 2, respectively, other light microscopic features of NASH were absent in the steatotic group (Table 1). Fibrosis, as assessed by Masson’s trichrome staining, was completely absent in all MCD-fed rats. In all of the control animals, liver histology was strictly normal (Figure S1).
Hemodynamic measurements
In the steatotic animals of series 1, the THPG was markedly elevated compared to controls, and the difference was highly significant: controls 2.3 ± 0.5 mmHg vs. steatosis 9.5 ± 0.5 mmHg (p < 0.001) (Table 1). These results, suggestive of an increased IHVR, were reproduced in series 2 where controls had a THPG of 3.0 ± 0.5 vs. 9.5 ± 0.8 mmHg in steatotic rats (p < 0.001) (Table 1).
In steatotic animals, MABP was significantly lower (98.7 ± 5.7 mmHg) compared to the control group (123.8 ± 1.8 mmHg) in series 1 (p < 0.005), which was confirmed in series 2. The pulse rate, which is already high in small rodents, did not change significantly throughout both the series (Table 1).
Organ bath experiments
Vascular contractility studies (series 1) and the effect of l-NAME
The potassium-induced contraction of the abdominal aorta was comparable between controls and animals with steatosis: 1.72 ± 0.19 vs. 1.43 ± 0.12 g, respectively (p = 0.213) (n = 12). The KCl-induced contraction could therefore be used as a 100% reference. Contractions were subsequently expressed as relative change of the KCl-induced pre-contraction (%).
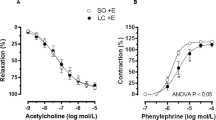
The Emax to PE was significantly lower in the abdominal aorta in rats with steatosis: 117.8 ± 3.8% vs. 150.8 ± 5.7% in controls (p < 0.001; Fig. 2a). The dose–response curve was shifted downwards, however, with no significant change in the pD2 value: 8.60 ± 0.79 in animals with steatosis vs. 7.99 ± 0.59 in controls (p = 0.546; Fig. 2a). No significant differences were demonstrated in the thoracic aorta (results not shown). ACh-induced relaxations were comparable between steatosis and controls, and confirmed endothelial integrity (results not shown).

Dose–response curves of abdominal aortic rings to phenylephrine (PE, dose range 3 × 10−9–3 × 10−3 M) of control and steatotic animals (n = 12 in each group). PE phenylephrine, l-NAME Nω-nitro-l-arginin methyl ester. a The maximal contraction response Emax to PE was significantly lower in the abdominal aorta in steatosis compared to controls (nonlinear regression (curve fit), p < 0.0001). There was no significant change in the pD2 value. b The time controls confirmed the hyporesponsiveness (i.e., a lower Emax) in rats with steatosis compared to controls. l-NAME did not significantly alter the response to PE both in animals with steatosis and in controls at the level of Emax or pD2. c Indomethacin shifted the curves significantly downwards both in controls and in rats with steatosis. In the control group the shift of the curve was more pronounced compared to steatosis (p < 0.001 for the maximum contraction). Both effects of steatosis as the effect of indomethacin were highly significant (p < 0.001), as was the interaction (p < 0.001) demonstrating that the effect of indomethacin is significantly modulated by the presence or absence of steatosis. d Piroxicam (Px) significantly reduced Emax both in controls (from 145.6 ± 42.7% to 80.6 ± 29.7%, p < 0.001) and in steatosis (from 82.1 ± 33.9% to 51.3 ± 22.7%, p = 0 < 0.05) and the effect was significantly more pronounced in controls than in steatosis (mean decrease 44.7 ± 4.5% in controls vs. 31.9 ± 2.9% in steatosis, p < 0.05). Px did not cause a significant shift of the pD2. e SC560 had no effect on vascular reactivity, both in control and in rats with steatosis. f NS398 reduced Emax in controls (p < 0.001) and in steatosis (p < 0.05) and the effect was significantly more pronounced in controls than in steatosis (mean decrease 46.6 ± 4.4% vs. 19.9 ± 2.6%, p < 0.001)
Subsequently, the effect of l-NAME on the PE-induced contraction was examined. The time controls confirmed the hyporesponsiveness (i.e., a lower Emax) in rats with steatosis compared to controls (Fig. 2b). l-NAME did not significantly alter the response to PE both in animals with steatosis and in controls, both at the level of Emax or pD2 (Fig. 2b).
Vascular contractility studies (series 2) and the effect of l-NAME and indomethacin
The difference in Emax between animals with steatosis and control animals was reconfirmed: 96.4 ± 2.3% vs. 149.9 ± 3.6%, respectively, p < 0.001. Again, there was no significant difference in pD2 and l-NAME did not alter the response to PE (data not shown).
Indomethacin shifted the curves significantly downwards both in controls and in rats with steatosis (Fig. 2c). In the control group, however, the shift of the curve was more pronounced as compared to rats with steatosis. This differential effect of indomethacin between control rats and rats with steatosis was significant (maximal contraction 84.9 ± 3.2% in steatosis vs. 61.3 ± 7.6% in controls, p < 0.001), meaning that not only steatosis or indomethacin per se were of influence, but also the effects of indomethacin were modulated by the presence of steatosis. Two-way ANOVA for repeated measurements showed highly significant results for both steatosis (p < 0.001) and the effect of indomethacin (p < 0.001), with a highly significant interaction between these factors (p < 0.001). This analysis hence not only confirms that the maximum contraction is significantly influenced by the presence of steatosis and by the addition of indomethacin, but, as the interaction is significant, it also shows that the effect of indomethacin is significantly modulated by the presence or absence of steatosis.
Vascular contractility studies (series 3) and the effect of Px, SC560, and NS398
As the results of series 2 showed a significant effect of indomethacin, the effect of another non-specific COX inhibitor was studied. Series 3 confirmed the downward shift of the contraction curve in rats with steatosis compared to control rats. Emax was significantly lower in steatosis (p = 0.001), pD2 was not significantly different. The values were comparable to those of series 2.
The addition of Px significantly reduced Emax both in controls (from 145.6 ± 42.7% to 80.6 ± 29.7%, p < 0.001) and in steatosis (from 82.1 ± 33.9% to 51.3 ± 22.7%, p < 0.05) and the effect was significantly more pronounced in controls than in steatosis (mean decrease 44.7 ± 4.5% in controls vs. 31.9 ± 2.9% in steatosis, p < 0.05). Px did not cause a significant shift of the pD2 (Fig. 2d).
SC560, a selective COX-1 inhibitor, had no effect on vascular reactivity, both in control and in rats with steatosis (Fig. 2e). However, NS398, a selective COX-2 inhibitor, reduced Emax in controls (p < 0.001) and in steatosis (p < 0.05) and the effect was significantly more pronounced in controls than in steatosis (mean decrease 46.6 ± 4.4% vs. 19.9 ± 2.6%, p < 0.001; Fig. 2f).
Liver perfusion experiments
In all experiments, the THPG in steatosis was significantly increased compared to controls (without the addition of any compounds at the different flows tested) (Fig. 3).

Flow–pressure curve of liver perfusion in control (n = 11) and steatotic (n = 8) animals without the addition of compounds. The transhepatic pressure gradient (THPG) in steatosis (5.41 ± 0.26 mmHg at 30 mL/min) was significantly increased compared to controls (4.36 ± 0.23 mmHg at 30 ml/min, p < 0.01). Data were analyzed using the generalized estimating equation model; significances between control and steatotic livers are demonstrated
Dose–response curves to the alpha-1-agonist Mx demonstrated a significantly increased sensitivity and responsiveness to Mx in livers of steatotic rats compared to the control group, with a pD2 of 4.76 in control livers and 5.00 in steatotic livers (p < 0.001) and an Emax of 8.23 mmHg in controls and 9.79 mmHg in steatosis, respectively (Fig. 4a). The THPG remained significantly elevated when Mx was added to the perfusate, both in control animals and steatotic animals at all flows (Fig. 4b). The change in THPG was, in line with the dose–response experiments, significantly more pronounced in steatosis compared to controls (steatosis + 5.10 ± 0.57 mmHg vs. controls + 3.10 ± 0.42 mmHg at 30 mL/min, p < 0.001; Fig. 4c). Dose–response curves to prazosin, used to detect the potential underlying presence of alpha-1-vasoconstriction, showed no significant effects on the THPG in both control and steatotic livers (data not shown). Therefore, consecutive flow–pressure experiments were not performed with prazosin.

Dose–response and flow–pressure curve to methoxamine (Mx) in liver perfusion experiments. a Dose–response curve of the transhepatic pressure gradient (THPG) to Mx in control (n = 7) and steatotic (n = 8) animals. There is a significantly increased sensitivity and responsiveness to Mx in steatotic livers compared to the control group, with a pD2 of 4.76 in control livers and 5.00 in steatotic livers (p < 0.001). Data were analyzed using the generalized estimating equation model; significances between control and steatotic livers are demonstrated. b Flow–pressure curve of the THPG in control (n = 8) and steatotic (n = 6) animals with the addition of Mx, causing a highly significant increased THPG, both in control animals and steatotic animals at all flows. Data were analyzed using the generalized estimating equation model; significances between THPG with or without Mx are demonstrated. c Flow–pressure curve of the relative change in THPG (b) in control (n = 8) and steatotic (n = 6) animals after the addition of Mx. The change in THPG was significantly more pronounced in steatosis compared to controls (steatosis + 5.10 ± 0.57 mmHg vs. controls + 3.10 ± 0.42 mmHg at 30 mL/min, p < 0.001). Data were analyzed using the generalized estimating equation model; significances between THPG with or without Mx are demonstrated
ET-1 induced a dose-dependent increase of the THPG in both controls and steatosis, with significantly increased sensitivity and responsiveness to ET-1 in steatosis (20.26 ± 1.31 mmHg at 3 × 10−10 M) compared to controls (14.90 ± 1.35 mmHg at 3 × 10−10 M, p < 0.001; Fig. 5a). Flow–pressure experiments confirmed the hyperreactivity to ET-1 in steatotic livers with a more rapid and higher increase in THPG and the maximum THPG was reached at significantly lower flows compared to controls (controls 15.33 ± 1.10 mmHg, steatosis 23.83 ± 0.65 mmHg at 30 mL/min, p < 0.001; Fig. 5b).

Dose–response and flow–pressure curve to endotheline-1 (ET-1) in liver perfusion experiments. a Dose–response curve of the transhepatic pressure gradient (THPG) to ET-1 in control (n = 7) and steatotic (n = 8) animals. There is a significantly increased sensitivity and responsiveness to ET-1 in steatotic livers compared to the control group, with respectively an increase in THPG of 13.03 ± 1.43 mmHg compared to 6.59 ± 1.67 mmHg at 3 × 10−10 M ET-1, p < 0.001. Data were analyzed using the generalized estimating equation model; significances between control and steatotic livers are demonstrated. b Flow–pressure curve of the THPG in control (n = 8) and steatotic (n = 7) animals with the addition of ET-1. Steatotic livers demonstrate a more rapid and a higher increase in THPG compared to controls, while the maximum THPG was reached a significantly lower flows (controls 15.3 ± 1.1 mmHg at 30 mL/min, steatosis 23.8 ± 0.6 mmHg at 30 mL/min, p < 0.001). Data were analyzed using the generalized estimating equation model; significances between THPG with or without ET-1 are demonstrated
Blocking NO production by l-NNA induced a significant increase of the THPG in steatosis at higher flows (45–50 mL/min) (11.07 ± 0.50 saline to 13.89 ± 1.20 mmHg l-NNA at 50 mL/min, p < 0.05), while the THPG remained unaltered in controls (9.45 ± 0.55 saline to 9.88 ± 0.60 mmHg l-NNA at 50 mL/min, p = 0.57; Fig. 6).

Flow–pressure curve of the transhepatic pressure gradient (THPG) in control (n = 8) and steatotic (n = 7) animals with the addition of Nω-nitro-l-arginine (l-NNA), inducing a significant increase of the THPG in steatosis at higher flows (45–50 mL/min) (11.07 ± 0.50 saline to 13.89 ± 1.20 mmHg l-NNA at 50 mL/min, p < 0.05). The THPG remained unaltered in controls (9.45 ± 0.55 saline to 9.88 ± 0.60 mmHg l-NNA at 50 mL/min, p = 0.57). Data were analyzed using the generalized estimating equation model; significances between THPG with or without l-NNA are demonstrated
To differentiate between a difference in NO production, or reactivity of the hepatic contractile elements to NO, the THPG was also analyzed using, respectively, ACh and SNP. ACh decreased the THPG dose-dependently in both groups, but this decrease was more pronounced in steatosis (controls from 4.87 ± 0.29 in saline to 4.51 ± 0.29 mmHg in 10−6 M ACh, p < 0.001; steatosis from 6.51 ± 0.54 in saline to 6.01 ± 0.54 mmHg in 10−6 M ACh, p < 0.001; Fig. 7a). Flow–pressure curves did not show significant shifts following the addition of ACh (Fig. 7b).

Dose–response and flow–pressure curve to acetylcholine (ACh) in liver perfusion experiments. a Dose–response curve of the transhepatic pressure gradient to ACh in control (n = 8) and steatotic (n = 7) animals. The transhepatic pressure gradient (THPG) was decreased dose-dependently in both groups (controls 4.87 ± 0.29 in saline to 4.51 ± 0.29 mmHg in 10−6 M ACh, p < 0.001; steatosis 6.51 ± 0.54 in saline to 6.01 ± 0.54 mmHg in 10−6 M ACh, p < 0.001), but this decrease was more pronounced in steatosis. Data were analyzed with using the generalized estimating equation model; significances between control and steatotic THPG are demonstrated. b Flow–pressure curve of the THPG in control and steatotic animals (n = 8 per group) with the addition of acetylcholine. There was no significant shift of the curves on the addition of acetylcholine. Data were analyzed using the generalized estimating equation model
Constructing dose–response curves with SNP, a decrease in the THPG in both groups was observed. When compared to saline, this decrease was significantly more pronounced in controls compared to steatosis (controls from 6.66 ± 0.53 in saline to 5.21 ± 0.32 mmHg in 10−3 M SNP vs. steatosis from 7.18 ± 0.64 in saline to 6.40 ± 0.48 mmHg in 10−3 M SNP, p < 0.05 (Fig. 8a). Flow–pressure curves did not show significant shifts on addition of this NO donor in both groups (Fig. 8b).

Dose–response and flow–pressure curve to sodium nitroprusside (SNP) in liver perfusion experiments. a Dose–response curve of the transhepatic pressure gradient (THPG) to SNP in control (n = 6) and steatotic (n = 5) animals. SNP decreased the THPG in both groups, but this decrease was significantly more pronounced in controls compared to steatosis (in steatosis a decrease of 0.78 ± 0.33 mmHg in 10−3 M SNP compared to saline vs. a decrease of 3.00 ± 1.44 mmHg in 10−3 M SNP compared to saline in controls, p < 0.05). Data were analyzed using the generalized estimating equation model; significances between control and steatotic THPG are demonstrated. b Flow–pressure curve of the THPG in control and steatotic animals (n = 8 per group) with the addition of SNP. There are no significant shifts in both groups. Data were analyzed using the generalized estimating equation model
Discussion
In this study we investigated both systemic and hepatic hemodynamics in severe steatosis and showed that the PP and THPG are significantly elevated. This confirms previous data demonstrating the presence of portal hypertension in NAFL, both in animal models [15, 22, 30] and in patients [13, 14], making it a relevant finding as this might lead to a therapeutic target in early NAFLD. In our rats fed the MCD diet for 4 weeks, microscopy confirmed severe steatosis after 4 weeks of MCD diet, without the histological presence of inflammation or fibrosis. These data therefore imply that the presence of portal hypertension is induced by a steatosis-related mechanism and is an early event in NAFLD pathogenesis. Even though the MCD diet does not resemble the typical systemic metabolic alterations that are seen in patients with NAFLD, this model can be considered valid for intrahepatic alterations and extrahepatic vascular alterations [31].
Hemodynamic assessment showed signs of a hyperdynamic systemic circulation in our animal model as classically seen in portal hypertension: a decrease in MABP, due to decreased vascular resistance, and a compensatory increase in cardiac pulse rate [32]. The latter, however, was not demonstrated in our animal model, though might partly be due to the already high baseline pulse rate in rodents. In a former set of experiments, the splanchnic blood flow itself was measured and shown to be increased [18]. We therefore conclude that steatosis per se induces changes in systemic hemodynamics resembling those seen in cirrhosis [16].
In organ bath experiments, we found a reduced response to PE in the abdominal aorta of rats with severe steatosis. This hyporesponsiveness was not related to altered contractile capabilities or structural changes of the vascular smooth muscle layer, demonstrated by unaltered potassium-induced vasoconstriction, nor to confounding factors, demonstrated by the use of potassium-induced contraction as a 100% reference value. A test for relaxation on administration of ACh confirmed endothelial integrity in steatosis, as endothelial damage might also contribute to differences in vasopressor response. Hence the altered response to PE depends on other vasoactive modifiers.
Most published papers favor a crucial role for NO in portal hypertension-associated vascular hyporeactivity of the splanchnic inflow [33, 34], but this has not always been confirmed [35]. In our experiments, non-specific NOS inhibition did not significantly alter the vascular response, suggesting that NO does not play a major role in vascular hyporeactivity in our steatosis model. Therefore the mechanism of arterial vasodilation in steatosis-induced portal hypertension might differ from the mechanism in liver cirrhosis.
Other potentially interesting mediators of vascular tone are eicosanoids, which are vasoactive products formed by COX. The COX enzyme catalyzes the formation of, among others, prostacyclin (PGI2) and TXA2 out of arachidonic acid. PGI2 acts as a vasodilator, while TXA2 acts as a vasoconstrictor via its receptors on vascular smooth muscle cells. PGI2 has been implicated in the pathogenesis of the hyperdynamic systemic circulation, vascular hyporesponsiveness, and the rise in PP seen in portal hypertension [36]. Therefore, we studied the effect of COX inhibition on the abdominal aorta. Non-selective COX inhibition diminished the vascular response, more pronounced in controls than in steatosis. Furthermore, selective inhibition of COX-1 (constitutional) showed no effects, while COX-2 (inducible) inhibition completely mimicked the results of indomethacin and Px. We therefore conclude that the observed COX-mediated effects are entirely COX-2-related. Hence, we hypothesize that COX-2 mediated vasoconstrictive eicosanoids, potentially TxA2, play an important role in the regulation of the normal vascular tone. In severe steatosis, however, TxA2 availability might be reduced, explaining the vascular hyporesponsiveness that we observed and the relative larger decrease in controls compared to steatosis.This reduced COX-mediated vasoconstriction in steatosis is different from other models of portal hypertension where NO plays the major role to regulate the inflow [36]. An upregulation of COX enzymes, inducing vasodilation via PGI2, was seen in cirrhosis [37]. The observations in our steatosis model might be due to a reduced expression and/or activity of COX-2, or a change in the balance between COX-dependent vasoconstrictor and vasodilator eicosanoids. Now that the importance of a COX-2-related mechanism has been demonstrated, further study is warranted to elucidate the exact mechanisms of these changes.
The THPG measured either in vivo in the hemodynamic studies as well as in the in situ ex vivo liver perfusion experiments was significantly higher at baseline in steatotic livers compared to controls, reproducing previous findings [18, 23]. This implies an imbalance in the regulation of the intrahepatic vascular tone, similar to what is seen at the extrahepatic level and as is known in cirrhosis [16]. Hence, the intrahepatic vasculature was further studied.
Striking differences could be observed in reaction to alpha-1-adrenergic stimulation. Mx, an alpha-1 agonist like PE, induced vasoconstriction in both control and steatotic livers, with a significant hypersensitivity (pD2) and hyperresponsiveness (Emax) to Mx in steatosis compared to controls. Intrahepatic hyperresponsiveness to vasoconstrictors is in line with observations in other models of chronic liver disease. Graupera et al. [38] demonstrated COX-1-mediated hyperreactivity to Mx in cirrhotic livers and Laleman et al. [39] demonstrated hyperreactivity in cirrhotic livers that could be reversed by an NO-releasing COX inhibitor. In previous perfusion experiments with Mx, we demonstrated a dose-dependent increase of the THPG after the administration of Mx in control livers whereas this was barely the case in steatotic livers [18]. We presently hypothesize that this is the result of an initially present vasoconstriction in steatotic livers in the previous experiments, where we reported higher differences in basal conditions, disguising any response to the administration of vasoconstrictors. Alpha-1-adrenergic antagonism, to detect the potential underlying presence of alpha-1-vasoconstriction, did not alter the THPG in both control and steatotic livers.
Since potassium-induced receptor-independent vasoconstriction remained unaltered between experimental groups (data not shown) while vasoactive compounds caused different reactions, contractile elements of the intrahepatic vasculature appear to be intact in NAFL, pointing towards a difference in sensitivity or number of receptors that influence the vascular tone. As the endothelium has been implied to play the most important role in the regulation of vascular tone, NO-related mechanisms, which are a hallmark of endothelial function, were further investigated.
NO decreased the THPG in control and steatotic livers. On the one hand, after direct administration of NO, the decrease of THPG was less pronounced in steatotic animals as compared to controls. This suggests hyporeactivity to NO in steatosis. On the other hand, when NO production was stimulated by ACh, the THPG was lowered more in steatotic than in control animals, suggesting a potential adaptation mechanism with a more pronounced response to Ach receptors in steatosis as a (though insufficient) compensation for an increased vasoconstrictor mechanisms or the hyposensitivity to NO. Both the significant increase of the THPG after NOS inhibition (at higher flows) and the less pronounced effect of exogenous NO administration in steatosis compared to controls point towards an already more pronounced underlying NO-mediated vasodilation in steatosis, potentially masking any change in the THPG caused by the addition of NO in our model, and implying that other than the NO-mediated pathways are more likely involved in the observed increased IHVR.
These results are conflicting with observations in other studies in NAFL or with what is known in cirrhosis. We previously reported that the response to ACh in normal livers was impaired in severe steatosis, while the levels of endothelial NOS remained unaltered [18]. In an in situ ex vivo liver perfusion study of steatotic rat livers, decreased NOS activity was demonstrated [22] with subsequently decreased vasodilation in response to ACh, before the development of inflammation or fibrosis [16]. Moreover, Gonzalez-Paredes et al. [23] recently demonstrated decreased levels of NO in steatotic livers compared to controls. Besides, vasodilation was reduced after blockade of NOS, but was unaltered in steatosis.
Statins, frequently used in patients with NAFLD patients, have been shown to have beneficial effects on the PP in cirrhosis [40, 41] that are potentially explained by NO stimulation and reduced fibrogenesis and angiogenesis [42,43,44]. Furthermore, oral administration of liver-specific NO-donor NCX-1000 to biliary cirrhotic rats has been demonstrated to decrease the PP, with no effects on the systemic blood pressure due to a high first-pass effects [45]. Even better results were achieved by oral administration of AVE 9488 in billiary cirrhotic rats, which enhances transcription of NOS, in which not only PP was lowered but also splanchnic vascular resistance was increased [46]. However, so far statins, CCX-1000 nor AVE 9488 have been tested in early NAFLD.
ET-1 can act as both a vasodilator and vasoconstrictor. In the liver, its effects are mainly vasoconstrictive, mediated by the ETA receptor on hepatic stellate cells [16]. An increased response to ET-1 was already demonstrated in cirrhosis [47, 48]. Overexpression of endothelins has been suggested to be the cause, as the hepatic concentration of ET-1 and the ET receptor expression appear to be increased both in human and experimental cirrhosis [20]. The increased expression of ET-1 was also demonstrated in NAFLD-related fibrosis [49], as well as in NAFL [18], while the level of ETA and ETB receptors seems to remain unaltered [18]. Thus, the sensitivity of ET-1 receptors and the concentration of ET-1 itself are most likely increased, explaining the hyperreactivity of the hepatic vasculature to ET-1, as observed in our study, and highlighing its potential role in the increased THPG in NAFLD.
In conclusion, our study reconfirms the presence of portal hypertension in NAFL(D), which can be seen as an early pathophysiologically meaningful event. In contrast to cirrhosis, NO-mediated mechanisms seem to be less relevant, both extra- and intrahepatically. The arterial hyporeactivity appears to be related to a specific COX-2-related mechanism, while the increased IHVR appears to be related to hyperreactivity to vasoconstrictive mediators.
References
Paredes AH, Torres DM, Harrison SA. Nonalcoholic fatty liver disease. Clin Liver Dis. 2012;16:397–419.
EASL. EASL–EASD–EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64:1388–402.
Charlton MR, Burns JM, Pedersen RA, et al. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141:1249–53.
Haas JT, Francque S, Staels B. Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu Rev Physiol. 2016;78:181–205.
Nascimbeni F, Pais R, Bellentani S, et al. From NAFLD in clinical practice to answers from guidelines. J Hepatol. 2013;59:859–71.
Adams LA, Anstee QM, Tilg H, et al. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. 2017;66:1138–53.
Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–46.
Farrell GC, Teoh NC, McCuskey RS. Hepatic microcirculation in fatty liver disease. Anat Rec (Hoboken). 2008;291:684–92.
Chu MJJ, Dare AJ, Phillips ARJ, et al. Donor hepatic steatosis and outcome after liver transplantation: a systematic review. J Gastrointest Surg. 2015;19:1713–24.
Tashiro H, Kuroda S. Ischemia–reperfusion injury in patients with fatty liver and the clinical impact of steatotic liver on hepatic surgery. Surg Today. 2014;44:1611–25.
Hakamada K, Sasaki M, Takahashi K, et al. Sinusoidal flow block after warm ischemia in rats with diet-induced fatty liver. J Surg Res. 1997;70:12–20.
Seifalian AM, Chidambaram V, Rolles K, et al. In vivo demonstration of impaired microcirculation in steatotic human liver grafts. Liver Transpl Surg. 1998;4:71–77.
Francque S, Verrijken A, Mertens I, et al. Noncirrhotic human nonalcoholic fatty liver disease induces portal hypertension in relation to the histological degree of steatosis. Eur J Gastroenterol Hepatol. 2010;22:1449–57.
Francque S, Verrijken A, Mertens I, et al. Visceral adiposity and insulin resistance are independent predictors of the presence of non-cirrhotic NAFLD-related portal hypertension. Int J Obes. 2011;35:270–8.
Francque S, Wamutu S, Chatterjee S, et al. Non-alcoholic steatohepatitis induces non-fibrosis-related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2009;30:365–75.
Iwakiri Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int. 2012;32:199–213.
Mihmanli I, Kantarci F, Yilmaz MH, et al. Effect of diffuse fatty infiltration of the liver on hepatic artery resistance index. J Clin Ultrasound. 2005;33:95–99.
Francque S, Laleman W, Verbeke L, et al. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Invest. 2012;92:1428–39.
Oda M, Yokomori H, Han J. Regulatory mechanisms of hepatic microcirculation. Clin Hemorheol Microcirc. 2003;29:167–82.
Laleman W, Landeghem L, Wilmer A, et al. Portal hypertension: from pathophysiology to clinical practice. Liver Int. 2005;25:1079–90.
McCuskey RS, Ito Y, Robertson GR, et al. Hepatic microvascular dysfunction during evolution of dietary steatohepatitis in mice. Hepatology. 2004;40:386–93.
Pasarín M, La Mura V, Gracia-Sancho J, et al. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS ONE. 2012;7:e32785.
Gonzalez-Paredes FJ, Hernández Mesa G, Morales Arraez D, et al. Contribution of cyclooxygenase end products and oxidative stress to intrahepatic endothelial dysfunction in early non-alcoholic fatty liver disease. PLoS ONE. 2016;11:e0156650.
Miyao M, Kotani H, Ishida T, et al. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest. 2015;95:1130–44.
Leclercq IA. Pathogenesis of steatohepatitis: insights from the study of animal models. Acta Gastroenterol Belg. 2007;70:25–31.
Michielsen P, Boeckxstaens G, Sys SU, et al. The role of increased nitric oxide in the vascular hyporeactivity to noradrenalin in long-term portal hypertensive rats. J Hepatol. 1995;23:341–7.
Kanno T, Tsuchiya A, Shimizu T, et al. Indomethacin serves as a potential inhibitor of protein phosphatases. Cell Physiol Biochem. 2012;30:1014–22.
Bessems M, Hart NA, Tolba R, et al. The isolated perfused rat liver: standardization of a time-honoured model. Lab Anim. 2006;40:236–46.
Ferrigno A, Richelmi P, Vairetti M. Troubleshooting and improving the mouse and rat isolated perfused liver preparation. J Pharmacol Toxicol Methods. 2013;67:107–14.
Francque SM, Verrijken A, Mertens I, et al. Noninvasive assessment of nonalcoholic fatty liver disease in obese or overweight patients. Clin Gastroenterol Hepatol. 2012;10:1162–8.
Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44.
García-Pagán JC. Complications of cirrhosis. I. Portal hypertension in cirrhosis. J Hepatol. 2012;57:458–61.
Abraldes JG, Iwakiri Y, Loureiro-Silva M, et al. Mild increases in portal pressure upregulate vascular endothelial growth factor and endothelial nitric oxide synthase in the intestinal microcirculatory bed, leading to a hyperdynamic state. Am J Physiol Gastrointest Liver Physiol. 2006;290:G980–7.
Serna E, Mauricio MD, Lluch P, et al. Basal release of nitric oxide in the mesenteric artery in portal hypertension and cirrhosis: role of dimethylarginine dimethylaminohydrolase. J Gastroenterol Hepatol. 2013;28:880–6.
Colle IO, De Vriese AS, Van Vlierberghe HR, et al. Vascular hyporesponsiveness in the mesenteric artery of anaesthetized rats with cirrhosis and portal hypertension: an in-vivo study. J Gastroenterol. 2004;16:139–45.
Colle I, Geerts AM, Van Steenkiste C, et al. Hemodynamic changes in splanchnic blood vessels in portal hypertension. Anat Rec (Hoboken). 2008;291:699–713.
Hennenberg M, Trebicka J, Sauerbruch T, et al. Mechanisms of extrahepatic vasodilation in portal hypertension. Gut. 2008;57:1300–14.
Graupera M, March S, Engel P, et al. Sinusoidal endothelial COX-1-derived prostanoids modulate the hepatic vascular tone of cirrhotic rat livers. Am J Physiol Gastrointest Liver Physiol. 2005;288:G763–70.
Laleman W, Van Landeghem L, Van der Elst I, et al. Nitroflurbiprofen, a nitric oxide-releasing cyclooxygenase inhibitor, improves cirrhotic portal hypertension in rats. Gastroenterology. 2007;132:709–19.
Abraldes JG, Rodríguez-Vilarrupla A, Graupera M, et al. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J Hepatol. 2007;46:1040–6.
Abraldes JG, Albillos A, Bañares R, et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology. 2009;136:1651–8.
Zafra C, Abraldes JG, Turnes J, et al. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology. 2004;126:749–55.
Wang W, Zhao C, Zhou J, et al. Simvastatin ameliorates liver fibrosis via mediating nitric oxide synthase in rats with non-alcoholic steatohepatitis-related liver fibrosis. PLoS ONE. 2013;8:1–11.
Chang C-C, Wang S-S, Hsieh H-G, et al. Rosuvastatin improves hepatopulmonary syndrome through inhibition of inflammatory angiogenesis of lung. Clin Sci (Lond). 2015;129:449–60.
Fiorucci S, Antonelli E, Brancaleone V, et al. NCX-1000, a nitric oxide releasing derivative of ursodeoxycholic acid, ameliorates portal hypertension and lowers norepinephrine-induced intrahepatic resistance in the isolated and perfused rat liver. J Hepatol. 2003;39:932–9.
Biecker E, Trebicka J, Kang A, et al. Treatment of bile duct-ligated rats with the nitric oxide synthase transcription enhancer AVE 9488 ameliorates portal hypertension. Liver Int. 2008;28:331–8.
Elliot AJ, Vo LT, Grossman VL, et al. Endothelin-induced vasoconstriction in isolated perfused liver preparations from normal and cirrhotic rats. J Gastroenterol Hepatol. 1997;12:314–8.
Shibamoto T, Kamikado C, Koyama S. Increased sinusoidal resistance is responsible for the basal state and endothelin-induced venoconstriction in perfused cirrhotic rat liver. Pflug Arch Eur J Physiol. 2008;456:467–77.
Chan C-C, Lee K-CL, Lin H-C, et al. Increased expression of hepatic endothelin-1 during fibrosis progression in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2014;1:1–7.
Acknowledgements
W.J.K. and S.M.F. received funding from the Fund for Scientific Research (FWO) Flanders (11J9513N, 1802154N).
Disclaimer
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Van der Graaff, D., Kwanten, W.J., Couturier, F.J. et al. Severe steatosis induces portal hypertension by systemic arterial hyporeactivity and hepatic vasoconstrictor hyperreactivity in rats. Lab Invest 98, 1263–1275 (2018). https://doi.org/10.1038/s41374-017-0018-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-017-0018-z
This article is cited by
-
The role of adipose tissue and subsequent liver tissue hypoxia in obesity and early stage metabolic dysfunction associated steatotic liver disease
International Journal of Obesity (2024)
-
Interventional Management of Portal Hypertension in Cancer Patients
Current Oncology Reports (2022)
-
Role of liver sinusoidal endothelial cells in liver diseases
Nature Reviews Gastroenterology & Hepatology (2021)
-
The pathophysiological basis of vascular disease
Laboratory Investigation (2019)
-
TGR(mREN2)27 rats develop non-alcoholic fatty liver disease-associated portal hypertension responsive to modulations of Janus-kinase 2 and Mas receptor
Scientific Reports (2019)
