
Abstract
Sex hormone receptors are expressed throughout the vasculature and play an important role in the modulation of blood pressure in health and disease. The functions of these receptors may be important in the understanding of sexual dimorphism observed in the pathophysiology of both hypertension and vascular ageing. The interconnectivity of these factors can be exemplified in postmenopausal females, who with age and estrogen deprivation, surpass males with regard to hypertension prevalence, despite experiencing significantly less disease burden in their estrogen replete youth. Estrogen and androgen receptors mediate their actions via direct genomic effects or rapid non-genomic signaling, involving a host of mediators. The expression and subtype composition of these receptors changes through the lifespan in response to age, disease and hormonal exposure. These factors may promote sex steroid receptor-mediated alterations to the Renin–Angiotensin–Aldosterone System (RAAS), and increases in oxidative stress and inflammation, thereby contributing to the development of hypertension and vascular injury with age.
Similar content being viewed by others
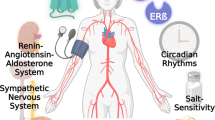


Introduction
Hypertension is the leading modifiable risk factor resulting in cardiovascular disease and mortality worldwide [1]. Ageing is an important mediator in the development of hypertension and contributes significantly to the rising prevalence of this condition [2]. However, the effects of age on blood pressure are not uniform between sexes. Males experience higher rates of hypertension compared with females, until the sixth decade of life, where thereafter this condition is more prevalent in the latter [3]. Therefore, although blood pressure and hypertension rates increase with age, this interaction is not consistent between males and females.
Despite this, there are no sex specific recommendations for treating hypertension in international guidelines [4, 5]. In the landmark Systolic Blood Pressure Intervention Trial (SPRINT), there was no evidence of differences in target or treatment choice between males and females, although this may be a consequence of being underpowered to detect such differences [6,7,8]. Females only comprised approximately 36% of the SPRINT cohort, which is in keeping with the chronic underrepresentation of this sex in cardiovascular trials. The exclusion of participants under 50 years ensured mostly postmenopausal females would have been recruited and therefore it is uncertain whether pharmacologic management of hypertension should differ premenopausal females compared to age-matched males. Importantly, although antihypertensive medication are generally used more in females, only 44.8% achieve blood pressure control versus 51.1% of treated males [9]. As a consequence, the age, sex, and hormonal status of individuals, which are paramount in the development of hypertension, are too often ignored to the detriment of patient care.
Vascular ageing describes the progressive decline in endothelial function, vascular remodeling, inflammation, and increased arterial stiffness [10]. Processes responsible for this include activation of proinflammatory pathways, oxidative stress, cell senescence, and the instigation of a vascular smooth muscle cell (VSMC) proliferative phenotype. These processes are also present in the development of hypertension and, as a consequence, are closely related and indeed reciprocal in that vascular ageing may propagate the pathophysiology of hypertension and vice versa. Notably, these processes also appear to be modulated by sex.
The means by which sex interacts with blood pressure and vascular ageing are complex and may result from a multitude of hormonal, chromosomal, or even psychosocial factors [11]. Sex steroids, and the receptors through which they act, are emerging as important mediators in the promotion and maintenance of sexual divergence in blood pressure regulation across the lifespan, and the development of vascular injury with age. In this narrative review we evaluate the relationship between estrogen and androgen hormone receptors, hypertension, and vascular ageing.
Blood pressure, vascular health, and sex steroids across the lifespan
The blood pressure of males and females are equivalent in childhood, however, rapidly rise and exhibit sexual dimorphism during and after puberty, which coincides with the advent of increased sex hormone secretion and function [12]. The influence of estrogen on female blood pressure can be observed during the menstrual cycle, where blood pressure inversely relates to circulating estrogen levels [13]. Increases in blood pressure are more evident in males, at least until later in life, resulting in males having significantly higher blood pressure than age-matched female counterparts [14]. In a meta-analysis of 23 studies including 3476 non-hypertensive participants, 24-h systolic and diastolic blood pressure was 6 and 4 mmHg higher in males than females, respectively [15].
However, females do exhibit a sharper incline in blood pressure, commencing and persisting from their third decade compared to males [16]. In the United States between 2013 and 2016, the prevalence of hypertension in females and males per age group in the National Health and Nutrition Examination Survey (NHANES) was 13% versus 25.7% (20–34 years), 31.6% versus 42.5% (35–44 years), 49.7% versus 56.3% (45–54 years), and 63.9% versus 66.4% (55–64 years) [17]. Following this timepoint, which would be consistent with the onset of the menopause and loss of estrogen, females consistently demonstrate a higher prevalence of hypertension than males (Fig. 1).

Ageing is associated with reductions in sex hormone levels that may facilitate alterations in blood pressure regulation and promote hypertension and vascular ageing. Testosterone declines by ~1% per year in males over 30 years, while almost a fifth of males over 60 years of age have testosterone levels below normal range values for young males [18, 19]. Although not formally studied in the vasculature, this would be expected to reduce androgen receptor (AR) expression. In a prospective study of males over the age of 50, total testosterone was inversely associated with systolic and diastolic blood pressure and increased risk of death [20]. Similarly, low testosterone is associated with increased pulse wave velocity in older hypogonadal males [21]. Interestingly, this was partially reversed with testosterone supplementation. Data from randomized controlled trials regarding the impact of testosterone on blood pressure are lacking, however, in observational studies of older hypogonadal males, testosterone therapy resulted in decreases in blood pressure [22, 23].
In females, the acceleration in cardiovascular risk and vascular dysfunction following the menopause, and reduction in endogenous estrogen, suggests that the interaction between age and estradiol levels may promote vascular vulnerability. However, in a sub-analysis of the Women’s Health Initiative (WHI) study, conjugated equine estrogen appeared to increase the risk of developing hypertension in older postmenopausal females, which decreased following the discontinuation of this treatment [24]. This effect may be limited to this dose or formulation, as in the Kronos Early Estrogen Prevention Study transdermal or lower doses of conjugated equine estrogen did not alter blood pressure [25].
Taken together, these data suggest that sex steroids are important mediators of blood pressure, however, the effect that these hormones elicit may differ according to the stage of life of an individual.
Cardiovascular sex steroid receptors
Estrogen receptors
Sex steroid receptors are expressed throughout the vasculature and sex steroids act through their receptors via genomic and non-genomic mechanisms. Estrogen receptors (ERs) are expressed in endothelial and VSMCs and their actions in these tissues that modulate vascular tone are numeorus [26]. The primary physiological estrogen is 17β-estradiol, which mediates direct genomic signaling, where it binds to cytosolic ER, ERα, and Erβ. These in turn dimerize and translocate to the nucleus where they bind to estrogen response elements (ERE). Alternatively, binding may occur on the activator protein-1 and specificity protein-1, which reside on the promoter of estrogen responsive genes and may modulate transcriptional changes [27]. Estrogen can also bind to membrane-bound ERs (ERα, ERβ, and G protein coupled estrogen receptor), which promote intracellular second messenger signaling, via MAPK/ERK/PI3K/cAMP, that indirectly modulates gene expression and facilitates rapid changes that may alter blood pressure such as increasing NO bioavailability and promoting vasodilatation.
The RAAS plays an important role in the vasculature aberrations that occur with ageing and hypertension and is also modulated by sex hormones [10]. Baseline levels of renin, plasma renin activity and aldosterone are elevated during the luteal phase of the menstrual cycle, where estrogen levels are high compared to the follicular phase [28]. Despite humoral activation of RAAS, mean arterial blood pressure during the luteal phase was not maintained during orthostatic stress, suggesting that estrogen may downregulate tissue responses to RAAS components either through direct ER signaling or through NO-mediated vasodilatation.
ERα appears to be pivotal in the relationship between estrogen and RAAS mediators (Table 1). In the juxtaglomerular cells, ERα directly binds to the ERE in the renin enhancer gene that is required for basal renin expression [29]. Importantly, angiotensin (Ang) II induced hypertension is increased in ovariectomized ERα knockout female mice, compared with intact wild-type [30]. In premenopausal females, an ERα mediated increase in Ang-(1-7) and angiotensin-converting enzyme (ACE) 2 activity promotes a vasodilatory phenotype [31]. The vasodilatory effect of Ang-(1-7) is lost in elderly female mice and restored with estrogen replacement [32]. The combination of ageing and estrogen loss in females may therefore blunt the protective effect of this RAAS component and promote hypertension.
ERα consists of six domains containing two independent activation functions, AF-1 and AF-2. Models of ERα inactivation (ER−/−) and selective inactivation of nuclear ERα actions, through activating function 2 (AF20) deletions, or membrane-initiated ERα actions via point mutations of the palmitoylable Cys451(C451A), have elucidated pathways by which estrogen elicits its cardioprotective effect [33]. In mice treated with Ang II, increased systolic blood pressure, ventricle weight, and vascular contractility were evident in ERα−/− and AF20 mice compared to either wild-type or C451A mice. Moreover, renal inflammation and oxidative stress were increased in old hypertensive ER−/− and AF20 mice, compared to old hypertensive wild-type and C451A mice. Therefore, nuclear ERα-AF2, and not extra-nuclear ERα signaling, appears to play a protective role in Ang-II dependent hypertension and target organ damage in ageing mice.
There is also evidence for the role of ERβ in the modulation of blood pressure, however, the mechanisms underpinning these actions are less clear (Table 1). ERβ-deficient mice develop abnormalities in VSMC ion channel function and age associated hypertension [34]. Ligand-mediated activation of ERβ also promotes reductions in blood pressure in spontaneously hypertensive rats (SHR) [35]. Interestingly, direct activation of ERβ was found to be more potent than stimulation through the nonselective use of 17-β estradiol. Moreover, in humans polymorphisms in ERβ have been found to be associated with salt-sensitive blood pressure and hypertension [36, 37].
Importantly, ER expression and the balance of ER subtypes can change with ageing and prolonged estrogen deficiency, which in turn alters responses to estrogen [38]. In a small sample of postmortem coronary arteries, VSMC ER expression was lower in postmenopausal versus premenopausal females, while atherosclerosis lowered ER expression independent of menopausal state [39]. Animal studies have demonstrated that endothelial ERα expression is downregulated and endothelial NO signaling is impaired following extended periods of estrogen deprivation [40]. This does not recover following the reintroduction of this sex hormone. In endothelial cells obtained from peripheral veins, ERα expression fluctuated throughout the menstrual cycle in response to estrogen, and was reduced in estrogen-deficient postmenopausal females [41]. Age-related methylation of ER promotor regions, histone deacetylation, and inhibition of membrane localization via posttranslational modifications may mediate senescence-related regulation of ERα expression and function [42]. Conversely, in uterine arteries of postmenopausal females there is a progressive increase in ERβ expression [38]. Consequently, alterations in ER subtype expression with age and declining estrogen levels, may mediate a shift in ERα: ERβ receptor ratios and promote an adverse vascular phenotype and contribute to the development of hypertension and vascular injury.
Androgen receptors
Testosterone and its more potent metabolite, dihydrotestosterone (DHT), are ligands of the AR. Much like the ER, the AR is expressed in both endothelial and VSMCs. The AR consists of a 110 kDa protein receptor with three major functional regions for transactivation, a DNA binding domain and a hormone binding domain [43]. Two AR variants have been discovered, AR-A and AR-B, with the latter predominating in the most tissues where both receptors are expressed [44]. The exact role of these receptor subtypes has yet to be elucidated.
Unbound cytosolic ARs are co-localized with a number of chaperones, such as heat shock proteins and cytoskeletal elements [44]. After binding, the receptor undergoes a conformational change, resulting in a dissociation of these chaperones and promotes AR dimerization and nuclear translocation, where its interactions with androgen response elements (ARE) to modulate genomic responses [45]. Alternatively, non-genomic testosterone effects are mediated by membrane-bound ARs and act via multiple pathways including PKA, PKC, and MAPK [46]. Through these non-genomic pathways, testosterone can stimulate rapid vasodilatation via endothelium dependent and independent mechanisms [47]. The former result from increased NO bioavailability via AR-mediated eNOS activation, and the release of vasodilating factors into the VSMCs.
Testosterone increases renin levels and expression/activity of ACE and AT1R, while downregulating AT2R, thereby favouring a vasoconstrictor pathway [48]. In models of hypertension, such as in New Zealand genetically hypertensive rats, androgens enhance vascular responsiveness to Ang II [49]. The interactions between these factors may therefore be important in the development of hypertension. Indeed, Ang II induced vascular contraction appears dependent on androgen status. Chronic testosterone deficiency via castration ameliorates Ang II induced increases in blood pressure [50]. Conversely, the hypertensive effect of Ang II is exaggerated in males with intact testis and castrated males that received exogenous testosterone replacement. Therefore, testosterone may modulate the development and maintenance of Ang II induced hypertension and increased vascular contractility to pressors. Interestingly, testosterone supplements in young non-hypogonadal male SHR resulted in increases in blood pressure, which is mediated via RAAS. However, when administered to older SHRs a decrease in blood pressure was observed [51]. Consequently, both the testosterone status, which may alter AR expression, and the age of recipient may influence the blood pressure response to this hormone.
Molecular mechanisms of sex steroid mediated hypertension and vascular ageing
In addition to direct effects of sex hormones on vasodilation via the NO system, or indirectly via the RAAS, sex steroids may modulate a number of mechanisms evident in the development of both hypertension and vascular ageing.
Oxidative stress
Oxidative stress plays a central role in the development of vascular ageing in hypertension. In rat coronary arterioles, both age and loss of circulating estrogens, as a consequence of ovariectomy, reduce NO bioavailability. Importantly, dilatation of these arterioles are highly dependent on this mechanism. Impairment in reactive oxygen species (ROS) regulation, especially O2-, appears to modulate this decrease in NO-mediated dilation as a consequence of age or estrogen deficiency [52]. Decreases in Cu/Zn superoxide dismutase expression in both aged and ovariectomized rats were observed. This was restored following estrogen replacement in young rats. This phenomenon has also been observed in mice, where the aortic contractile response to thromboxane A2 is modulated by the interaction between NO synthase (NOS) and cyclooxygenase (COX) pathways [53]. This interaction is dependent again on age and estrogen status and promoted by COX-mediated generation of superoxide that decreases NO bioavailability. Of note, increases in the ERβ: ERα ratio that appear to occur with ageing are associated with increased oxidative stress [54].
The relationship between testosterone and redox status is also complex. The AR modulates increased expression of a number of pro-oxidant enzymes such as NAD(P)H oxidases, xanthine oxidases and COX-2. Furthermore, AR may increase the transcription of genes related to the c-Src and PI3K/Akt pathways, which also promote ROS generation [55]. In a rat model of Ang II induced hypertension, ROS generation was increased by testosterone, only in hypertensives through phosphorylation of c-Src, an upstream regulator of NADPH oxidase [56]. However, these effects appear to be determined by testosterone status and ageing. In a rat model of ageing and testosterone deprivation, a decrease in antioxidant haeme oxygenase activity was observed and reversed with testosterone supplementation [57]. Moreover, testosterone also induces AR-mediated mitochondrial-associated ROS generation and apoptosis in VSMCs [58]. Orchiectomized male rats receiving testosterone replacement demonstrated an improved cardiovascular redox state, thereby reversing elevations in lipid peroxidation and nitrotyrosine [59]. In line with this, low testosterone levels are associated with enhanced oxidative stress and in males with type 2 diabetes and a mean age of over 50 years [60], however, it is unclear whether testosterone supplementation is capable of restoring this balance.
Inflammation
Inflammation plays a significant role in both the pathogenesis of hypertension and cardiovascular ageing. Via the ERs, estrogen has demonstrated the capacity to reduce the inflammatory response by negatively modulating proinflammatory mediator expression, which likely contributes to the cardioprotective role of this sex hormone [61]. Following balloon injury of the right carotid artery of ovariectomized rats, estradiol significantly reduced the expression of adhesion modules (P-selectin, vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule-1 (ICAM-1)), chemoattractants (cytokine-induced neutrophil chemoattractant 2β (CINC-2β), monocyte chemoattractant protein 1 (MCP-1)), and proinflammatory cytokines (IL-1 and IL-6).
However, these effects may also be age-dependent. In murine VSMCs and bone marrow-derived macrophages, the estrogen-mediated reduction in inflammatory response to C-reactive protein occurred only in female cells derived from young and not aged mice [62]. Similarly, in uterine arteries of postmenopausal females, ageing was associated with a switch from an anti-inflammatory to proinflammatory profile [38]. In particular, an increased correlation was observed MCP-1 and the adhesion molecules soluble VCAM-1 and ICAM-1 with ageing. These data further corroborate the hypothesis of the influence of altered ER subtype ratio promoting an adverse vascular phenotype, as increased ERβ expression with ageing was observed.
The immunomodulatory role of androgens, and in particular testosterone, has long been theorized due to the greater incidence of immune-mediated diseases in females and androgen deficient males [63]. Evidence exploring the relationship of between testosterone, hypertension and vascular ageing is limited, however, inference from other vascular models pertains to the immunomodulatory role of the AR in this context. In a murine inflammatory abdominal aortic aneurysm (AAA) model, castration promoted AAA formation via the expansion of inflammatory macrophages and IL-6 and IL-1β upregulation [64]. Conversely, following the administration of testosterone, AAA formation was found to be inhibited by the amelioration of macrophage-mediated inflammatory responses. In human endothelial cells the AR mediates the downregulation of adhesion molecules, chemokines and proteases induced by TNFα, following exposure to DHT [65].
It has been recently demonstrated that testosterone may provide a protective effect on vascular ageing by improving vascular remodeling through the Growth arrest-specific protein 6/Axl pathway, which has been implicated in cell survival, adhesion, migration and inflammatory cytokine release [66]. These findings are consistent with the results of a randomized control trial of testosterone replacement in older hypogonadal males, where testosterone was found to reduce TNFα, IL-1β and increase IL-10, thereby promoting an anti-inflammatory state [67].
Beyond the vasculature
Beyond the evidence provided in this review, sex hormones may play a wider role in blood pressure regulation through central nervous system, renal involvement and effects on cardiac output and temporal peripheral resistance. For instance, ERs are expressed in the subfornical organ, the paraventricular nucleus and the rostral ventral lateral medulla [68]. These are key regions that regulate sympathetic nerve activity (SNA), which when increased has been implicated as a primary precursor of hypertension. Young premenopausal females have been shown to have lower SNA, than do males of the same age, whereas postmenopausal females have resting muscle SNA that is similar to those of age-matched males [69, 70]. In postmenopausal females long term transdermal estrogen administration decreased SNA and was associated with significant reduction in 24-h ambulatory BP [71, 72]. Similarly, ARs are widely expressed in the central nervous system, however, their role, or indeed the influence of fluctuating sex steroid receptor expression, in the central nervous system in modulating SNA is unclear. Ultimately, a multi-system approach will be required to develop a comprehensive understanding of the role of these receptors in the development of hypertension, and how their age-related plasticity may modulate this relationship.
Sex-dependent hormonal action
Another factor that must be considered is the sex-dependent role of vascular androgen and ERs. For instance, it has been demonstrated that raised free androgen index (i.e. the ratio of circulating testosterone to sex hormone binding globulin) in postmenopausal females is associated with hypertension and vascular ageing [73]. Therefore, although one system of sex steroid receptor may predominate in a particular sex, both are likely to be of physiological significance, particularly if the other is perturbed. However, the definitive role of ARs in females and ERs in males with respect to the development of hypertension and vascular ageing has not yet been ellucidated [47, 74].
This putative interaction may be of particular importance to the long-term vascular health of transgender individuals receiving gender-affirming hormonal therapy. There is currently insufficient data to advise the impact of sex steroids on blood pressure in this population, however, some studies suggest a higher rate of hypertension [75, 76]. It remains unclear to what effect the administration of physiological concentrations of estrogen or testosterone has in natal males and females, respectively, on sex steroid receptor expression or function. This has the added complexity that unless doses of these hormones are adjusted, age-relative supraphysiological levels will be achieved in older individuals, and the long-term vascular sequelae of this is uncertain [77].
Conclusions
Mechanisms whereby sex steroid receptors mediate shared processes in both hypertension and vascular ageing are becoming evident. A common theme in the evidence provided is that both the sex hormone status of an individual and their physiological age are important determinants of their response to sex steroid administration. Estrogen, despite eliciting a number of cardioprotective effects in females in youth, may facilitate vascular injury later in life following periods of deprivation. The plasticity of receptor subtype expression in ageing estrogen-deficient females appears to be of particular importance in this population. Conversely, the increased cardiovascular risk in males elicited by testosterone rises further through the relative hypogonadism and age, and is potentially rescued through subsequent re-exposure. It is clear that much work is required to understand the mechanisms by which sex steroid receptors modulate the development of hypertension and vascular ageing, and how these relationships can be exploited to prevent such conditions.
References
Yusuf S, Joseph P, Rangarajan S, Islam S, Mente A, Hystad P, et al. Modifiable risk factors, cardiovascular disease, and mortality in 155 722 individuals from 21 high-income, middle-income, and low-income countries (PURE): a prospective cohort study. Lancet. 2020;395:795–808.
Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, et al. Global disparities of hypertension prevalence and control. Circulation. 2016;134:441–50.
Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart Disease and Stroke Statistics—2018 Update A Report From the American Heart Association. Circulation. 2018;137:67–492.
Williams B, Mancia G, Spiering W, Rosei EA, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. 2018;39:3021–104.
Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Himmelfarb CD, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task F. Hypertension. 2018;71:E13–E115.
Wright JT, Williamson JD, Whelton PK, Snyder JK, Sink KM, Rocco MV, et al. A Randomized Trial of Intensive versus Standard Blood-Pressure Control. N. Engl J Med. 2015;373:2103–16.
Foy CG, Lovato LC, Vitolins MZ, Bates JT, Campbell R, Cushman WC, et al. Gender, blood pressure, and cardiovascular and renal outcomes in adults with hypertension fromthe Systolic Blood Pressure InterventionTrial. J Hypertens. 2018;36:904–15.
Delles C, Currie G. Sex differences in hypertension and other cardiovascular diseases. J Hypertens. 2018;36:768–70.
Gu Q, Burt VL, Paulose-Ram R, Dillon CF. Gender differences in hypertension treatment, drug utilization patterns, and blood pressure control among US adults with hypertension: Data from the National Health and Nutrition Examination Survey 1999-2004. Am J Hypertens. 2008;21:789–98.
Harvey A, Montezano AC, Touyz RM. Vascular biology of ageing-Implications in hypertension. J Mol Cell Cardiol. 2015;83:112–21.
Connelly PJ, Azizi Z, Alipour P, Delles C, Pilote L, Raparelli V. The importance of gender to understand sex differences in cardiovascular disease. Can J Cardiol. 2021;37:699–710.
Jackson LV, Thalange NKS, Cole TJ. Blood pressure centiles for Great Britain. Arch Dis Child. 2007;92:298–303.
Caroccia B, Seccia TM, Barton M, Rossi GP. Estrogen signaling in the adrenal cortex: implications for blood pressure sex differences. Hypertension. 2016;68:840–8.
Colafella KMM, Denton KM. Sex-specific differences in hypertension and associated cardiovascular disease. Nat Rev Nephrol. 2018;14:185–201.
Staessen J, Fagard R, Lijnen P, Thijs L, Van Hoof R, Amery A. Mean and range of the ambulatory pressure in normotensive subjects from a meta-analysis of 23 studies. Am J Cardiol. 1991;67:723–7.
Ji H, Kim A, Ebinger JE, Niiranen TJ, Claggett BL, Bairey Merz CN, et al. Sex differences in blood pressure trajectories over the life course. JAMA Cardiol. 2020;5:255–62.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics—2020 update: a report from the American Heart Association. Circulation. 2020;141:E139–E596.
Matsumoto AM. Andropause: clinical implications of the decline in serum testosterone levels with aging in men. J Gerontol. 2002;57:M76–99.
Harman SM, Metter EJ, Tobin JD, Pearson J, Blackman MR. Longitudinal effects of aging on serum total and free testosterone levels in healthy men. J Clin Endocrinol Metab. 2001;86:724–31.
Laughlin GA, Barrett-Connor E, Bergstrom J. Low serum testosterone and mortality in older men. J Clin Endocrinol Metab. 2008;93:68–75.
Yaron M, Greenman Y, Rosenfeld JB, Izkhakov E, Limor R, Osher E, et al. Effect of testosterone replacement therapy on arterial stiffness in older hypogonadal men. Eur J Endocrinol. 2009;160:839–46.
Traish AM, Haider A, Haider KS, Doros G, Saad F. Long-term testosterone therapy improves cardiometabolic function and reduces risk of cardiovascular disease in men with hypogonadism. J Cardiovasc Pharm Ther. 2017;22:414–33.
Yassin DJ, Doros G, Hammerer PG, Yassin AA. Long-term testosterone treatment in elderly men with hypogonadism and erectile dysfunction reduces obesity parameters and improves metabolic syndrome and health-related quality of life. J Sex Med. 2014;11:1567–76.
Swica Y, Warren MP, Manson JE, Aragaki AK, Bassuk SS, Shimbo D, et al. Effects of oral conjugated equine estrogens with or without medroxyprogesterone acetate on incident hypertension in the Women’s Health Initiative hormone therapy trials. Menopause. 2018;25:753–61.
Harman SM, Black DM, Naftolin F, Brinton EA, Budoff MJ, Cedars MI, et al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women. Ann Intern Med. 2014;161:249.
Khalil RA. Estrogen, vascular estrogen receptor and hormone therapy in postmenopausal vascular disease. Biochem Pharm. 2013;86:1627–42.
Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ. 2017;8:33.
Chidambaram M, Duncan JA, Lai VS, Cattran DC, Floras JS, Scholey JW, et al. Variation in the renin angiotensin system throughout the normal menstrual cycle. J Am Soc Nephrol. 2002;13:446–52.
Lu KT, Keen HL, Weatherford ET, Sequeira-Lopez MLS, Gomez RA, Sigmund CD. Estrogen receptor α is required for maintaining baseline renin expression. Hypertension. 2016;67:992–9.
Xue B, Pamidimukkala J, Lubahn DB, Hay M. Estrogen receptor-α mediates estrogen protection from angiotensin II-induced hypertension in conscious female mice. Am J Physiol Hear Circ Physiol. 2007;292:1770–6.
Medina D, Mehay D, Arnold AC. Sex differences in cardiovascular actions of the renin–angiotensin system. Clin Auton Res. 2020;30:393–408.
Costa-Fraga FP, Goncalves GK, Souza-Neto FP, Reis AM, Capettini LAS, Santos RAS, et al. Age-related changes in vascular responses to angiotensin-(1-7) in female mice. J Renin-Angiotensin-Aldosterone Syst. 2018;19. https://doi.org/10.1177/1470320318789332.
Guivarc’h E, Favre J, Guihot AL, Vessières E, Grimaud L, Proux C, et al. Nuclear activation function 2 estrogen receptor α attenuates arterial and renal alterations due to aging and hypertension in female mice. J Am Heart Assoc. 2020;9:e013895.
Zhu Y, Bian Z, Lu P, Karas RRH, Bao L, Cox D, et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science. 2002;295:505–8.
Jazbutyte V, Arias-Loza PA, Hu K, Widder J, Govindaraj V, von Poser-Klein C, et al. Ligand-dependent activation of ER lowers blood pressure and attenuates cardiac hypertrophy in ovariectomized spontaneously hypertensive rats. Cardiovasc Res. 2007;77:774–81.
Ogawa S, Emi M, Shiraki M, Hosoi T, Ouchi Y, Inoue S. Association of estrogen receptor β (ESR2) gene polymorphism with blood pressure. J Hum Genet. 2000;45:327–30.
Manosroi W, Tan JW, Rariy CM, Sun B, Goodarzi MO, Saxena AR, et al. The association of estrogen receptor-β gene variation with salt-sensitive blood pressure. J Clin Endocrinol Metab. 2017;102:4124–35.
Novella S, Heras M, Hermenegildo C, Dantas AP. Effects of estrogen on vascular inflammation: a matter of timing. Arterioscler Thromb Vasc Biol. 2012;32:2035–42.
Losordo DW, Kearney M, Kim EA, Jekanowski J, Isner JM. Variable expression of the estrogen receptor in normal and atherosclerotic coronary arteries of premenopausal women. Circulation. 1994;89:1501–10.
Pinna C, Cignarella A, Sanvito P, Pelosi V, Bolego C. Prolonged ovarian hormone deprivation impairs the protective vascular actions of estrogen receptor α agonists. Hypertension. 2008;51:1210–7.
Gavin KM, Seals DR, Silver AE, Moreau KL. Vascular endothelial estrogen receptor α is modulated by estrogen status and related to endothelial function and endothelial nitric oxide synthase in healthy women. J Clin Endocrinol Metab. 2009;94:3513–20.
Somani YB, Pawelczyk JA, De Souza MJ, Kris-Etherton PM, Proctor DN. Aging women and their endothelium: probing the relative role of estrogen on vasodilator function. Am J Physiol Hear Circ Physiol 2019;317:H395–H404.
Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23:175–200.
Boese AC, Kim SC, Yin K-JJ, Lee J-PP, Hamblin MH. Sex differences in vascular physiology and pathophysiology: estrogen and androgen signaling in health and disease. Am J Physiol Heart Circ Physiol. 2017;313:H524–H545.
Chistiakov D, Myasoedova V, Melnichenko A, Grechko A, Orekhov A. Role of androgens in cardiovascular pathology. Vasc Health Risk Manag. 2018;14:283–90.
Lopes RAM, Neves KB, Carneiro FS, Tostes RC. Testosterone and vascular function in aging. Front Physiol. 2012;3:89.
Moreau KL, Babcock MC, Hildreth KL. Sex differences in vascular aging in response to testosterone. Biol Sex Differ. 2020;11:18.
Te Riet L, Van Esch JHM, Roks AJM, Van Den Meiracker AH, Danser AHJ. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res. 2015;116:960–75.
Song J, Kost CK, Martin DS. Androgens augment renal vascular responses to ANG II in New Zealand genetically hypertensive rats. Am J Physiol Integr Comp Physiol. 2006;290:R1608–R1615.
Mishra JS, More AS, Gopalakrishnan K, Kumar S. Testosterone plays a permissive role in angiotensin II-induced hypertension and cardiac hypertrophy in male rats. Biol Reprod. 2019;100:139–48.
Dalmasso C, Patil CN, Cardozo LLY, Romero DG, Maranon RO. Cardiovascular and metabolic consequences of testosterone supplements in young and old male spontaneously hypertensive rats: Implications for testosterone supplements in men. J Am Heart Assoc. 2017;6:e007074.
Kang LS, Chen B, Reyes RA, LeBlanc AJ, Teng B, Mustafa SJ, et al. Aging and estrogen alter endothelial reactivity to reactive oxygen species in coronary arterioles. Am J Physiol Circ Physiol. 2011;300:H2105–H2115.
Vidal-Gómez X, Novella S, Pérez-Monzó I, Garabito M, Dantas AP, Segarra G, et al. Decreased bioavailability of nitric oxide in aorta from ovariectomized senescent mice: role of cyclooxygenase. Exp Gerontol. 2016;76:1–8.
Novensà L, Novella S, Medina P, Segarra G, Castillo N, Heras M, et al. Aging negatively affects estrogens-mediated effects on nitric oxide bioavailability by shifting ERα/ERβ balance in female mice. PLoS One. 2011;6:e25335.
Cruz-Topete D, Dominic P, Stokes KY. Uncovering sex-specific mechanisms of action of testosterone and redox balance. Redox Biol. 2020;31:101490.
Chignalia AZ, Schuldt EZ, Camargo LL, Montezano AC, Callera GE, Laurindo FR, et al. Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c-Src-dependent pathways. Hypertension. 2012;59:1263–71.
Szabó R, Börzsei D, Kupai K, Hoffmann A, Gesztelyi R, Berkó AM, et al. Spotlight on a new heme oxygenase pathway: testosterone-induced shifts in cardiac oxidant/antioxidant status. Antioxidants. 2019;8:288.
Lopes RAM, Neves KB, Pestana CR, Queiroz AL, Zanotto CZ, Chignalia AZ, et al. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement. Am J Physiol Circ Physiol. 2014;306:H1485–H1494.
Eleawa SM, Sakr HF, Hussein AM, Assiri AS, Bayoumy NMK, Alkhateeb M. Effect of testosterone replacement therapy on cardiac performance and oxidative stress in orchidectomized rats. Acta Physiol. 2013;209:136–47.
Rovira-Llopis S, Bañuls C, de Marañon AM, Diaz-Morales N, Jover A, Garzon S, et al. Low testosterone levels are related to oxidative stress, mitochondrial dysfunction and altered subclinical atherosclerotic markers in type 2 diabetic male patients. Free Radic Biol Med. 2017;108:155–62.
Miller AP, Feng W, Xing D, Weathington NM, Blalock JE, Chen YF, et al. Estrogen modulates inflammatory mediator expression and neutrophil chemotaxis in injured arteries. Circulation. 2004;110:1664–9.
Bowling MR, Xing D, Kapadia A, Chen YF, Szalai AJ, Oparil S, et al. Estrogen effects on vascular inflammation are age dependent: role of estrogen receptors. Arterioscler Thromb Vasc Biol. 2014;34:1477–85.
Cutolo M, Wilder RL. Different roles for androgens and estrogens in the susceptibility to autoimmune rheumatic diseases. Rheum Dis Clin North Am. 2000;26:825–39.
Son B-K, Kojima T, Ogawa S, Akishita M. Testosterone inhibits aneurysm formation and vascular inflammation in male mice. J Endocrinol. 2019;241:307–17.
Norata GD, Tibolla G, Seccomandi PM, Poletti A, Catapano AL. Dihydrotestosterone decreases tumor necrosis factor-α and lipopolysaccharide-induced inflammatory response in human endothelial cells. J Clin Endocrinol Metab. 2006;91:546–54.
Chen YQ, Zhou HM, Chen FF, Liu YP, Han L, Song M, et al. Testosterone ameliorates vascular aging via the Gas6/Axl signaling pathway. Aging. 2020;12:16111–25.
Malkin CJ, Pugh PJ, Jones RD, Kapoor D, Channer KS, Jones TH. The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J Clin Endocrinol Metab. 2004;89:3313–8.
Hay M. Sex, the brain and hypertension: brain oestrogen receptors and high blood pressure risk factors. Clin Sci. 2016;130:9–18.
Matsukawa T, Sugiyama Y, Watanabe T, Kobayashi F, Mano T. Gender difference in age-related changes in muscle sympathetic nerve activity in healthy subjects. Am J Physiol. 1998;275:R1600–4.
Ettinger SM, Silber DH, Gray KS, Smith MB, Yang QX, Kunselman AR, et al. Effects of the ovarian cycle on sympathetic neural outflow during static exercise. J Appl Physiol. 1998;85:2075–81.
Vongpatanasin W, Tuncel M, Mansour Y, Arbique D, Victor RG. Transdermal estrogen replacement therapy decreases sympathetic activity in postmenopausal women. Circulation. 2001;103:2903–8.
O’Sullivan AJ, Crampton LJ, Freund J, Ho KKY. The route of estrogen replacement therapy confers divergent effects on substrate oxidation and body composition in postmenopausal women. J Clin Investig. 1998;102:1035–40.
Georgiopoulos GA, Lambrinoudaki I, Athanasouli F, Armeni E, Rizos D, Kazani M, et al. Free androgen index as a predictor of blood pressure progression and accelerated vascular aging in menopause. Atherosclerosis. 2016;247:177–83.
Morselli E, Santos RS, Criollo A, Nelson MD, Palmer BF, Clegg DJ. The effects of oestrogens and their receptors on cardiometabolic health. Nat Rev Endocrinol. 2017;13:352–64.
Connelly PJ, Marie Freel E, Perry C, Ewan J, Touyz RM, Currie G, et al. Gender-affirming hormone therapy, vascular health and cardiovascular disease in transgender adults. Hypertension. 2019;74:1266–74.
Connelly PJ, Clark A, Touyz RM, Delles C. Transgender adults, gender-affirming hormone therapy and blood pressure: a systematic review. J Hypertens. 2021;39:223–30.
Gooren LJ, T’Sjoen G. Endocrine treatment of aging transgender people. Rev Endocr Metab Disord. 2018;19:253–62.
Ober C, Loisel DA, Gilad Y. Sex-specific genetic architecture of human disease. Nat Rev Genet. 2008;9:911–22.
Acknowledgements
The authors are grateful to the British Heart Foundation (Centre of Research Excellence Award, RE/18/6/34217) for funding our research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Connelly, P.J., Casey, H., Montezano, A.C. et al. Sex steroids receptors, hypertension, and vascular ageing. J Hum Hypertens 36, 120–125 (2022). https://doi.org/10.1038/s41371-021-00576-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41371-021-00576-7
This article is cited by
-
Genetic hypogonadal mouse model reveals niche-specific influence of reproductive axis and sex on intestinal microbial communities
Biology of Sex Differences (2023)
-
Clinical utility of lipid ratios as potential predictors of metabolic syndrome among the elderly population: Birjand Longitudinal Aging Study (BLAS)
BMC Geriatrics (2023)
-
Arterial stiffness and cardiometabolic health in omnivores and vegetarians: a cross-sectional pilot study
BMC Research Notes (2022)
-
Rural-urban difference in the prevalence of hypertension in West Africa: a systematic review and meta-analysis
Journal of Human Hypertension (2022)
-
Estrogen-mediated mechanisms in hypertension and other cardiovascular diseases
Journal of Human Hypertension (2022)
