
Abstract
Background
Genetic screening for youth with obesity in the absence of syndromic findings has not been part of obesity management. For children with early onset obesity, genetic screening is recommended for those having clinical features of genetic obesity syndromes (including hyperphagia).
Objectives
The overarching goal of this work is to report the findings and experiences from one pediatric weight management program that implemented targeted sequencing analysis for genes known to cause rare genetic disorders of obesity.
Subjects/Methods
This exploratory study evaluated youth tested over an 18-month period using a panel of 40-genes in the melanocortin 4 receptor pathway. Medical records were reviewed for demographic and visit information, including body mass index (BMI) percent of 95th percentile (%BMIp95) and two eating behaviors.
Results
Of 117 subjects: 51.3% were male; 53.8% Hispanic; mean age 10.2 years (SD 3.8); mean %BMIp95 157% (SD 29%). Most subjects were self- or caregiver-reported to have overeating to excess or binge eating (80.3%) and sneaking food or eating in secret (59.0%). Among analyzed genes, 72 subjects (61.5%) had at least one variant reported; 50 (42.7%) had a single variant reported; 22 (18.8%) had 2–4 variants reported; most variants were rare (<0.05% minor allele frequency [MAF]), and of uncertain significance; all variants were heterozygous. Nine subjects (7.7%) had a variant reported as PSCK1 “risk” or MC4R “likely pathogenic”; 39 (33.3%) had a Bardet-Biedl Syndrome (BBS) gene variant (4 with “pathogenic” or “likely pathogenic” variants). Therefore, 9 youth (7.7%) had gene variants previously identified as increasing risk for obesity and 4 youth (3.4%) had BBS carrier status.
Conclusions
Panel testing identified rare variants of uncertain significance in most youth tested, and infrequently identified variants previously reported to increase the risk for obesity. Further research in larger cohorts is needed to understand how genetic variants influence the expression of non-syndromic obesity.
Similar content being viewed by others


Introduction
Obesity is a heterogeneous disorder that develops from a complex interplay of risk factors, which include environmental, biologic, genetic, and social determinants of health [1]. Twin studies have estimated the heritability of obesity to be between 40 and 75% [2]. Genome wide association studies (GWAS) have demonstrated that polygenic obesity is caused by the cumulative influence of many genes whose effect is amplified in an obesogenic environment [1, 3, 4]. In the United States (US), 7.9% of children and adolescents aged 2–19 years have severe obesity, which has been defined as having a body mass index (BMI) ≥ 120th percent of the 95th BMI percentile (%BMIp95) for age and sex [5]. Children with severe obesity are at increased risk for cardio-metabolic and psychological comorbidities [6, 7].
Genetic testing for children with syndromic early onset obesity, such as Prader-Willi or Bardet-Biedl syndromes (BBS) [8, 9] may occur at early ages, however, genetic screening for children with obesity in the absence of syndromic features has not been a routine part of pediatric weight management (PWM) care. Rare genetic forms of obesity in children are poorly understood and likely underdiagnosed [10], despite being responsible for 5–7% of early onset obesity in children [10, 11]. Because genetic variants may influence physiological pathways involved in energy homeostasis, it is important to consider polygenic and monogenic forms of obesity in children with early onset obesity and hyperphagia [12, 13]. In children with early onset obesity, genetic screening is recommended for those who have clinical features of genetic obesity syndromes (including hyperphagia) and/or a family history of severe obesity [13, 14]. If the child presents with developmental delay, dysmorphic features, hormonal deficiencies (e.g., hypogonadism, adrenal insufficiency), congenital anomalies, or vision loss, the threshold for genetic testing should be lowered [14]. Children with underlying genetic disorders may be at greater risk for significant sequelae requiring targeted, thorough, and specialized approaches [15]. The aims of this study are to: (1) describe the findings and experiences from one PWM program implementing targeted sequencing analysis for genes known to cause rare genetic disorders of obesity; (2) provide insight for PWM programs considering genetic testing; and (3) identify areas for future research.
Subjects and methods
This exploratory study reports on 117 youth evaluated for rare disorders of obesity using genetic screening. The study included youth seen at a PWM program over 18 months (July 2019 – December 2020). The hospital institutional review board (IRB) determined this study to be secondary research exempt from consent.
PWM program clinicians include medical providers, registered dieticians, and social workers. Approximately 2/3 of the patients receiving care in the program have severe obesity [16]. During the initial visit, detailed family history, including parent-reported weight and height or obesity status are collected. Parental consanguinity is not routinely assessed.
For the first 7 months of the study, providers queried families about child eating behaviors, and caregivers completed a paper survey used in the clinical assessment of hyperphagia or disordered eating (Table 1). For the last 9 months of the study, some patients received care remotely via telemedicine due to COVID-19 risks, so responses were collected through provider interview. After resuming in person visits, parents additionally completed questions 3–7 (Table 1) on a paper survey. Hyperphagia is difficult to assess and measure, and a single definition is evolving in the literature [17]. Patient and/or family reports of frequently feeling hungry were recorded. Genetic testing was offered based on results of the eating behavior screen, concerns raised during the visit, child weight gain patterns, and family history. Study activity tracked which PWM provider tested subjects. Providers were interviewed at the end of data collection to assess their perceptions of and rationale for offering testing.
Individual electronic medical records were reviewed for demographic information (sex, parental-reported race/ethnicity) and visit information (child physical exam, age and %BMIp95). Subjects with %BMIp95 ≥ 120% were considered to have severe obesity [5]. Progress notes were reviewed as well as patient/parent responses to eating behavior questions. Subjects with records affirming “Eats in secret or sneaks food” were reported in the variable sneaking food or eating in secret. Subjects with records affirming “Continues to eat even though he/she is not hungry”, “Eats to the point of stomach pain or vomiting” or having binge eating behaviors were reported in the variable overeating to excess or binge eating. Race and ethnicity information were categorized into 4 groups: non-Hispanic Black (hereafter, Black); Hispanic; non-Hispanic White (hereafter, White); and non-Hispanic Other (hereafter, Other). Youth in the Other race and ethnicity grouping were of Asian and multiracial ancestry. Self-reported weight and height of biological parents were obtained from records and BMI was calculated when data were available. Parental BMI ≥ 30 kg/m2 or noted as having obesity were recorded to have obesity [18].
The clinic completed testing at no cost to patients through a program sponsored by Rhythm Pharmaceuticals (Boston, MA, USA), a company developing drug therapy for treatment of rare disorders of obesity [19]. Buccal swab or saliva samples were collected and sent to a clinical lab for analysis (PreventionGenetics, Marshfield, WI, USA). Reports were sent to the providers, who shared results with families. The program offers free testing to parents and siblings of youth to clarify inheritance and family phenotype patterns, for example for youth having a variant of uncertain significance in a gene known to cause autosomal dominant (AD) disease or when two changes to the same gene known to cause autosomal recessive (AR) pathology are found.
Clinical indications for testing include youth ≤18 years with BMI ≥ 97th percentile for age and sex. This panel sequences 40 genes for which obesity is a common feature, including genes in the melanocortin 4 receptor (MC4R) pathway [10, 20], including 22 Bardet-Biedl (BBS) genes (Table 2) [21]. Methods used in the testing process can be found at the PreventionGenetics Website [22].
The genetic testing reports issued to the clinical team include the following information: the specific gene(s) involved; Online Mendelian Inheritance in Man (OMIM) number; mode of inheritance established for pathogenic variants in that gene to cause disease (autosomal recessive [AR], autosomal dominant [AD], both AR/AD, or unknown); genetic variant (i.e., nucleotide change with predicted amino acid sequence change); clinical variant identification number (ClinVar ID) [23] highest allele frequency among any population gnomAD;[24] in silico missense predictions; and variant interpretation. The specific in silico algorithms used by the lab are Polymorphism Phenotyping V-2 (PolyPhen-2), Sorting Intolerant From Tolerant (SIFT), Mutation Taster, and Functional Analysis Through Hidden Markov Models (FATHMM) [25, 26]. Per American College of Medical Genetics guidelines [27], gene variants are interpreted as “pathogenic”, “likely pathogenic”, “variant of uncertain significance” (VUS), “likely benign”, and “benign”. The “likely benign” and “benign” variants are not listed in the reports. The testing lab applies the classification of “risk” alleles for variants that are common in the general population, but have been shown to be a risk factor for obesity in laboratory and population/family studies as well as by expert consensus of well-established low penetrance variants reported in ClinVar [28].
Reports from genetic testing for each subject (and family members if tested) were reviewed and collated. To explore differences in evaluated characteristics, subjects were placed into four groups based on known inheritance patterns for target genes and risk/pathogenicity: (1) negative (no variant identified); (2) subjects with a variant reported as PCSK1 “risk” or MC4R “likely pathogenic”; (3) subjects having at least one variant in a gene known to cause disease in AD or AD/AR inheritance patterns with or without other gene variants (excluding group 2); and (4) subjects with variants in genes known to cause disease in AR inheritance patterns only (excluding group 2).
We report descriptive data overall and by gene variant group for categorical variables, including child sex, age group, race, and ethnicity group, two eating behavior variables (sneaking food or eating in secret and overeating to excess or binge eating), and biological maternal and paternal obesity status, and for the continuous variables %BMIp95 and age. Fisher’s Exact and Kruskal-Wallis tests, as appropriate, were applied to evaluate between group differences. Analyses were done using IBM SPSS Statistics, version 27.
Results
Overview, subject characteristics, and provider response
During the 18-month study period, 117 children with obesity and hyperphagia were screened for rare genetic causes of obesity. Two subjects were siblings. About half of subjects were male (51.3%), half were Hispanic (53.8%), mean age was 10.2 years (SD, 3.8), mean %BMIp95 was 157% (SD 29), and almost all subjects (95.7%) had severe obesity (Table 3). No subjects had syndromic findings on physical exam. Seventy-two subjects (61.5% of individuals tested) had at least one variant reported among the analyzed genes; 50 (42.7% of individuals tested; 69.4% of those with a variant) had a single variant, and 22 (18.8% of the total tested; 30.1% of those with a variant) had more than one variant reported. Seventeen youth had 2 variants, 3 youth had 3 variants, and 2 had 4 variants reported (Table 4). All variants found were heterozygous with no complex heterozygosity. Of the 5 providers in the PWM, the majority (78%) of testing was offered by one provider with the remaining distributed among the other providers. Providers reported feeling unprepared to explain the results to families, had concerns about the added workload of contacting families with results, and thought that results would not influence care.
Reported variants
Among 72 subjects with a variant identified, 93 unique variants were reported in 34 different genes (Supplementary Table 1). Among those screened, variants were found almost equally among all age groups (Table 3). Approximately one-quarter of the sample were aged 2–6 years, but among children in this age group 22/29 (75%) had a variant reported. Among older children, just over half had a variant reported. There was a significant difference in groups by race and ethnicity (p = 0.019), with 66.7% of White youth in the group having a PSCK1 “risk” or MC4R likely pathogenic variant. Frequency of White ethnicity among other groups ranged from 5.0% to 31.1%. Many children were reported to be sneaking food or eating in secret (59.0%) and most (80.3%) were overeating to excess or binge eating. Most of the youth had a biological parent with obesity. Frequencies were similar across the gene groupings for sex, eating behavior variables, and parental obesity status (Table 3). All groups had similar %BMIp95.
Nine youth (7.7%) had the same variant reported as PCSK1 “risk” (see Supplementary Table 1) [29,30,31]. One youth with a PCSK1 “risk” variant also had an MC4R variant reported as “likely pathogenic”. Among these 9 youth, 2 (22.2%) had a single variant, 5 (55.6%) had 2 variants, 1 (11.2 %) subject had 3 and 1 (11.2 %) subject had 4 variants. Other reported variants of PCSK1 (n = 4) were VUS. Detailed information (e.g., ClinVar ID, known inheritance mode for which that gene causes disease, nucleotide alteration and amino acid change, etc.) on each gene variant per subject can be found in Supplementary Table 1.
There were 20 youth (Table 4) who had at least one heterozygous VUS in genes associated with AD or AD/AR inheritance in Mendelian forms of obesity. Seven of these youth had more than one VUS including 5 with one or more variants in genes that cause disease through AR inheritance; 2 of these 5 youth had a Bardet-Biedl syndrome (BBS) gene variant. Of the 20 youth in this group, 14 youth had variants in genes associated previously with AD inheritance, including BDNF, GNAS, KSR2, MC3R, NCOA1, NTRK2, RAI1, SH2B1, and SIM1 (Table 4, Supplementary Table 1). MC4R, and POMC are associated with both AD and AR disease. Variants in MC4R (n = 6) and POMC (n = 4) were reported for 11 youth: including one MC4R “likely pathogenic” variant noted above.
We had 22 subjects whose immediate families qualified for genetic testing at no cost and counseling under the sponsored program [21]. Families of 14 subjects completed testing of one or more additional family members, 3 declined and 5 were not offered as they were lost to follow-up.
There were 43 youth (Table 4) with at least one variant in a gene that causes obesity disorders through AR inheritance, including 5 youth with two or more variants in genes known to cause AR disease. In addition, 31 of these 43 youth had one or more BBS gene variants (26 had a single BBS variant, 5 had two BBS variants). Twenty-nine youth had other gene variants in non-BBS AR disease genes (ADCY3, ALMS1, CPE, LEPR, and PCSK1). These include the PCSK1 variants assessed as “risk” noted above. Six individuals had an ALMS1 variant, all reported as VUS. Two subjects with ALMS1 variants also had a LEPR variant and one subject with an ALMS1 variant also had 2 BBS gene variants (Table 4). CPE variants (n = 3) were reported for 3 youth; all reported as VUS.
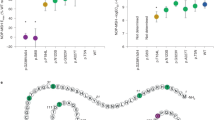
There were 39 youth (33%) with a BBS gene variant across the whole sample. There were a total of 43 variants reported in 16 different genes of the 22 tested that have been associated with BBS [9], an AR condition (BBIP1, BBS1, BBS9, BBS10, BBS12, CEP290, IFT74, IFT172, IFT174, LZTFL1, MKKS, MKS1, SDCCAG8, TRIM32, TTC8, WDPCP) (Table 2). One subject had a single heterozygous variant in BBS10 assessed as “pathogenic” and 3 subjects had a single heterozygous variant assessed as “likely pathogenic” (WDPCP, BBS12, CEP290). There were 3 youth identified to have 2 BBS gene variants within the same BBS gene (Table 4). To confirm carrier status and segregation of variants, testing of family members was recommended; only 2 of 3 families completed testing. One youth with 2 BBIP1 variants assessed as VUS inherited both in cis from a single parent with obesity, ruling out AR inheritance. Similarly, the youth with two BBS9 variants inherited both from a single parent with obesity.
Discussion
Main findings
This study describes the findings and experiences of targeted sequencing analysis for genes that cause rare disorders of obesity in youth with obesity and hyperphagia in a PWM program. Among youth screened, 61.5% (n = 72) had a variant fulfilling objective criteria reported. Fifty subjects (42.7%) had one variant and 22 subjects (18.8%) had multiple variants. Most variants were of uncertain significance. Only 9 subjects (7.7%) had a “risk”, or “likely pathogenic” variant interpreted as increasing risk for obesity. Serra-Juhe et al. [32], reported that ~5% (23/463) of children with early onset obesity had a likely pathogenic variant contributing to obesity risk. Loid et al. [33], identified pathogenic/likely pathogenic variants in 8% (7/92) of subjects with severe early-onset obesity before age 10 years. Variants in several genes known to cause rare forms of monogenic obesity have also been shown to contribute to polygenic obesity [34, 35]. Notably, variant pathogenicity interpretation can evolve over time; new clinical, genetic, or functional data may influence pathogenicity calls and result in differences between labs. For example, the genetics lab used for this study reported the PCSK1 (ClinVar ID 14040) variant as “risk” and the LEPR (ClinVar ID 631614) variant as uncertain which is discrepant from current ClinVar reports [29, 30, 36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51] (Supplementary Table 2), highlighting the importance of variant reanalysis. Without extensive efforts to provide genetic and functional evidence of variant effect, such as segregation analysis to establish inheritance patterns, more extensive examination of phenotypic characteristics, assessment in physiologically relevant experimental systems, and transcriptomic analysis of subject biospecimens, interpretation of whether these variants of uncertain significance do or do not impart risk for obesity is limited.
MC4R pathogenic variants have been reported as the most common cause of early onset obesity with prevalence rates up to 7% in primarily White European populations [1, 11]. However, among the 6 subjects who had a MC4R variant in our sample, only one youth (1%) had a “likely pathogenic” variant. This discrepancy in prevalence may reflect differences in frequencies between racial and ethnic groups as our subjects were 29% White [52, 53], or may be due to the modest size of our study cohort. A total of 9 (7.7%) youth had genetic changes (PCSK1 “risk”, MC4R “likely pathogenic”) established as increasing risk for obesity. There were 4 youth (3.4%) identified as carriers for BBS (“pathogenic” or “likely pathogenic”: WDPCP, BBS12, BBS10, CEP290). One of these 4 youth also had a PCSK1 “risk” gene in addition to being a carrier for a BBS gene variant.
There were 39 (33.3%) youth with a rare (MAF < 0.5%) heterozygous BBS gene variant, including 4 with “pathogenic” or “likely pathogenic” variants. This equates to a BBS gene variant carrier frequency of ~1 in 29. Assuming an estimated incidence of 1/100,000 for BBS, Sapp, et al. [54], estimated carrier frequency for pathogenic variants in 12 different BBS loci ranging from 1 in 250 to 1 in 2,200. BBS is a clinically heterogeneous disorder hallmarked by obesity, polydactyly, retinitis pigmentosa, renal anomalies, and learning difficulties [55]. It is also genetically heterogeneous, following a predominantly autosomal recessive inheritance pattern of at least 27 genes that play crucial roles in ciliary function [55,56,57]. There have been varying reports of BBS gene carrier status affecting risk for obesity [58]. Pathogenic alteration in BBS genes decreases leptin sensitivity, increases LEPR, and causes overactivity of the LEPR signaling pathway leading to leptin resistance [34, 35]. Balanced leptin signaling is needed for a normal satiety response [59, 60]. Heterozygous carriers of pathogenic BBS genes variants are expected to lack syndromic and phenotypic characteristics, but it is possible that monoallelic variants may contribute to increased risk for obesity in certain contexts [35, 61]. Our preliminary data suggest a potential enrichment of rare protein-coding variants in BBS genes compared to the general population. Notably, the BBS gene variants identified in our cohort are rare when compared to ethnically-matched populations in gnomAD (9% of variants ≤0.5% and >0.1% MAF; 66% of variants ≤0.1% and <0.001%); and 25% of variants ≤ 0.001% MAF; (Supplementary Table 1). Although we recognize that a majority of variants were interpreted as VUS, previous reports have shown that rare allele frequency correlates with a greater likelihood for variant pathogenicity [62]. Even so, family member testing, expanded testing to include all causal BBS and/or known ciliopathy genes, targeted clinical evaluation for clinical characteristics associated with BBS [55], and expanded cohort size are needed to formally substantiate the clinical significance of observations from this study [63].
Everyone in our cohort with variants in genes associated with AD or AR/AD inherited disease and the two youth with 2 BBIP1 and 2 BBS12 variants qualified for family testing though not all were completed due to difficulties in follow-up. For individuals with variants in genes associated with AD inheritance, family studies are indicated to delineate inheritance. For example, for individuals harboring GNAS variants, family testing and testing for calcium metabolism disorders may be indicated [64]. Individuals with some CPE variants are at increased risk for early onset type 2 diabetes [65], and those with variants of KSR2 may have severe insulin resistance that responds well to metformin [66]. Thus, identification of variants associated with rare causes of obesity may provide insight into clinical care and further evaluation. The high frequency of obesity among family members in our cohort suggest further data on inheritance patterns within families may be helpful.
Lessons learned and recommendations
At this PWM program gene variants were common in our cohort, though most were VUS. Several subjects had more than one gene variant reported. Our current understanding is limited regarding how gene variants, regardless of significance, influence obesity and hyperphagia [67]. Findings from his study suggest that identification of particular variants which may be contributing to the child’s obesity can help guide more personalized obesity care and help identify patients who may or may not respond to drug therapies or bariatric surgery [9, 15, 68].
We identified differences in how testing was employed across providers. While some providers routinely offered testing others were more selective. Development of clinical tools to increase provider confidence in offering testing and sharing the results may be helpful. Innovative ways to collaborate with colleagues in genetics, including genetic counselors as ad hoc consults embedded in PWM programs may prove to be both time and cost effective.
While information gained from genetic testing did not lead to a specific diagnosis, the structured hyperphagia assessment was helpful for providers to understand what families are experiencing. Currently there is not a standardized way to assess for hyperphagia and more research is needed [17]. Likewise, most genetic variants found were VUS, which meant providers and families were left with test results that were indeterminant based on current evidence. Though inconclusive results may not change current treatment, providers can communicate how our understanding of genetic data is evolving. Future research partnerships are needed to understand provider and family concerns regarding the testing process, their understanding of the results, and their perceptions of how the results may influence their decision-making regarding treatment and management.
We provided free genetic screening through the sponsoring lab [21], otherwise testing would have been costly for the families and the healthcare facility. This suggests that it may be important to develop comprehensive diagnostic and management strategies for youth with hyperphagia that include testing for genetic disorders [8, 15]. These strategies should include insurance coverage for genetic screening and genetic counseling given the potential for ambiguous results.
Limitations
The gene panel used for this study [21] is not comprehensive and other genes not evaluated may be contributing to null findings [69]. The reporting of the risk for obesity associated with each variant may differ between clinical labs. Due to the small sample size and lack of comparison of our findings to larger population database phenotypic and genotypic findings, it is not possible to determine if the subjects tested have a higher or lower prevalence of gene variants associated with obesity. Our findings can neither establish nor refute causality for any of the variants, nor was that the intent of this study. GWAS, mutational burden analysis, and functional studies to understand the impact of rare variants are required to understand significance, if any, of many of the variants found. Our study did not routinely include family phenotype or testing which is needed to establish causality of specific variants. These evaluations were beyond the scope of this clinical study.
A series of 9 questions were used to assess hyperphagia. Though not validated, they are the result of clinical experience in this field. A validated hyperphagia questionnaire has been used in studies of children with Prader-Willi [17], but translation of this to the clinical PWM setting has not been reported. Only children with obesity and hyperphagia were tested, so the frequency of genetic variants among youth receiving care at this PWM program with obesity not assessed as having hyperphagia is not known. Nor did we collect data on other characteristics which have been associated with increased risk of obesity (such as neuro-developmental concerns and medications). Information was not collected to track youth offered genetic screening, but whose caregiver refused. Finally, interruptions/alterations in care due to COVID-19 restrictions and access to clinic resources contributed to a non-uniform application of testing by clinic providers.
Conclusions
Our study reports findings and experiences of genetic testing for genes in the MC4R pathway among youth with obesity and hyperphagia being seen at a PWM program. Implementation of testing identified VUS in most youth tested, and variants previously identified to increase risk for obesity were infrequently found. Thus, providers and families were left with indeterminant test results based on the current state of knowledge. Further research in larger cohorts, which include familial and functional studies, is needed as to better understand how genetic variants associated with risk for obesity influence the expression of non-syndromic obesity. In addition, inclusion of provider, patient/family experiences with genetic testing and how it may impact treatment should be considered.
Data availability
The datasets generated during and/or analyzed during the current study are not publicly available due to IRB restrictions but are available from the corresponding author on reasonable request and approval of the IRB of record.
References
Thaker VV. Genetic and epigentic causes of obesity. Adolesc Med State Art Rev. 2017;28:379–405.
Wardle J, Carnell S, Haworth CM, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87:398–404.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
Yengo L, Sidorenko J, Kemper KE, Zheng, Wood AR, Weedon MN, et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27:3641–9.
Skinner AC, Ravanbakht SN, Skelton JA, Perrin EM, Armstrong SC. Prevalence of obesity and severe obesity in US children, 1999–2016. Pediatr. 2018;141:e20173459.
Rankin J, Matthews L, Cobley, Han A, Sanders R, Wiltshire HD, et al. Psychological consequences of childhood obesity: psychiatric comorbidity and prevention. Adolesc Health Med Ther. 2016;7:125–46.
Skinner AC, Perrin EM, Moss LA, Skelton JA. Cardiometabolic risks and severity of obesity in children and young adults. N Engl J Med. 2015;373:1307–17.
Koves IH, Roth C. Genetic and syndromic causes of obesity and its management. Indian J Pediatr. 2018;85:478–85.
Huvenne H, Dubern B, Clément K, Poitou C. Rare genetic forms of obesity: clinical approach and current treatments in 2016. Obes Facts. 2016;9:158–73.
Eneli I, Xu J, Webster M, McCagg A, Van Der Ploeg L, Garfield AS, et al. Tracing the effect of the melanocortin-4 receptor pathway in obesity: study design and methodology of the TEMPO registry. Appl Clin Genet. 2019;12:87–93.
Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95.
Ramachandrappa S, Farooqi IS. Genetic approaches to understanding human obesity. J Clin Invest. 2011;121:2080–6.
Styne DM, Arslanian SA, Connor EL, Farooqi IS, Murad MH, Silverstein JH, et al. Pediatric obesity-assessment, treatment, and prevention: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2017;102:709–57.
Dayton K, Miller J. Finding treatable genetic obesity: strategies for success. Curr Opin Pediatr. 2018;30:526–31.
Fox CK, Gross AC, Bomberg EM, Ryder JR, Oberle MM, Bramante CT, et al. Severe obesity in the pediatric population: current concepts in clinical care. Curr Obes Rep. 2019;8:201–9.
Gorecki MC, Feinglass JM, Binns HJ. Characteristics associated with successful weight management in youth with obesity. J Pediatr. 2019;212:35–43.
Heymsfield SB, Avena NM, Baier L, Baier P, Brantley P, Bray GA, et al. Hyperphagia: current concepts and future directions proceedings of the 2nd international conference on hyperphagia. Obesity (Silver Spring). 2014;22:S1–s17.
Hales CM, Fryar CD, Carroll MD, Freedman DS, Ogden CL Trends in obesity and severe obesity prevalence in US youth and adults by sex and age, 2007-2008 to 2015-2016. JAMA. 2018;319(16):1723–5.
Rhythm Pharmaceuticals. ND [Available from: https://www.rhythmtx.com/.]
Mutch DM, Clément K. Unraveling the genetics of human obesity. PLoS Genet. 2006;2:e188.
Uncommon Obesity ND [Available from: https://uncommonobesity.com.]
Prevention Genetics. ND [Available from: https://preventiongenetics.com.]
ClinVar Available from: https://www.ncbi.nlm.nih.gov/clinvar/.
gnomAD Genome Aggregation Database Available from: https://gnomad.broadinstitute.org/
Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37:235–41.
Pshennikova VG, Barashkov NA, Romanov GP, Teryutin FM, Solov’ev AV, Gotovtsev NN, et al. Comparison of Predictive In Silico Tools on Missense Variants in GJB2, GJB6, and GJB3 Genes Associated with Autosomal Recessive Deafness 1A (DFNB1A). ScientificWorldJournal. 2019;2019:5198931.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Group CLPRAW. Recommended terminology when descibing variants with decreased penetrance for Mendelian conditions 2020 [updated February 18, 2020]. Available from: https://www.clinicalgenome.org/site/assets/files/4531/clingenrisk_terminology_recomendations-final-02_18_20.pdf.
Mbikay M, Sirois F, Nkongolo KK, Basak A, Chrétien M. Effects of rs6234/rs6235 and rs6232/rs6234/rs6235 PCSK1 single-nucleotide polymorphism clusters on proprotein convertase 1/3 biosynthesis and activity. Mol Genet Metab. 2011;104:682–7.
Nead KT, Li A, Wehner MR, Neupane B, Gustafsson S, Butterworth A, et al. Contribution of common non-synonymous variants in PCSK1 to body mass index variation and risk of obesity: A systematic review and meta-analysis with evidence from up to 331 175 individuals. Hum Mol Genet. 2015;24:3582–94.
Shabana, Hasnain S. Prevalence of POMC R236G mutation in Pakistan. Obes Res Clin Pract. 2016;10:S110–s116.
Serra-Juhe C, Martos-Moreno GA, Bou de Pieri F, Flores R, Chowen JA, Perez-Jurado LA, et al. Heterozygous rare genetic variants in non-syndromic early-onset obesity. Int J Obes. 2020;44:830–941.
Loid P, Mustila T, Mäkitie RE, Viljakainen H, Kämpe A, Tossavainen P, et al. Rare variants in genes linked to appetite control and hypothalamic development in early-onset severe obesity. Front Endocrinol (Lausanne). 2020;11:81.
Day SE, Muller YL, Koroglu C, Kobes S, Wiedrich K, Mahkee D, et al. Exome Sequencing of 21 Bardet-Biedl syndrome (BBS) genes to identify obesity variants in 6,851 American Indians. Obesity (Silver Spring). 2021;29:748–54.
Guo DF, Rahmouni K. Molecular basis of the obesity associated with Bardet-Biedl syndrome. Trends Endocrinol Metab. 2011;22:286–93.
Albuquerque D, Estévez MN, Víbora PB, Giralt PS, Balsera AM, Cortés PG, et al. Novel variants in the MC4R and LEPR genes among severely obese children from the Iberian population. Ann Hum Genet. 2014;78:195–207.
Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356:237–47.
Kimber W, Peelman F, Prieur X, Wangensteen T, O’Rahilly S, Tavernier J, et al. Functional characterization of naturally occurring pathogenic mutations in the human leptin receptor. Endocrinol. 2008;149:6043–52.
Stoetzel C, Laurier V, Davis EE, Muller J, Rix S, Badano JL, et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nature Genet. 2006;38:521–4.
Mary L, Chennen K, Stoetzel C, Antin M, Leuvrey A, Nourisson E, et al. Bardet-Biedl syndrome: Antenatal presentation of forty-five fetuses with biallelic pathogenic variants in known Bardet-Biedl syndrome genes. Clin Genet. 2019;95:384–97.
Pawlik B, Mir A, Iqbal H, Li Y, Nürnberg G, Becker C, et al. A novel familial BBS12 mutation associated with a mild phenotype: implications for clinical and molecular diagnostic strategies. Mol Syndromol. 2010;1:27–34.
Sanchez-Navarro I, da Silva LRJ, Blanco-Kelly F, Zurita O, Sanchez-Bolivar N, Villaverde C, et al. Combining targeted panel-based resequencing and copy-number variation analysis for the diagnosis of inherited syndromic retinopathies and associated ciliopathies. Sci Rep. 2018;8:5285.
Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, et al. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 2010;329:1337–40.
Saari J, Lovell MA, Yu HC, Bellus GA. Compound heterozygosity for a frame shift mutation and a likely pathogenic sequence variant in the planar cell polarity—ciliogenesis gene WDPCP in a girl with polysyndactyly, coarctation of the aorta, and tongue hamartomas. Am J Med Genet. 2015;167a:421–7.
Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet A. 2016;48:648–56.
Lubrano-Berthelier C, Le Stunff C, Bougnères P, Vaisse C. A homozygous null mutation delineates the role of the melanocortin-4 receptor in humans. J Clin Endocrinol Metab. 2004;89:2028–32.
Stutzmann F, Tan K, Vatin V, Dina C, Jouret B, Tichet J, et al. Prevalence of melanocortin-4 receptor deficiency in Europeans and their age-dependent penetrance in multigenerational pedigrees. Diabetes. 2008;57:2511–8.
Stanikova D, Surova M, Ticha L, Petrasova M, Virgova D, Huckova M, et al. Melanocortin-4 receptor gene mutations in obese Slovak children. Physiol Res. 2015;64:883–90.
Fan ZC, Tao YX. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J Cell Mol Med. 2009;13:3268–82.
Benzinou M, Creemers JW, Choquet H, Lobbens S, Dina C, Durand E, et al. Common nonsynonymous variants in PCSK1 confer risk of obesity. Nature Genet. 2008;40:943–5.
Pickett LA, Yourshaw M, Albornoz V, Chen Z, Solorzano-Vargas RS, Nelson SF, et al. Functional consequences of a novel variant of PCSK1. PLoS One. 2013;8:e55065.
Namjou B, Stanaway IB, Lingren T, Mentch FD, Benoit B, Dikilitas O, et al. Evaluation of the MC4R gene across eMERGE network identifies many unreported obesity-associated variants. Int J Obes. 2021;45:155–69.
De Rosa MC, Chesi A, McCormack S, Zhou J, Weaver B, McDonald M, et al. Characterization of rare variants in MC4R in African American and Latino children with severe early-onset obesity. J Clin Endocrinol Metab. 2019;104:2961–70.
Sapp JC, Nishimura D, Johnston JJ, Stone EM, Héon E, Sheffield VC, et al. Recurrence risks for Bardet-Biedl syndrome: implications of locus heterogeneity. Genet Med. 2010;12:623–7.
Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21:8–13.
Forsyth R, Gunay-Aygun M Bardet-Biedl Syndrome Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle. Copyright © 1993-2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 2020.
Niederlova V, Modrak M, Tsyklauri O, Huranova M, Stepanek O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum Mutat. 2019;40:2068–87.
Suspitsin EN, Imyanitov EN. Bardet-Biedl syndrome. Mol Syndromol. 2016;7:62–71.
Zhao S, Zhu Y, Schultz RD, Li N, He A, Zhang Z, et al. Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab. 2019;30:706–19. e6
LeDuc CA, Leibel RL. Auto-regulation of leptin neurobiology. Cell Metab. 2019;30:614–6.
Stryjecki C, Alyass A, Meyre D. Ethnic, and population differences in the genetic predisposition to human obesity. Obes Rev. 2018;19:62–80.
Kobayashi Y, Yang S, Nykamp K, Garcia J, Lincoln SE, Topper SE. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017;9:13.
Muller J, Stoetzel C, Vincent MC, Leitch CC, Laurier V, Danse JM, et al. Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet. 2010;127:583–93.
Grüters-Kieslich A, Reyes M, Sharma A, C. Demirci C, DeClue TJ, Lankes E, et al. Early-onset obesity: unrecognized first evidence for GNAS mutations and methylation changes. J Clin Endocrinol Metab. 2017;102:2670–7.
Chen H, Jawahar S, Qian Y, Duong Q, Chan G, Parker A, et al. Missense polymorphism in the human carboxypeptidase E gene alters enzymatic activity. Hum Mutat. 2001;18:120–31.
Pearce LR, Atanassova N, Banton MC, Bottomley B, van der Klaauw AA, Revelli JP, et al. KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell. 2013;155:765–77.
Bouchard C. Genetics of obesity: What we have learned over decades of research. Obesity (Silver Spring). 2021;29:802–20.
Cooiman MI, Alsters SIM, Duquesnoy M, Hazebroek EJ, Meijers-Heijboer HJ, Chahal H, et al. Long-term weight outcome after bariatric surgery in patients with melanocortin-4 receptor gene variants: a case-control study of 105 patients. Obes Surg. 2022;32:837–44.
Ayers KL, Glicksberg BS, Garfield AS, Longerich S, White JA, Yang P, et al. Melanocortin 4 receptor pathway dysfunction in obesity: atient stratification aimed at MC4R agonist treatment. J Clin Endocrinol Metab. 2018;103:2601–12.
Acknowledgements
EED is funded by grants from the National Institute of Health R01 HD042601 and R01 DK072301. We would like to thank Anne M. McRae, MMS, CGC, PA-C for sharing considerable time in reviewing and editing this manuscript.
Author information
Authors and Affiliations
Contributions
KR and HB conceptualized the project, conducted data collection and analysis, and wrote the initial manuscript draft. All authors (AA, KS, MQ, CM, SN, EED) were involved in the writing, review and editing of the manuscript and had final approval of the submitted and published versions. Specifically, KR: Conceptualization, methodology, validation, investigation, writing original draft, project administration, AA: writing-review & editing, KS: investigation, writing-review & editing, MQ: investigation, writing-review & editing, CM: writing-review & editing, SN: writing-review & editing, EED: writing review & editing, HB: Conceptualization, methodology, validation, investigation, formal analysis, writing-original draft, supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest and have not received any compensation from PreventionGenetics or Rhythm Pharmaceuticals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Roberts, K.J., Ariza, A.J., Selvaraj, K. et al. Testing for rare genetic causes of obesity: findings and experiences from a pediatric weight management program. Int J Obes 46, 1493–1501 (2022). https://doi.org/10.1038/s41366-022-01139-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-022-01139-7
