
Abstract
Recent advances have been made in understanding the nature of placental dysfunction causing preeclampsia, and other hypertensive disorders of pregnancy. The contribution of animal studies in the understanding of the effects of inadequate placentation on blood pressure and other target organs will be explored in this review. This will include technical aspects of animal studies in pregnancy, as well as the translation of data regarding newly discovered pathological pathways, in particular the angiogenic pathway, into targets in clinical practice.
Similar content being viewed by others

Background
Preeclampsia and the other hypertensive disorders of pregnancy remain a major global health burden and contribute to 15% worldwide of all maternal deaths and between 5 and 25% of 5.5 million stillbirths and neonatal deaths annually [1]. Strategies to reduce mortality have focused on improved perinatal care, allowing a safer delivery at any gestation. Early risk stratification, however, will achieve the greatest impact on improving poor outcomes by allowing preventative strategies to be implemented thus ensuring timely diagnosis and treatment is directed to those still developing preeclampsia. This is especially true in developing countries [1] where the rates of preeclampsia are the highest juxtaposed to the least resources. Emerging novel treatments such as small-interfering RNA (siRNA) target pathways that indicate placental dysfunction, and the maternal responses to that placental change. This article will review the contribution of animal models to recent developments in diagnostics, prevention, and treatment of preeclampsia, the most severe of the hypertensive disorders in pregnancy.
Technical aspects of animal studies in preeclampsia
Ultimately, blood pressure changes are a requisite measure of the impact of placental dysfunction in preeclampsia. Therefore, whole animal models are required, and accurate measurement of blood pressure must be achieved. Species used to model preeclampsia have included mice, rats, dogs, sheep, rhesus monkeys and baboons, with mice and rats the most commonly used animals for study. The main disadvantage of most animal species relates to the ability to translate research findings to human pregnancy given differences in reproductive and placental physiology. Preeclampsia only occurs spontaneously in humans and apes, and has been reported in a baboon with twins.
Non-human primates
The placental structure of non-human primates (NHP) most closely reflects that of the human (i.e., is haemomonochorial) and thus it is possible to do proof-of-principle studies using these animals. NHP including rhesus monkeys (Macaca mulatta) and baboons (Papio anubis and Papio hamadryas) have been used to model preeclampsia, predominantly by initiating uteroplacental ischaemia via surgical ligation of placental blood supply. However, NHP have long gestation periods, singleton pregnancies and require specialised management, housing, and husbandry, all of which are limitations of working with this species despite their physiological advantages.
Mice
The advantages of studying pregnancy in mice include their ease of handling and housing, and short gestation which reduces the time (and expense) of studies [2]. Large litter size has the advantage of allowing concurrent assessment of any differing effects of the intervention on male and female offspring [2]. Detailed understanding of pregnant mouse physiology also allows contextual interpretation of findings in mouse models. However, the relevance of mouse models to human pregnancy is limited as placental development and physiology differs significantly. Abnormal spiral artery remodelling in mice does not lead to placental insufficiency or abnormal blood pressure regulation whereas abnormal uterine spiral artery remodelling in human preeclamptic placentae is widely thought to precede the development of the maternal syndrome of preeclampsia. To establish the human uteroplacental circulation, extravillous trophoblast invasion begins early and by 8 weeks of gestation endovascular trophoblasts have already invaded the inner part of the decidua basalis. By contrast, in mice, the invasion of trophoblast cells into the decidua basalis begins in the second half of pregnancy and completes during late gestation [3]. The murine artery vascular endothelium is not completely replaced by the trophoblast, and the trophoblast per se does not extend into the myometrium as it does in humans [3].
Blood pressure measurement
Identification of hypertension in animals is reliant on accurate blood pressure readings. The majority of studies of hypertension in mice employ tail-cuff measurements despite the known limitations of this indirect method. Tail-cuff measurements which rely on sensor detection of blood flow rather than detection of the pulse are particularly unreliable [4]. Tail-cuff measurements may not be accurate at extremes of blood pressure and restraint affects blood pressure results, even in expert hands [4] but relative changes in blood pressure can likely still be detected, especially if differences expected are large (15–20 mmHg). Direct blood pressure measurement in the form of intra-arterial catheters is the common direct method used in rats. Although accurate and able to detect small changes in blood pressure, this method is complicated by the need for anaesthesia close to the time of recording (usually approximately 24 hours), by restraint, as well as by complications related to any surgical procedure. General anaesthesia may also impact upon the circadian rhythm and hence the blood pressure of rodents.
Continuous undisturbed intra-arterial radiotelemetry is the gold standard and allows unrestrained measurement of the blood pressure. This technique is used in baboons as well as rodents. It is less commonly used due to the expense and technical difficulty of surgical insertion. Care regarding data continuity and line integrity (i.e., potential clotting or migration of the transducing catheter) needs to be undertaken. Other disadvantages include occlusion of the vessel at times, resulting in either stroke (in the case of carotid placement) or limb ischaemia (in the case of femoral placement), wound infection and pressure injury due to the presence of the transmitter which, especially in the case of rodents, is large relative to their body size.
Demonstrating multi-organ and systemic endothelial dysfunction is also critical for validating any model of preeclampsia. This is often indirectly inferred by the presence of proteinuria, which is linked to histological evidence of endotheliosis in renal tissue. Dynamic studies demonstrating endothelial dysfunction can be linked to changes in other clinically relevant target organs (e.g., liver and brain). Opportunities to study dynamic physiological responses such as flow mediated dilatation are only possible in whole animals. Individual organ pharmacological responses may not represent the same findings as those that are elicited in a whole organism circumstance where organ interactions occur.
Modelling the pathophysiology of hypertensive disorders of pregnancy
There is a multitude of established experimental preeclampsia models. While in vitro experiments provide the opportunity for pathway analysis, they cannot examine blood pressure and multiple organ responses. Animal models of preeclampsia broadly include those induced by inflammation, placental ischaemia, angiogenic imbalance often due to changes in soluble fms-like tyrosine kinase-1 (sFLT-1), manipulation of various aspects of the immune system, inhibition of nitric oxide pathways and genetic manipulations such as of the renin angiotensin aldosterone system (RAAS) which have been summarised in other reviews that the reader is referred to [5].
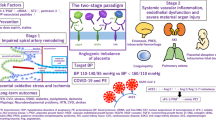
The pathogenesis of preeclampsia is thought to occur in two stages, stage 1 being abnormal placentation, followed by stage 2, the maternal syndrome resulting from the dissemination of placental factors which cause systemic endothelial dysfunction. Placental dysfunction is demonstrated in the first trimester of pregnant women who subsequently develop preeclampsia or intrauterine growth restriction by abnormal uterine artery resistive indices and placental markers such as placental growth factor (PlGF) and pregnancy-associated placenta protein-A (PAPP-A). However, the majority of animal models do not replicate the primary event in preeclampsia pathogenesis and preventative therapies aimed at targeting this process are therefore difficult to study. The commonly used models induce sudden placental ischaemia well after the placenta is established or infuse the animal with substances produced by the abnormal placenta. Models that may result from a primary defect in placentation include the STOX1 (storkhead box-1) transgenic mouse model [6] which overexpresses a transcription factor affecting trophoblast proliferation and migration, however, in this model, severe hypertension develops almost at the onset of pregnancy. Rodents with chronic hypertension, including BPH/5 mice [7] and Dahl salt-sensitive rats [8], can also develop features of superimposed preeclampsia during pregnancy which may possibly be related to abnormal placentation or the underlying cause of chronic hypertension. This is shown in Fig. 1.

Animal models of preeclampsia according to physiological stage of preeclampsia being replicated.
Important more recent refinements to existing animal models include early- and late-onset experimental preeclampsia induced by specifically timed administrations of L-NAME (an inhibitor of nitric oxide synthase) in rats [9], as most animal models have not been able to replicate these clinical phenotypes. Overexpression of the primate-specific sFLT-1 e15a isoform localised to the placenta [10] by implantation of murine blastocysts harvested from mice transduced with lentiviral vectors into pseudopregnant female mice is also a significant development as sFLT-1 production is localised to the placenta unlike in adenovirus-sFLT-1 rodent models. Other experimental preeclampsia models including those induced by infusion of arginine vasopressin and knockout of heme oxygenase-1 are discussed in the reviews mentioned above.
Long-term consequences of preeclampsia
Animal models in preeclampsia have also been useful for demonstrating the long-term consequences of the disease. It is well known that the risk of cardiovascular disease is increased after women develop preeclampsia, but it is difficult to determine whether this results from the effects of overlapping risk factors for preeclampsia and cardiovascular disease, or from a direct effect of preeclampsia itself. Several different animal models of experimental preeclampsia have shown that preeclampsia itself has an adverse effect on overall cardiovascular health. A rat model of experimental preeclampsia induced by constriction of the aorta showed increased susceptibility to hypertension and retinal vessel damage [11]. A mouse model induced by adenovirus overexpression of sFLT-1 similarly showed a different response to vascular injury when compared with normal pregnant mice [12]. The STOX1 transgenic mouse model of preeclampsia shows the abnormal cardiac function and cardiac fibrosis 8 months after pregnancy (corresponding to 20 years in human life) when compared with wild-type mice [13]. The long-term consequences of preeclampsia are remote from the preeclampsia thus making it difficult to determine which women diagnosed with preeclampsia are most at risk and at what point screening for cardiovascular disease would be beneficial. Animal models allow a clearer understanding of the subtle, longer-term effects of preeclampsia on the cardiovascular system, with one group using speckle tracking echocardiography in transgenic rats at 4 weeks post-partum which corresponds to 2 years post-partum in women [14]. This demonstrated myocardial wall thickening with a reduction in ejection fraction and global longitudinal strain in both preeclamptic women and an animal model of PE as compared with normal controls.
Phenotype of experimental preeclampsia in non-human primates
In NHP, experimental preeclampsia has been induced by both placental ischaemia [15], infusion of TNF-α [16] from mid-gestation and IL-10 inhibition [17]. The uteroplacental ischaemia (UPI) model of experimental preeclampsia in NHP replicates a mild syndrome of late-onset preeclampsia. Ligation of the uterine artery of the Papio hamadryas after the second trimester at 135 days of a normal 182 days duration pregnancy leads to an increase in systolic blood pressure of 16.4 ± 1.7 mmHg and diastolic of 18.5 ± 2.4 mmHg, proteinuria (61.2 ± 15.3 mg/mmol Cr) and elevation of bioactive factors thought to be responsible for these clinical manifestations such as elevated sFLT-1 and decreased PlGF. As early placental development is unaffected in both TNF-α and UPI induced experimental preeclampsia, the establishment of a model of early-onset experimental preeclampsia in NHP was demonstrated using anti-IL-10 antibodies and also TNF-α. Inhibition of IL-10 causes a mild elevation of blood pressure but the infusion of TNF-α at 45 days gestation was unsuccessful in reproducing an early-onset preeclampsia model instead resulting in abortion of fetuses within 2 weeks [16].
Translation of preclinical studies to clinical practice
The presentation of preeclampsia in women is heterogenous with varying severity of maternal and fetal involvement at different stages of gestation. This is likely due to the complex interaction between pre-existing maternal health conditions, genetic susceptibility, the placenta as well as fetal susceptibility. Animal models may be able to replicate specific aspects of the pathophysiology but ultimately cannot predict the action of potential therapeutic agents in women. This limitation has been demonstrated both by varying responses to treatment in different animal models and by current attempts at the translation of preclinical studies to human trials where treatment efficacy has thus far been elusive and is discussed further below.
The oxidative stress and nitric oxide pathway
Oxidative stress may potentially influence the pathogenesis of preeclampsia in both stage 1 and stage 2 of the pathogenesis of preeclampsia. In early placental development, the clearance of trophoblast plugs within maternal spiral arteries results in the rapid increase in oxygen tension in placental tissues. It is hypothesised that this increases oxidative stress and an imbalance of redox signalling may hamper the subsequent trophoblast invasion of maternal uterine arteries or ‘second wave’ of placental development. A poorly developed placenta may then, under oxidative stress, produce many other bioactive compounds that cause endothelial dysfunction. Endothelial nitric oxide synthase is uncoupled by oxidative stress leading to reduced nitric oxide but also produces radical nitrogen species, further contributing to redox imbalance.
Despite the likely pathogenic role of oxidative stress in preeclampsia, therapies aimed at modulating oxidative stress by increasing the activity of enzymatic (e.g., heme-oxygenase) and non-enzymatic antioxidants (e.g., vitamin C and E, glutathione replacement and replacement of micronutrients such as selenium) have largely been unsuccessful in human studies even though various preclinical studies showed promise. For example, esomeprazole was shown to upregulate heme-oxygenase but in the setting in of the treatment of established preeclampsia, esomeprazole did not prolong pregnancy. Women diagnosed with early-onset preeclampsia between 26 to 31 + 6 weeks gestational age who were given esomeprazole 40 mg daily extended their pregnancy by 8.3 days as opposed to 11.4 days in the placebo group but this was not statistically significant (p = 0.31) and there was also no effect on individual features of preeclampsia such as hypertension, proteinuria and other organ dysfunction. Other antioxidants such as Tempol (4-hydroxy-2,2,6,6-tetramethyl piperidin-1-oxyl), a superoxide dismutase mimetic and MitoTempo (a mitochondria-specific superoxide dismutase mimetic) suggest improvement in feto-placental function in a variety of animal models with an increase in fetal weight but have not progressed further to human studies. Tempol is currently being studied in phase 1 and 2 trials for various conditions including chemotherapy side effects and COVID-19 infection.
Sildenafil, a phosphodiesterase-5 inhibitor, was demonstrated in a number of animal models—reviewed in a meta-analysis by Paauw et al. [18]—to attenuate the features of fetal growth restriction and preeclampsia. In these preclinical studies, varying species were used with models induced by L-NAME (a nitric oxide synthase inhibitor) where sildenafil may be expected to be efficacious, and other means both with and without evidence of placental dysfunction. Although these data suggested a physiological basis for sildenafil therapy and small studies in pregnant women showed promising results, subsequent large multi-centre studies conducted in multiple countries showed no benefit in fetal growth restriction (an ultrasound measurement of the fetal abdominal circumference (AC) < 3rd percentile for gestational age or estimate of fetal weight (EFW) < 5th percentile at 20 + 0-27 + 6 weeks, or an ultrasound EFW < 700 grams using Hadlock C formula) with the potential for harm.
The angiogenic pathway
The discovery of angiogenic pathway abnormalities and their contribution to the pathogenesis of preeclampsia have revolutionised early prediction, diagnosis and raised possibilities for both treatment and prevention.
In 2003, the Karumanchi group realised the importance of sFLT-1 and other angiogenic factors in the pathogenesis of preeclampsia. The potential predictive and diagnostic value of patterns of circulating angiogenic factors identified in preeclamptic women was supported by the validation lent by animal models. Sprague-Dawley rats were injected with an adenovirus expressing a truncated version of murine sFLT-1 and this induced hypertension and proteinuria in both pregnant and non-pregnant rats. While the changes in non-pregnant rats indicated that pregnancy was not a requirement for the development of the clinical syndrome, that sFLT-1 was elevated in other established models of experimental preeclampsia [15] and supported the strength of these initial findings.
At present, angiogenic factors have a role in the early prediction of preeclampsia and risk stratification for women with suspected preeclampsia. The Fetal Medicine Foundation early prediction model combining PlGF at 11–13 weeks’ gestation with maternal history factors, maternal biophysical markers (uterine artery Doppler, and maternal blood pressure) and maternal biochemical markers (PAPP-A) [19] has a 91% detection rate (presuming a 5% false-positive rate) for early-onset preeclampsia. For women with suspected preeclampsia, PlGF [20] or the sFLT-1:PlGF ratio [21] are being used to risk-stratify patients and guide frequency of follow-up.
Treatment and prevention of preeclampsia by manipulation of these angiogenic factors is an area of continued research. In established preeclampsia, neutralisation of circulating sFLT-1 or redressing the imbalance of sFLT-1:PlGF has been proposed as a treatment strategy. Initial demonstrations that this may be an effective strategy included studies showing abrogation of preeclampsia manifestations by recombinant vascular endothelial growth factor (VEGF) treatment in rats with experimental preeclampsia induced by recombinant sFLT-1 [22] and placental ischaemia [23]. Since then, parenteral PlGF in various animal models—adenoviral overexpression of sFLT-1 in mice [12], reduced uterine perfusion pressure (RUPP) in rats [24] and UPI in NHP [25] have been successful in attenuating hypertension and other manifestations of experimental preeclampsia. Attempts to prevent experimental preeclampsia in animal studies by priming with PlGF itself before the placental insult have not changed the trajectory of the disease [26]. Yet, it remains unclear how exactly exogenous angiogenic factors exert these findings.
In RUPP rats administered PlGF and VEGF [26], free or unbound circulating sFLT-1 was shown to be decreased suggesting binding of administered PlGF to sFLT-1 rendering it inert, but the reduction of sFLT-1 expression could not be excluded. In NHP treated with PlGF, total (both free and bound) sFLT-1 concentrations temporarily increased in treated animals, then were similar to pre-treatment levels despite reductions in blood pressure. In the mouse model of placenta confined lentivirus expression of sFLT-1 e15a, simultaneous lentivirus induced expression of PGF resulted in normalisation of blood pressure and reduction of total circulating sFLT-1 concentration, suggesting that sFLT-1 was destabilised in some way [10]. Thus, while ligands for sFLT-1 may attenuate the manifestations of preeclampsia it is unclear whether this is due to inactivation or disruption of sFLT-1, or reduction in sFLT-1 expression.
The relevance of angiogenic factors in mouse pregnancy as compared to human pregnancy is not well understood however, the magnitude of angiogenic factor concentrations is substantially different. In humans, sFLT-1 concentration at the completion of pregnancy is in the order of 1000 pg/mL and PlGF concentration is approximately 100 pg/mL. In mice, plasma sFLT-1 concentrations are markedly higher than PlGF with sFLT-1 concentrations of 25 ng/mL while PlGF is similar to that in humans at 25 pg/mL. Calculation of a sFLT-1:PlGF ratio, therefore, yields values in the thousands. It has been argued, that as sFLT-1 concentrations in mice far exceed mouse concentrations of PlGF and VEGF-A the role of sFLT-1 may differ between humans and mice and affect the translatability of results. Rats however have a comparable ratio of sFLT-1:PlGF to humans.
There also remains much to discover about the role of PlGF in both mouse and human pregnancy. The pattern of circulating PlGF in mouse pregnancy is similar to that of humans. In mice, the peak in PlGF occurs slightly earlier at approximately mid-gestation as compared to the peak in humans at 30 weeks gestation. While PlGF knockout mice display some abnormalities in placentation with decreased branching of utero- and feto-placental vessels there is no difference in trophoblast invasion [27] and PlGF knockout mice are fertile. Furthermore, humans express 4 isoforms of PlGF but mice only express one isoform corresponding to PlGF-2 in humans. PGF is secreted as a glycosylated homodimer and PlGF-1 and −3 are diffusible isoforms whereas PlGF-2 and PlGF-4 have heparin-binding domains. The presence of a heparin-binding domain suggests that PlGF-2 and −4 remain cell membrane-associated and act in an autocrine fashion while the diffusible forms of PlGF probably affect targets in a paracrine manner. A recent study found that while catechol-O-methyltransferase (COMT) deficient mice develop features of preeclampsia, breeding these mice with PlGF knockout mice unexpectedly resulted in the reduction of hypertension rather than exacerbation of the COMT experimental preeclampsia model as anticipated. In pregnant women, however, low PlGF in early gestation is strongly associated with subsequent development of placental disorders.
Recently, a number of medications are being studied for repurposing as preeclampsia treatments on the basis of their ability to reduce sFLT-1 or increase PlGF expression (in addition to other proposed effects) including esomeprazole, pravastatin and metformin. It has been demonstrated that esomeprazole reduces sFLT-1 expression in a number of in-vitro and ex-vivo experiments. In the sFLT-1 e15a lentivirus mouse model, esomeprazole at equivalent human doses decreased blood pressure substantially but the mechanism by which this was achieved was uncertain as sFLT-1 was unchanged, presumably because lentiviral expression could not be modified. Unfortunately, a large study of 120 women with pre-term preeclampsia randomised to either placebo or esomeprazole from the point of diagnosis [28] did not reduce sFLT-1 nor prolong gestation.
Pravastatin has been shown to be effective in preventing preeclampsia in different animal models including the sFLT-1 e15a lentivirus mouse model, C1q knockout mouse model and RUPP rats. A potential mechanism of pravastatin is increasing PlGF expression and reducing circulating sFLT-1. A small drug safety trial involving 20 women with a history of severe early-onset preeclampsia [29] suggested that the promising findings in animal studies may be replicated in humans, but this study did not show that sFLT-1 and PlGF were statistically significantly altered by pravastatin. The Statins to Ameliorate Preeclampsia trial has also shown no significant difference in sFLT-1 concentrations and no difference in length of pregnancy with similar maternal and fetal outcomes thus far, albeit this study is underpowered. Interestingly, aspirin, the only preventative therapy currently available has been observed in a number of human studies to increase PlGF [30].
Removal of circulating sFLT-1 by apheresis in women with established diagnosis of preeclampsia may potentially prolong the duration of pregnancy but the efficacy of this novel treatment remains uncertain in the absence of controlled studies. Although peripheral sFLT-1 concentrations were substantially reduced by apheresis (17–34%), sFLT-1 levels quickly rebounded. Targeting the source of sFLT-1 by direct reduction of sFLT-1 expression using targeted molecular manipulation of placental production using small-interfering RNA (siRNA) [31] has been recently examined in the UPI model in NHP. This reduces sFLT-1 expression by approximately 50% for up to 4 weeks. This is the first time that a biological agent targeting the placental source as the cause of endothelial dysfunction has been tried. The potential clinical application of a siRNA-based approach would need to ensure placental binding, fetal safety and limit any off-target effects of siRNA in vascular beds unaffected by preeclampsia.
Therapies that appear promising in preclinical studies have so far been unsuccessful and this apparent lack of efficacy may be related to a variety of reasons including inadequate dosage due to possible species difference in metabolism, suboptimal timing and individual responses to therapy. As the multitude of studies and meta-analyses of aspirin can attest, dosage and timing are extremely important and aspirin resistance may also play a role. The risk of preeclampsia is increased by presence of the metabolic syndrome and both maternal genetic predisposition to hypertension [32] and fetal DNA variants near the FLT-1 gene which are likely present in the placenta [33]. These factors unique to each patient may also determine response to therapy.
That attempts at translation are thus far fruitless may also highlight some limitations in the animal models used for the study of preeclampsia. In pregnant women, the difference in sFLT-1 concentration between women with and without preeclampsia is approximately 2-fold but may vary substantially with a subset of women having ‘non-angiogenic preeclampsia’ [34]. In animal models, the change in sFLT-1 required to recapitulate the findings of preeclampsia also varies widely. In NHP, UPI results in a variable increase in circulating sFLT-1 (sFLT-1 = 13223.3 + /− 2175 pg/ml at peak in UPI vs 1939.6 + /− 885.4 pg/mL at peak in control animals) with placental expression of sFLT-1 increasing markedly by 20 fold. In RUPP rats plasma sFLT-1 may increase 8-fold (660 + /− 270 versus 82 + /− 26 pg/mL [23]) and this varies from study to study even within the same research laboratory (2910 + /− 499 vs 1071 + /− 300 pg/mL [35]). This likely relates to the degree of ischaemia induced in these models (which cannot always be investigator controlled due to natural anatomical variation) and the response to various treatments may be dependent on the severity of experimental preeclampsia phenotype achieved.
Conclusion
Animal studies in hypertensive disorders of pregnancy have been vital to furthering the understanding of pathogenic pathways. The effect of various therapies on blood pressure in models of experimental preeclampsia needs to also be supported by evidence of improvement of feto-placental function or endothelial dysfunction but sadly, even when dramatic improvements in multiple facets are demonstrated, translation to clinical practice is still to be realised. This is not dissimilar to the success rates in other fields such as oncology where many potential therapeutic agents with strong evidence to support their efficacy in preclinical studies have failed in human studies. However, unlike other diseases, a disease of pregnancy cannot be studied without a whole animal model. Future preclinical studies of therapeutic agents such as the siRNA-based approach used by Turanov et al [31] should continue to examine the efficacy in different animal models and analyse the impact of treatments on multiple pathophysiological pathways of preeclampsia. As it is well known that preeclampsia is multifactorial, combined use of therapies targeting different pathways may be the means to move forward.
References
Say L, Chou D, Gemmill A, Tunçalp Ö, Moller A-B, Daniels J, et al. Global causes of maternal death: a WHO systematic analysis. Lancet Glob Health. 2014;2:e323–e33.
Dilworth MR, Sibley CP. Review: transport across the placenta of mice and women. Placenta. 2013;34:S34–9.
Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placentae. Placenta. 2002;23:3–19.
Whitesall S, Hoff J, Vollmer A, D’Alecy L. Comparison of simultaneous measurement of mouse systolic arterial blood pressure by radiotelemetry and tail-cuff methods. Am J Physiol. 2004;55:H2408–H15.
Gatford KL, Andraweera PH, Roberts CT, Care AS. Animal models of preeclampsia: causes, consequences, and interventions. Hypertension. 2020;75:1363–81.
Doridot L, Passet B, Mehats C, Rigourd V, Barbaux S, Ducat A, et al. Preeclampsia-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension. 2013;61:662–8.
Heyward CY, Sones JL, Lob HE, Yuen LC, Abbott KE, Huang W, et al. The decidua of preeclamptic-like BPH/5 mice exhibits an exaggerated inflammatory response during early pregnancy. J Reprod Immunol. 2017;120:27–33.
Turbeville HR, Taylor EB, Garrett MR, Didion SP, Ryan MJ, Sasser JM. Superimposed preeclampsia exacerbates postpartum renal injury despite lack of long-term blood pressure difference in the Dahl Salt-sensitive rat. Hypertension. 2019;73:650–8.
Soobryan N, Murugesan S, Phoswa W, Gathiram P, Moodley J, Mackraj I. The effects of sildenafil citrate on uterine angiogenic status and serum inflammatory markers in an L-NAME rat model of pre-eclampsia. Eur J Pharmacol. 2017;795:101–7.
Kumasawa K, Ikawa M, Kidoya H, Hasuwa H, Saito-Fujita T, Morioka Y, et al. Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc Natl Acad Sci USA. 2011;108:1451–5.
Ramírez-Montero C, Lima-Gómez V, Anguiano-Robledo L, Hernández-Campos ME, López-Sánchez P. Preeclampsia as predisposing factor for hypertensive retinopathy: Participation by the RAAS and angiogenic factors. Exp Eye Res. 2020;193:107981.
Suzuki H, Ohkuchi A, Matsubara S, Takei Y, Murakami M, Shibuya M, et al. Effect of recombinant placental growth factor 2 on hypertension induced by full-length mouse soluble fms-like tyrosine kinase 1 adenoviral vector in pregnant mice. Hypertension. 2009;54:1129–35.
Miralles F, Collinot H, Boumerdassi Y, Ducat A, Duché A, Renault G, et al. Long-term cardiovascular disorders in the STOX1 mouse model of preeclampsia. Sci Rep. 2019;9:11918.
Kräker K, Schütte T, O’Driscoll J, Birukov A, Patey O, Herse F, et al. Speckle tracking echocardiography: new ways of translational approaches in preeclampsia to detect cardiovascular dysfunction. Int J Mol Sci. 2020;21:1162.
Makris A, Thornton C, Thompson J, Thomson S, Martin R, Ogle R, et al. Uteroplacental ischemia results in proteinuric hypertension and elevated sFLT-1. Kidney Int. 2007;71:977–84.
Sunderland NS, Thomson SE, Heffernan SJ, Lim S, Thompson J, Ogle R, et al. Tumor necrosis factor alpha induces a model of preeclampsia in pregnant baboons (Papio hamadryas). Cytokine. 2011;56:192–9.
Orange S, Rasko JE, Thompson JF, Vaughan J, Olive E, Pedler M, et al. Interleukin-10 regulates arterial pressure in early primate pregnancy. Cytokine. 2005;29:176–85.
Paauw ND, Terstappen F, Ganzevoort W, Joles JA, Gremmels H, Lely AT. Sildenafil during pregnancy: a preclinical meta-analysis on fetal growth and maternal blood pressure. Hypertension. 2017;70:998–1006.
Akolekar R, Syngelaki A, Sarquis R, Zvanca M, Nicolaides KH. Prediction of early, intermediate and late pre‐eclampsia from maternal factors, biophysical and biochemical markers at 11–13 weeks. Prenat Diagnosis. 2011;31:66–74.
Chappell LC, Duckworth S, Seed PT, Griffin M, Myers J, Mackillop L, et al. Diagnostic accuracy of placental growth factor in women with suspected preeclampsia: a prospective multicenter study. Circulation. 2013;128:2121–31.
Zeisler H, Llurba E, Chantraine F, Vatish M, Staff AC, Sennström M, et al. Predictive value of the sFlt-1:PlGF ratio in women with suspected preeclampsia. N. Engl J Med. 2016;374:13–22.
Li Z, Zhang Y, Ying Ma J, Kapoun AM, Shao Q, Kerr I, et al. Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension 2007;50:686–92.
Gilbert JS, Verzwyvelt J, Colson D, Arany M, Karumanchi SA, Granger JP. Recombinant vascular endothelial growth factor 121 infusion lowers blood pressure and improves renal function in rats with placentalischemia-induced hypertension. Hypertension 2010;55:380–5.
Spradley FT, Tan AY, Joo WS, Daniels G, Kussie P, Karumanchi SA, et al. Placental growth factor administration abolishes placental ischemia-induced hypertension. Hypertension. 2016;67:740–7.
Makris A, Yeung KR, Lim SM, Sunderland N, Heffernan S, Thompson JF, et al. Placental growth factor reduces blood pressure in a uteroplacental ischemia model of preeclampsia in nonhuman primates. Hypertension. 2016;67:1263–72.
Chau K, Bobek G, Xu B, Stait-Gardner T, Price W, Hennessy A, et al. Effect of placental growth factor in models of experimental pre-eclampsia and trophoblast invasion. Clin Exp Pharmacol Physiol. 2020;47:49–59.
Ratsep MT, Carmeliet P, Adams MA, Croy BA. Impact of placental growth factor deficiency on early mouse implant site angiogenesis. Placenta. 2014;35:772–5.
Cluver CA, Hannan NJ, van Papendorp E, Hiscock R, Beard S, Mol BW, et al. Esomeprazole to treat women with preterm preeclampsia: a randomized placebo controlled trial. Am J Obstet Gynecol. 2018;219:388.e1–e17.
Costantine MM, Cleary K, Hebert MF, Ahmed MS, Brown LM, Ren Z, et al. Safety and pharmacokinetics of pravastatin used for the prevention of preeclampsia in high-risk pregnant women: a pilot randomized controlled trial. Am J Obstet Gynecol. 2016;214:720 e1–e17.
Shanmugalingam R, Wang X, Motum P, Fulcher I, Lee G, Kumar R, et al. Clinical influence of nonadherence with prophylactic aspirin in preventing preeclampsia in high-risk pregnancies: a multicenter, prospective, observational cohort study. Hypertension. 2020;75:1125–32.
Turanov AA, Lo A, Hassler MR, Makris A, Ashar-Patel A, Alterman JF, et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat Biotechnol. 2018;36:1164–73.
McGinnis R, Steinthorsdottir V, Williams NO, Thorleifsson G, Shooter S, Hjartardottir S, et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat Genet. 2017;49:1255–60.
Steinthorsdottir V, McGinnis R, Williams NO, Stefansdottir L, Thorleifsson G, Shooter S, et al. Genetic predisposition to hypertension is associated with preeclampsia in European and Central Asian women. Nat Commun. 2020;11:5976.
Rana S, Schnettler WT, Powe C, Wenger J, Salahuddin S, Cerdeira AS, et al. Clinical characterization and outcomes of preeclampsia with normal angiogenic profile. Hypertens Pregnancy. 2013;32:189–201.
Xiang QQ, Yang Z, Huai J, Wang GJ. Different effects of pravastatin on sFlt-1, PlGF and VEGF in different preeclampsia-like mouse models. Zhonghua fu Chan ke za zhi. 2019;54:601–7.
Author information
Authors and Affiliations
Contributions
KC wrote the manuscript with significant input from MW, AH and AM. All authors provided critical feedback on the final revision. Progress in preeclampsia: the contribution of animal models.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chau, K., Welsh, M., Makris, A. et al. Progress in preeclampsia: the contribution of animal models. J Hum Hypertens 36, 705–710 (2022). https://doi.org/10.1038/s41371-021-00637-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41371-021-00637-x
This article is cited by
-
Developmental expression of catecholamine system in the human placenta and rat fetoplacental unit
Scientific Reports (2024)
