
Abstract
Overweight and obesity are major risk factors for diabetes, cardiovascular disease, and lung disease. These diseases are the most commonly reported health conditions that predispose individuals with SARS-CoV-2 infection to require hospitalization including intensive care unit admissions. The innate immune response is the host’s first line of defense against a human coronavirus infection. However, most coronaviruses are armed with one strategy or another to overcome host antiviral defense, and the pathogenicity of the virus is related to its capacity to suppress host immunity. The multifaceted nature of obesity including its effects on immunity can fundamentally alter the pathogenesis of acute respiratory distress syndrome and pneumonia, which are the major causes of death due to SARS-CoV-2 infection. Elevated circulating leptin concentrations are a hallmark of obesity, which is associated with a leptin-resistant state. Leptin is secreted by adipocytes in proportion to body fat and regulates appetite and metabolism through signaling in the hypothalamus. However, leptin also signals through the Jak/STAT and Akt pathways, among others, to modulate T cell number and function. Thus, leptin connects metabolism with the immune response. Therefore, it seems appropriate that its dysregulation would have serious consequences during an infection. We propose that leptin may be the link between obesity and its high prevalence as a comorbidity of the SARS-CoV-2 infection. In this article, we present a synthesis of the mechanisms underpinning susceptibility to respiratory viral infections and the contribution of the immunomodulatory effects of obesity to the outcome.
Similar content being viewed by others
Introduction
Worldwide obesity has tripled in <50 years. In 2016, more than 1.9 billion adults were overweight or obese [1]. Obesity reduces lifespan by 9–13 years, and in the United States (US) alone, more than 300,000 deaths annually are linked to obesity [2,3,4,5]. Excess body weight increases the risk of cardiovascular disease, type 2 diabetes, hypertension, dyslipidemia, obstructive sleep apnea, and some cancers [2,3,4]. Innovative medicine and public health measures reduced mortality from the pandemics of infection and increased lifespan, setting the stage for gradual displacement by epidemics of obesity and chronic disease [6, 7]. While this is true for the most part, it lulled us into a false sense of security that infectious diseases are no longer the threat to life. The 60.8 million cases and 12,469 deaths from the 2009 influenza (H1N1) pdm09 virus in the US alone [8], notwithstanding, our preparedness for a novel infectious disease outbreak was clearly abysmal.
SARS-CoV-2 is a novel coronavirus (CoV) that originated in China toward the end of 2019. The virus has wreaked havoc worldwide, especially in the US and Europe. Reports from China and Italy show that advancing age and the presence of one or more underlying health conditions are risk factors for increased severity of the disease [9]. In the US, the most commonly reported health conditions that predisposed infected individuals to require hospitalization including intensive care unit admissions were diabetes, chronic lung disease, and cardiovascular disease [9]. However, 89% of US adults who have diabetes are also overweight or have obesity [10]. The prevalence of cardiovascular disease in the US is 56.9%. Overweight and obesity that account for 69% of the US population are major risk factors for cardiovascular disease [11]. Chronic lung disease affects 9.1% of the US population, and obesity is a risk factor for the development of a number of respiratory diseases including pneumonia and acute respiratory distress syndrome (ARDS) [12].
Following the H1N1 pandemic in 2009, obesity was recognized for the first time as an independent risk factor for increased disease severity and death due to influenza [13, 14]. In California, 51% of the 534 cases of H1N1 in adults included individuals with obesity, and 61% of the mortality occurred in these individuals [15]. In an analysis of 5700 patients from among the confirmed SARS-CoV-2 cases admitted to hospitals belonging to Northwell Health (the largest academic health system in New York), between March 1, 2020 and April 4, 2020, hypertension (56.6%), obesity (41.7%), and diabetes (33.8%) were the most common comorbidities [16].
Obesity appears to predispose patients with SARS-CoV-2 infection to increased severity of the disease [17]. At a single center in France, 47.6% of patients admitted to the intensive care unit (ICU) had a body mass index (BMI) > 30 kg/m2 and 28.2% had a BMI > 35 kg/m2 [18]. Reports from ICUs in two hospitals in Vitoria (Spain) identified obesity as the most common comorbidity, accounting for 48% of the admissions due to SARS-CoV-2 [19]. In patients with SARS-CoV-2 admitted to the ICU of six University hospitals in New York, there was a significant inverse relationship between BMI and age. Advancing age increases the risk for severe illness from SARS-CoV-2-, but in younger patients, those with severe forms of the infection were more likely to have obesity [20].
The multifaceted nature of obesity including its effects on immunity can fundamentally alter the pathogenesis of ARDS and pneumonia, which are the major causes of death due to SARS-CoV-2 infection [21]. The inevitable question that arises is: How do the metabolic alterations of obesity affect the immune response to SARS-CoV-2? In this article, we present a synthesis of the mechanisms underpinning susceptibility to respiratory viral infections and the contribution of the immunomodulatory effects of obesity to the outcome.
Detection of coronavirus by host
In humans, angiotensin-converting enzyme 2 (ACE2) is the receptor for SARS-CoV-2. The viral spike protein (S) binds to the receptor prior to activation and initial entry into primary target cells. The type II membrane serine protease (TMPRSS2) is the main host protease that mediates viral entry and activation [22]. Evolving literature suggests that in SARS-CoV-2, there are other proteases that could mediate activation of the S protein [23, 24]. The innate immune response is the host’s first line of defense against a human CoV infection. Foreign patterns of invading microbes that are distinct from the host are detected by pattern recognition receptors such as Toll-like receptors and retinoic acid-inducible gene 1-like receptors. These pathogen-associated molecular patterns (PAMP) are usually biomolecules derived from the surface of the microbe or generated during its life cycle [25]. Upon recognition by PAMPs, inflammatory cytokines and antiviral interferons (IFNs) are produced. The three distinct types of IFNs include: type I IFNs (IFN-α and IFN-β), type II IFNs (IFN-γ), and type III IFNs (IFN-λ) [26]. Mounting evidence suggests that each type of IFN system has a functionally non-redundant role in the host defense or immunopathology [27,28,29].
The IFN-mediated signaling and transcriptional activation of cellular gene expression occurs when IFNs bind to their cell surface receptors thus activating the Jak-STAT pathway proteins. Upon stimulation, the signal transducer and activator of transcription (STAT) family of proteins, which are latent cytoplasmic transcription factors, are tyrosine phosphorylated by the Janus family of tyrosine kinase (Jak) enzymes to precipitate gene transcription [30]. The STAT-1 transcription factor was found to have a particularly important role in mediating the IFN-dependent biological responses including induction of the antiviral effects in the SARS-CoV pathogenesis [25]. Evidence from the activity of viral proteins encoded by CoV 229E suggested that type 1 interferon is a key component of the host antiviral defense [31]. However, gene enrichment analyses of differentially expressed transcripts from cell culture studies of SARS-CoV-2 infection conducted in human airway epithelial cells illustrate a diminished IFN-I and IFN-III expression and signaling biology. These results were corroborated in an in vivo longitudinal study in ferrets infected with SARS-CoV-2, and post mortem lung samples as well as serum from SARS-CoV-2 positive patients. Nevertheless, SARS-CoV-2 infection elicits a transcriptional response characterized by a unique gene signature enriched for cell death and leukocyte activation. Thus, the reduced IFN-1 and IFN-III response to SARS-CoV-2 notwithstanding, there is a robust chemotactic and inflammatory response [32].
Evasion of host antiviral response by CoVs
By impairing the activity of the Jak-STAT signaling pathway, CoVs can antagonize the IFN-mediated antiviral response. Among the multiple pathways that appear to be involved, mimicry is common, whereby viruses encode products that mimic cellular components of the IFN signal transduction pathway. These countermeasures to suppress host interferon production impair the development of an antiviral state [30]. In addition, CoVs can suppress or induce cell death and cytopathy. Viral gene products that function as suppressors of apoptosis extend the time following infection to produce new virions. Those that function as inducers facilitate the release and dissemination of progeny virions from the infected host [30].
Pyroptosis is a form of cell death mediated by CoVs that results from an exuberant proinflammatory cytokine release [31]. Mortality in patients with SARS-CoV is associated with high levels of proinflammatory cytokines in the lower respiratory tract [33]. The NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome is an oligomeric complex that is a critical component of the host antiviral response. This complex promotes interleukin 1β (IL-1β), secretion and induces pyroptosis. PAMPS generated during viral replication trigger the NLRP3 inflammasome-dependent antiviral immune response that targets viral replication. Aberrant NLRP3 inflammasome activation or chronic inflammation can lead to severe pathological injury. Viruses have evolved elaborate strategies to either evade NLRP3 activation, which would facilitate viral replication, or promote its activation to initiate a highly pathological inflammatory response. Although most CoVs are armed with one strategy or another to thwart host antiviral defense, the pathogenicity of the virus is related to its capacity to suppress host immunity [31]. In addition to pathogen prevalence and virulence, host factors are the critical elements affecting the outcome of pulmonary viral infections [34].
Growing evidence suggests that CoVs have evolved to leverage features of the human IFN pathway to promote their pathogenesis [35,36,37]. Analyses of single-cell RNA sequencing data sets of tissues that would potentially harbor SARS-CoV-2, show that in human airway epithelial cells, IFN-I, and to some extent IFN II, upregulate ACE2 expression [38]. Angiotensin-converting enzyme (ACE), a key component of the renin–angiotensin system, cleaves angiotensin I to generate angiotensin II, whereas ACE2 inactivates angiotensin II and is a negative regulator of the system. The actions of angiotensin II drive acute lung injury through various mechanisms, including increased vascular permeability [39]. In an influenza infection, angiotensin II levels in humans and mice rise and ACE2 provides protection to tissues by reducing the amounts of angiotensin II [40]. Binding of SARS-CoV-S to mouse ACE2 in vivo reduced ACE2 expression leading to acute acid-aspiration-induced lung failure [41]. Therefore, ACE2 is critical for early tissue tolerance responses to respiratory infection [38]. When low levels of IFN-I are produced in response to SARS-CoV-2 infection, the protective effect of ACE2 is diminished. The net effect of diminished IFN-I expression may be protective or detrimental depending upon the stage of the infection, viral clade, cell subsets affected, and other factors such as age, gender, and comorbidities to name a few [38, 42,43,44].
Leptin and immune cell function
Leptin is an important mediator of pulmonary immunity and chronically elevated circulating leptin concentrations impair host pulmonary defenses [34, 45]. Leptin is produced by adipocytes and regulates satiety by binding to receptors in the hypothalamus. Leptin is also secreted by bronchial epithelial cells, type II pneumocytes, and lung macrophages [46]. Through receptors on leukocytes, leptin signals through many pathways including the Jak/STAT pathway to mediate immune cell number and function [47,48,49,50]. During an infection, T-cell activation is accompanied by high energy requirements to support biosynthesis of intracellular components [47]. Leptin is released by adipocytes in proportion to fat mass and is especially important for activated T-cells to upregulate glucose metabolism to meet the demands of the cell [51]. While early studies show that starvation and leptin deficiency are associated with decreased immune reactivity [52,53,54], hyperleptinemia has also been shown to have detrimental effects on the immune response [55, 56]. These studies clearly demonstrate that leptin is a key link connecting nutritional status and immune responses.
Individuals with leptin deficiency have reduced T-cell numbers, decreased CD4 + helper T-cells (Th, contributor to cellular, humoral, and mucosal immunity), increased proliferation of regulatory T-cells (Treg, suppressor of effector T-cells activation and excessive inflammatory responses) and aberrant cytokine production [47,48,49,50]. In starvation, falling leptin levels act as a peripheral signal for conservation of energy as reserves start to deplete. Energy for vital functions such as central nervous system metabolism take precedence and systems that are not of immediate necessity such as reproduction or a finely tuned cognate immune response are inhibited. An adaptive neuroendocrine response featuring activation of the hypothalamic–pituitary–adrenal axis and suppression of the thyroid and gonadal axes accompanies the nutritional deprivation of starvation [52, 57]. Leptin blunts these adaptive changes [58]. Furthermore, leptin biases the induction of Th cells toward the Th1 subset, which has a more proinflammatory response than the Th2 subset that has predominantly regulatory functions [52].
The interactions between the immune system and integrative neuroendocrine mechanisms affect host homeostasis. The metabolic derangements that occur when the immune system is activated during inflammatory and infective processes are well recognized [57]. Leptin regulates neuroendocrine and immune function, but this regulation occurs below a critical threshold leptin level reminiscent of chronic nutrient deprivation [53]. Thus, a severe energy deficit and leptin deficiency increase susceptibility to infectious diseases. The immune system can withstand acute starvation whereas chronic starvation alters the immune response and renders humans and mice susceptible to infections [59].
Leptin has also been shown to inhibit the apoptosis of cells of the immune system, and deficits in circulating leptin levels contribute to the defective immune response, in energy and nutrient deficiencies. Leptin administration inhibits baseline thymic apoptosis in young rats by 15–30%, acting through the insulin receptor substrate-1 (IRS-1) and AKT signaling pathways. Interestingly, the maturation process of young rats is accompanied by reduction in leptin receptor expression [54].
Leptin has an important role in the metabolic regulation of Treg cells. These cells produce high amounts of both leptin and its receptor (LepR), which are the stimulus for an autocrine inhibitory loop that constrains the proliferation of Treg cells. Therefore high leptin levels can promote hyporesponsiveness of Treg cells [60]. Similarly, leptin produced by Treg cells contributes to activation of the mammalian target of rapamycin (mTOR)-pathway in Treg cells. Overexpression of the leptin-mTOR pathway in freshly isolated human Treg cells leads to hyporesponsiveness and constrained proliferation [61]. As the activation of mTOR supports T-cell growth and function, the negative regulation seems counterintuitive. Therefore, there must be a dose and timing by which the intracellular metabolic state controls Treg cell responsiveness through the leptin-mTOR axis [61].
The relevance of the appropriate dose of leptin is also manifested in the regulation of suppressor of cytokine signaling 3 (SOCS3). The expression of SOCS3 that is a key negative regulator of cytokine signaling is induced by leptin signaling through the Jak/STAT pathway [62]. In human Treg cells, SOCS3 was found to be highly expressed in basal conditions, and acute stimulation promoted its increase consistent with the hyporesponsive state of the Treg cells. Leptin neutralization rescued Treg cells from their hyporesponsiveness [60]. Thus, high circulating leptin would likely have a detrimental effect on intracellular signaling and the response to an infection.
In a mouse model of diet-induced obesity, hyperleptinemia was associated with increased mortality, viral spread, and lung levels of proinflammatory cytokines including interleukin 6 (IL-6) and IL-1β, following infection with influenza (H1N1) pdm09 virus. Administration of anti-leptin antibody led to a decrease in the proinflammatory response and improved lung pathology and survival rate [55]. In individuals with obesity, SOCS3 basal mRNA expression in peripheral blood mononuclear cells increases in response to Toll-like-receptor ligand stimulation and is associated with a diminished IFN-I response compared with individuals without obesity [56]. Excess leptin secretion from adipocytes could have paracrine effects on T-cells and promote the development of systemic inflammation [47].
Obesity arises from a chronic energy imbalance and one of its characteristic features is a state of leptin resistance marked by persistent hyperleptinemia. The effects of leptin on metabolic complications in obesity are numerous [63,64,65,66,67]. Similarly, elevated leptin levels are present in diabetes [68, 69]. In patients with diabetes and ARDS, elevated leptin levels in bronchoalveolar fluid are associated with increased mortality [70].
Insulin receptor signaling
Upon activation T-cells use glycolysis in the presence of oxygen over more energy-efficient oxidative phosphorylation to produce the biosynthetic precursors required for rapid cell growth and proliferation. Activation of the phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR signaling pathway drives glucose metabolism and aerobic glycolysis. If glucose metabolism is insufficient, T-cells take on a state of hyporesponsiveness, which is known as anergy, or a state of non-responsivenes, which is termed exhaustion [47]. Although activation of the PI3K/Akt/mTOR pathway in T-cells primarily occurs through triggering of the T-cell receptor and CD28 co-stimulation [71], the insulin receptor has also been shown to have a role. Signaling downstream of the insulin receptor including the PI3K/Akt/mTOR signaling pathway is reduced in CD4+ T-cells from insulin receptor knockdown transgenic rats [72]. Type 2 diabetes and prediabetes are insulin resistant conditions that commonly co-exist with obesity [10]. In these subjects, impaired insulin receptor signaling may contribute to insufficient energy supply for effector T-cells to mount an effective response to infection.
Adipose tissue inflammation
Adipose tissue inflammation is a hallmark of obesity and a critical event leading to the development of metabolic syndrome, diabetes, and atherosclerotic cardiovascular disease [73]. The adaptive immune system has a key role in the initiation and maintenance of adipose tissue inflammation. Macrophage accumulation in adipose tissue provides a mechanism for adipocyte production of the proinflammatory cytokines thought to be related to many of the metabolic consequences of obesity [74]. However, CD8+ T-cell infiltration precedes macrophage accumulation and is essential for macrophage differentiation, activation, and migration. Adipose tissue contains the necessary stimuli for activating CD8+ T-cells to produce a local inflammatory cascade, without the need for a systemic increase in T-cells. Reduced numbers of CD4+ T-cells, which balance the immune response, and Treg cells known to have mitigating effects on the adaptive immune responses, can compound adipose tissue inflammatory processes in the development of obesity [75].
In humans with obesity, adipose tissue depots contain ~40% macrophages, which are important in immune function [74]. They are primary mediators of the innate immune response and have an important role in the adaptive immune response. Therefore, one might expect that individuals with obesity would have increased mortality when afflicted with ARDS, which is at least in part driven by a proinflammatory cytokine milieu. Reports that obesity is not associated with mortality in mechanically ventilated patients with acute lung injury, have given rise to the obesity paradox in ARDS [76, 77].
Obesity and mortality in SARS-CoV-2
The high mortality among patients with obesity who manifest SARS-CoV-2 has prompted the notion that SARS-CoV-2 has disproved the obesity paradox in ARDS [78]. However, based on evidence from patients with obesity and pneumonia, and rodent models of hyperleptinemia with and without obesity, high baseline levels of leptin drive immune defects. Impairments in neutrophil response, and an insufficient antiviral response predispose to increased susceptibility to, and severity of, respiratory infections [34, 45]. In non-obese patients with diabetes and ARDS, elevated leptin levels and diminished leptin receptor signaling were associated with poor clinical outcomes [70]. Elevated circulating plasma leptin levels in obesity are associated with reductions in levels of key biomarkers of inflammation and an attenuated inflammatory response in ARDS and pneumonia, which exacerbates the outcomes of pulmonary infections [34]. During an infection, T-cell activation is accompanied by high energy requirements to support biosynthesis of intracellular components [47]. From an evolutionary perspective, downregulation of nonessential and energy-consuming pathways such as immune cell activation is pragmatic [47]. As leptin is a link between metabolism and the immune response, it seems appropriate that its dysregulation would have serious consequences during an infection.
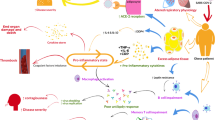
In SARS-CoV-2 infections, lymphopenia appears to be a consistent finding, and occurs in approximately 80% of patients. Patients who have succumbed to the infection exhibit a marked decline in the levels of circulating CD4+ and CD8+ T lymphocytes, and in lung tissue mononuclear cells such as macrophages predominate [79,80,81]. Lymphopenia and the predominance of innate immune macrophages are also a feature of the SARS-CoV infection, and this maybe a strategy adopted by the CoV to suppress host antiviral response [82]. Immune insufficiency or misdirection may increase viral replication or render its clearance ineffective, which can cause tissue damage, stimulation of further macrophage activation, and an uncontrolled loop of self-amplification. The resulting cytokine-storm syndrome can precipitate multi-organ failure [83]. By altering the metabolic environment, obesity and its attendant conditions of hyperleptinemia and insulin resistance disrupt T-cell function resulting in a suppressed T-cell response to infection [34, 84]. A schematic summary of the immune system activation and response to a viral infection is presented in Fig. 1.

Virion particles are illustrated by open pentagons. The three distinct types of IFNs include: type I IFNs (IFN-α and IFN-β), type II IFNs (IFN-γ), and type III IFNs (IFN-λ), All IFN types bind to distinct receptors but activate similar signaling pathways and transcriptional responses. The type I, type II, and type III IFN receptors are heterodimers composed of IFNAR1 and IFNAR2 subunits, IFNGR1 and IFNGR2 subunits, and IFNLR1 and IL10Rβ subunits, respectively. Viral-encoded products antagonize the IFN-signaling pathways and the biochemical activity of IFN-induced cellular proteins to thwart host antiviral defense. In activated lymphocytes, lipid oxidation is downregulated and glycolysis increases in the presence of oxygen, together with glutamine oxidation in order to produce the biosynthetic precursors required for rapid cell growth and proliferation. Leptin signals via the Jak/STAT and Akt pathways among others to mediate immune cell number and function. Leptin dysregulation in obesity has detrimental effects during an infection. LepR: leptin receptor.
Another feature of ARDS associated with SARS-CoV-2 is that it presents in an atypical form of preserved lung mechanics and severe hypoxemia [85]. In patients with obesity, the accumulation of fat in the mediastinum and in the abdominal and thoracic cavities decreases functional residual capacity by altering the mechanical properties of the chest wall. The diaphragm is elevated and its downward pressure is limited, which causes pleural pressure to increase [21]. In supine patients with abdominal obesity, diaphragmatic excursion decreases making ventilation difficult [86]. Prone positioning is recommended to improve oxygenation in patients with refractory hypoxemia due to SARS-CoV-2 ARDS [87], but the physical challenges and risks involved may be exacerbated in obesity.
Conclusions
The interplay between SARS-CoV-2 and host antiviral defense determines the course and pathogenesis of the viral infection. Obesity and elevated circulating leptin predispose patients to morbidity and mortality from the SARS-CoV-2 infection through a compromised immune response. This diminished immune response that fails to contain viral replication coupled with high levels of chemokines may explain why SARS-CoV-2 has taken its toll on individuals with obesity. Furthermore, obesity renders mechanical ventilation difficult and the mechanical effects of lung compression in obesity contribute to the respiratory symptoms [88].
Host antiviral response lies at the crux of the control of SARS-CoV-2 infection at both the cellular and organismal level. A weakened immune response can spark a series of events culminating in immunopathogenesis and a heightened cytokine release that can prove fatal. The high mortality rates from SARS-CoV-2 infection in individuals with obesity suggests that the metabolic consequences of obesity compromise host antiviral defenses. Elevated circulating leptin is a common feature of obesity and the mediating factor linking metabolism to immunity. Therefore, the role of leptin in the pathogenesis of SARS-CoV-2 bears investigation along with the innate immunomodulatory effects of viral proteins encoded by SARS-CoV-2 in patients with obesity.
The high pathogenicity of the virus in obesity could be the outcome of unique virus countermeasures to suppress host interferon production that interact with the metabolic milieu of obesity. Whether a small molecule NLRP3 inhibitor could constitute an approach to treat SARS-CoV-2 particularly in the state of chronic inflammation as exists in obesity warrants further investigation. How the proinflammatory state of adipose tissue in obesity might contribute to a low grade protracted activation of T-cells and their premature senescence marked by anergy and exhaustion also bears investigation. Importantly, in the development of vaccines, the immunocompromised state predicated by obesity may need to be considered in much the same way as advancing age.
References
WHO. Obesity and Overweight. 2018. https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. Accessed 5 April 2020.
Prospective Studies C, Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, et al. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373:1083–96.
Masters RK, Powers DA, Link BG. Obesity and US mortality risk over the adult life course. Am J Epidemiol. 2013;177:431–42.
Global BMIMC, Di Angelantonio E, Bhupathiraju Sh N, Wormser D, Gao P, Kaptoge S, et al. Body-mass index and all-cause mortality: individual-participant-data meta-analysis of 239 prospective studies in four continents. Lancet. 2016;388:776–86.
Pi-Sunyer X. The medical risks of obesity. Postgrad Med. 2009;121:21–33.
Omran AR. The epidemiologic transition: a theory of the epidemiology of population change. Milbank Mem Fund Q. 1971;49:509–38.
Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. N Engl J Med. 2017;376:1492.
CDC. 2009 H1N1 Pandemic (H1N1pdm09 virus). 2020. https://www.cdc.gov/flu/pandemic-resources/2009-h1n1-pandemic.html. Accessed 10 April 2020.
CDC. Preliminary Estimates of the Prevalence of Selected Underlying Health Conditions Among Patients with Coronavirus Disease 2019—United States, February 12–March 28, 2020. 2020. https://www.cdc.gov/mmwr/volumes/69/wr/mm6913e2.htm. Accessed 10 April 2020.
CDC. National Diabetes Statistics Report. 2020. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf. Accessed 6 April 2020.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics—2020 update: a report from the American Heart Association. Circulation. 2020;141:e139–e596.
American Lung Association. Estimated Prevalence and Incidence of Lung Disease. 2020. https://www.lung.org/research/trends-in-lung-disease/estimated-prevalence-and-incidence-of-lung-dis-(1)/methodology. Accessed 10 April 2020.
Milner JJ, Beck MA. The impact of obesity on the immune response to infection. Proc Nutr Soc. 2012;71:298–306.
Huttunen R, Syrjanen J. Obesity and the outcome of infection. Lancet Infect Dis. 2010;10:442–3.
Louie JK, Acosta M, Samuel MC, Schechter R, Vugia DJ, Harriman K, et al. A novel risk factor for a novel virus: obesity and 2009 pandemic influenza A (H1N1). Clin Infect Dis. 2011;52:301–12.
Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323:2052–2059.
Sattar N, McInnes IB, McMurray JJV. Obesity a risk factor for severe COVID-19 infection: multiple potential mechanisms. Circulation. 2020;142:4–6.
Simonnet A, Chetboun M, Poissy J, Raverdy V, Noulette J, Duhamel A. et al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity. 2020;28:1195–1199.
Barrasa H, Rello J, Tejada S, Martin A, Balziskueta G, Vinuesa C et al. SARS-Cov-2 in Spanish intensive care: early experience with 15-day survival in Vitoria. Anaesth Crit Care Pain Med. 2020. https://doi.org/10.1016/j.accpm.2020.04.001.
Kass DA, Duggal P, Cingolani O. Obesity could shift severe COVID-19 disease to younger ages. Lancet. 2020;395:1544–5.
Peters U, Suratt BT, Bates JHT, Dixon AE. Beyond BMI: obesity and lung disease. Chest. 2018;153:702–9.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80.e8.
Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562–9.
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742.
Wong LY, Lui PY, Jin DY. A molecular arms race between host innate antiviral response and emerging human coronaviruses. Virol Sin. 2016;31:12–23.
Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity. 2019;50:907–23.
Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14:315–28.
Broggi A, Granucci F, Zanoni I. Type III interferons: balancing tissue tolerance and resistance to pathogen invasion. J Exp Med. 2020;217:e20190295.
Jewell NA, Cline T, Mertz SE, Smirnov SV, Flano E, Schindler C, et al. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol. 2010;84:11515–22.
Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809.
Fung SY, Yuen KS, Ye ZW, Chan CP, Jin DY. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect. 2020;9:558–70.
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced Host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–45.e9.
He L, Ding Y, Zhang Q, Che X, He Y, Shen H, et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J Pathol. 2006;210:288–97.
Ubags ND, Stapleton RD, Vernooy JH, Burg E, Bement J, Hayes CM et al. Hyperleptinemia is associated with impaired pulmonary host defense. JCI Insight. 2016;1:e82101.
Fung TS, Liu DX. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol. 2019;73:529–57.
Mar KB, Rinkenberger NR, Boys IN, Eitson JL, McDougal MB, Richardson RB, et al. LY6E mediates an evolutionarily conserved enhancement of virus infection by targeting a late entry step. Nat Commun. 2018;9:3603.
Zhao X, Guo F, Liu F, Cuconati A, Chang J, Block TM, et al. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci USA. 2014;111:6756–61.
Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181:1016–35.e19.
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–6.
Zou Z, Yan Y, Shu Y, Gao R, Sun Y, Li X, et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat Commun. 2014;5:3594.
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9.
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19:181–93.
Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39:529–39.
Davidson S, Maini MK, Wack A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J Interferon Cytokine Res. 2015;35:252–64.
Kordonowy LL, Burg E, Lenox CC, Gauthier LM, Petty JM, Antkowiak M, et al. Obesity is associated with neutrophil dysfunction and attenuation of murine acute lung injury. Am J Respir Cell Mol Biol. 2012;47:120–7.
Mancuso P. Obesity and lung inflammation. J Appl Physiol. 2010;108:722–8.
MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–83.
Martin-Romero C, Sanchez-Margalet V. Human leptin activates PI3K and MAPK pathways in human peripheral blood mononuclear cells: possible role of Sam68. Cell Immunol. 2001;212:83–91.
Sanchez-Margalet V, Martin-Romero C. Human leptin signaling in human peripheral blood mononuclear cells: activation of the JAK-STAT pathway. Cell Immunol. 2001;211:30–6.
Matarese G, Moschos S, Mantzoros CS. Leptin in immunology. J Immunol. 2005;174:3137–42.
Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, Maciver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol. 2014;192:136–44.
Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901.
Chan JL, Matarese G, Shetty GK, Raciti P, Kelesidis I, Aufiero D, et al. Differential regulation of metabolic, neuroendocrine, and immune function by leptin in humans. Proc Natl Acad Sci USA. 2006;103:8481–6.
Mansour E, Pereira FG, Araujo EP, Amaral ME, Morari J, Ferraroni NR, et al. Leptin inhibits apoptosis in thymus through a janus kinase-2-independent, insulin receptor substrate-1/phosphatidylinositol-3 kinase-dependent pathway. Endocrinology. 2006;147:5470–9.
Zhang AJ, To KK, Li C, Lau CC, Poon VK, Chan CC, et al. Leptin mediates the pathogenesis of severe 2009 pandemic influenza A(H1N1) infection associated with cytokine dysregulation in mice with diet-induced obesity. J Infect Dis. 2013;207:1270–80.
Teran-Cabanillas E, Montalvo-Corral M, Caire-Juvera G, Moya-Camarena SY, Hernandez J. Decreased interferon-alpha and interferon-beta production in obesity and expression of suppressor of cytokine signaling. Nutrition. 2013;29:207–12.
Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev. 1996;17:64–102.
Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–2.
Matarese G, La Cava A, Sanna V, Lord GM, Lechler RI, Fontana S, et al. Balancing susceptibility to infection and autoimmunity: a role for leptin? Trends Immunol. 2002;23:182–7.
De Rosa V, Procaccini C, Cali G, Pirozzi G, Fontana S, Zappacosta S, et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity. 2007;26:241–55.
Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–41.
La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4:371–9.
Liu AG, Smith SR, Fujioka K, Greenway FL. The effect of leptin, caffeine/ephedrine, and their combination upon visceral fat mass and weight loss. Obesity. 2013;21:1991–6.
Galgani JE, Greenway FL, Caglayan S, Wong ML, Licinio J, Ravussin E. Leptin replacement prevents weight loss-induced metabolic adaptation in congenital leptin-deficient patients. J Clin Endocrinol Metab. 2010;95:851–5.
Simonds SE, Pryor JT, Ravussin E, Greenway FL, Dileone R, Allen AM, et al. Leptin mediates the increase in blood pressure associated with obesity. Cell. 2014;159:1404–16.
Kelly KR, Williamson DL, Fealy CE, Kriz DA, Krishnan RK, Huang H, et al. Acute altitude-induced hypoxia suppresses plasma glucose and leptin in healthy humans. Metabolism. 2010;59:200–5.
Barkoukis H, Marchetti CM, Nolan B, Sistrun SN, Krishnan RK, Kirwan JP. A high glycemic meal suppresses the postprandial leptin response in normal healthy adults. Ann Nutr Metab. 2007;51:512–8.
Friedman JM. Modern science versus the stigma of obesity. Nat Med. 2004;10:563–9.
Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–61.
Jain M, Budinger GR, Lo A, Urich D, Rivera SE, Ghosh AK, et al. Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-gamma. Am J Respir Crit Care Med. 2011;183:1490–8.
Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172:4661–5.
Fischer HJ, Sie C, Schumann E, Witte AK, Dressel R, van den Brandt J, et al. The insulin receptor plays a critical role in T cell function and adaptive immunity. J Immunol. 2017;198:1910–20.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808.
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20.
O’Brien JM Jr., Welsh CH, Fish RH, Ancukiewicz M, Kramer AM, National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome Network. Excess body weight is not independently associated with outcome in mechanically ventilated patients with acute lung injury. Ann Intern Med. 2004;140:338–45.
Anzueto A, Frutos-Vivar F, Esteban A, Bensalami N, Marks D, Raymondos K, et al. Influence of body mass index on outcome of the mechanically ventilated patients. Thorax. 2011;66:66–73.
Jose RJ, Manuel A. Does COVID-19 disprove the obesity paradox in ARDS? Obesity. 2020;6:1007.
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130:2620–9.
Wang F, Nie J, Wang H, Zhao Q, Xiong Y, Deng L, et al. Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J Infect Dis. 2020;221:1762–9.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62.
Chen J, Lau YF, Lamirande EW, Paddock CD, Bartlett JH, Zaki SR, et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J Virol. 2010;84:1289–301.
Liu PP, Blet A, Smyth D, Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation. 2020. https://doi.org/10.1161/CIRCULATIONAHA.120.047549.
Green WD, Beck MA. Obesity impairs the adaptive immune response to influenza virus. Ann Am Thorac Soc. 2017;14(Supplement_5):S406–S409.
Gattinoni L, Coppola S, Cressone M, Busana M, Rossi S, Chiumello D. Covid-19 does not lead to “typical” acute respiratory distress syndrome. Am J Respir Crit Care Med. 2020;201:1299–1300.
Dietz W, Santos-Burgoa C. Obesity and its implications for COVID-19 mortality. Obesity. 2020;28:1005.
Wilson KC, Chotirmall SH, Chunxue B, Rello J. Covid-19. Interim Guidance on Management Pending Empirical Evidence from an American Thoracic Society-Led International Task Force. 2020. https://www.thoracic.org/covid/covid-19-guidance.pdf. Accessed 20 April 2020.
Sharp JT, Henry JP, Sweany SK, Meadows WR, Pietras RJ. The total work of breathing in normal and obese men. J Clin Invest. 1964;43:728–39.
Acknowledgements
This study was supported in part by U54 GM104940 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH), which funds the Louisiana Clinical and Translational Science Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rebello, C.J., Kirwan, J.P. & Greenway, F.L. Obesity, the most common comorbidity in SARS-CoV-2: is leptin the link?. Int J Obes 44, 1810–1817 (2020). https://doi.org/10.1038/s41366-020-0640-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-020-0640-5
This article is cited by
-
Calorie restriction-induced leptin reduction and T-lymphocyte activation in blood and adipose tissue in men with overweight and obesity
International Journal of Obesity (2024)
-
Highlighting the idea of exerkines in the management of cancer patients with cachexia: novel insights and a critical review
BMC Cancer (2023)
-
Relationship between leptin and white blood cells: a potential role in infection susceptibility and severity—the Olivetti Heart Study
Internal and Emergency Medicine (2023)
-
Resident and migratory adipose immune cells control systemic metabolism and thermogenesis
Cellular & Molecular Immunology (2022)
-
Mechanisms contributing to adverse outcomes of COVID-19 in obesity
Molecular and Cellular Biochemistry (2022)
