
Abstract
Background
The early life environment may influence susceptibility to obesity and metabolic disease in later life through epigenetic processes. SLC6A4 is an important mediator of serotonin bioavailability, and has a key role in energy balance. We tested the hypothesis that methylation of the SLC6A4 gene predicts adiposity across the life course.
Methods
DNA methylation at 5 CpGs within the SLC6A4 gene identified from a previous methyl binding domain array was measured by pyrosequencing. We measured DNA methylation in umbilical cord (UC) from children in the Southampton Women’s Survey cohort (n = 680), in peripheral blood from adolescents in the Western Australian Pregnancy Cohort Study (n = 812), and in adipose tissue from lean and obese adults from the UK BIOCLAIMS cohort (n = 81). Real-time PCR was performed to assess whether there were corresponding alterations in gene expression in the adipose tissue.
Results
Lower UC methylation of CpG5 was associated with higher total fat mass at 4 years (p = 0.031), total fat mass at 6–7 years (p = 0.0001) and % fat mass at 6–7 years (p = 0.004). Lower UC methylation of CpG5 was also associated with higher triceps skinfold thickness at birth (p = 0.013), 6 months (p = 0.038), 12 months (p = 0.062), 2 years (p = 0.0003), 3 years (p = 0.00004) and 6–7 years (p = 0.013). Higher maternal pregnancy weight gain (p = 0.046) and lower parity (p = 0.029) were both associated with lower SLC6A4 CpG5 methylation. In adolescents, lower methylation of CpG5 in peripheral blood was associated with greater concurrent measures of adiposity including BMI (p ≤ 0.001), waist circumference (p = 0.011), subcutaneous fat (p ≤ 0.001) and subscapular, abdominal and suprailiac skinfold thicknesses (p = 0.002, p = 0.008, p = 0.004, respectively). In adipose tissue, methylation of both SLC6A4 CpG5 (p = 0.019) and expression of SLC6A4 (p = 0.008) was lower in obese compared with lean adults.
Conclusions
These data suggest that altered methylation of CpG loci within SLC6A4 may provide a robust marker of adiposity across the life course.
Similar content being viewed by others
Introduction
Obesity is a disorder of energy balance resulting from a combination of genetic, lifestyle and environmental factors, and is a major risk factor for non-communicable diseases including type-2 diabetes and cardiovascular disease. Polymorphisms in several genes have been linked to obesity [1,2,3], but to date genetic variation only accounts for a modest proportion (<10%) of obesity risk at the population level [4, 5]. Epidemiological and experimental studies suggest that susceptibility to adiposity and metabolic disease in later life is influenced by the early life environment [6]. In animal studies, variation in maternal body composition and diet leads to alterations in metabolism and body composition in the offspring [7], while in humans maternal obesity and excess gestational weight gain independently increase adiposity and risk of obesity in the infant, child and adult [8]. The mechanisms underlying these relationships are poorly understood, but modulation of the epigenome, in particular DNA methylation, is thought to be a key mechanism.
Epigenetic processes, which include DNA methylation, histone modifications and non-coding RNAs, induce heritable changes in gene expression without altering the gene sequence. In animal models, variations in the maternal diet induces epigenetic changes in a range of metabolic control genes, this is accompanied by changes in gene expression and metabolism in the offspring [9,10,11]. Human studies have also suggested an important role for epigenetic processes in mediating the effects of early life environment. For example, methylation of a CpG within the promoter of RXRA in umbilical cord predicted >25% of the variation in percent and absolute fat mass in children aged 9 years, with replication being found in a second independent cohort of 6 year old children [12]. In a separate study, methylation of specific CpG loci in the promoter of PGC1α at 5–7 years of age predicted adiposity year-on-year from 8 to 14 years [13]. These findings support the hypothesis that epigenetic marks induced during development make an important contribution to phenotype and suggest that detection of such marks in peripheral tissues may provide predictive markers of later phenotype.
To identify further epigenetic marks at birth associated with later adiposity, we previously carried out a discovery scan of DNA methylation in the promoters of all refseq genes in umbilical cord DNA using a methyl capture array (MBD-array). We aimed to identify methylation differences in umbilical cord DNA at birth associated with percentage fat mass in children aged 6 years. Our analyses identified 93 differentially methylated regions (DMRs) [14], including a DMR within the first intron of the SLC6A4 gene, which encodes the serotonin transporter. SLC6A4 has a major role in modulating the bioavailability of the neurotransmitter serotonin [15], which is synthesised both peripherally and in the central nervous system (CNS), and has a key role in modulating mood, anxiety, and energy homoeostasis [16, 17]. Polymorphisms within SLC6A4 have been associated with obesity in children [18] and adults [19], while differential methylation of the promoter region of SLC6A4 in peripheral blood has been associated with concurrent obesity in adults [20].
Given the central role that SLC6A4 has in energy homoeostasis, and the previous genetic and epigenetic associations with obesity, this study sought to examine the relationship between SLC6A4 DMR methylation in cord tissue at birth and adiposity in infancy and childhood in children from the Southampton Women’s Survey (SWS) cohort (n = 680). Here, we report an association between the methylation of CpG loci within the SLC6A4 gene at birth and measures of adiposity from birth through to 6 years of age. To assess whether this effect is sustained through the life course, we additionally measured SLC6A4 DMR methylation in peripheral blood mononuclear cells (PMBCs) from adolescents aged 17 years in the RAINE study and in subcutaneous adipose tissue in lean and obese adults from the BIOCLAIMS cohort. We found that SLC6A4 CpG methylation at Hg19 chr17:28561468 was associated with adiposity in adolescents (n = 812) and in adipose tissue from obese vs. lean adults (n = 81), suggesting that altered methylation of CpG loci within SLC6A4 may provide a robust marker of adiposity across the life course.
Materials and methods
Southampton Women’s Survey (SWS) Cohort
The SWS is a prospective mother-offspring cohort study that has assessed the diet, body composition, physical activity and social circumstances of non-pregnant women aged 20–34 years living in Southampton, UK. Comprehensive details of SWS have been published [21, 22]. Follow-up of the children and sample collection/analysis was carried out under Institutional Review Board approval (Southampton and South West Hampshire Research Ethics Committee) with written informed consent. Offspring adiposity was measured by dual-energy X-ray absorptiometry (DXA) at birth, 4 and 6–7 years of age and triceps skinfold thickness at birth, 6 months, 1, 2, 3 and 6–7 years of age. Table 1 shows cohort characteristics.
The West Australian Pregnancy Cohort (RAINE) Study
The RAINE Study enroled pregnant women ≤ 18 weeks gestation (1989–1991) through the antenatal clinic at King Edward Memorial Hospital and nearby private clinics in Perth, Western Australia. Detailed clinical assessments were performed at birth (n = 2868) and the children followed up at multiple time points including at 17 years of age, when a blood sample was taken, and waist/hip circumference, skinfold thickness and abdominal (subcutaneous and visceral) adipose thickness measurements made [23]. The Human Ethics Committees of King Edward Memorial Hospital and Princess Margaret Hospital approved all protocols (Table 1). Informed, written consent to participate in the study was obtained from the mother of each child at enrolment and at each subsequent follow-up.
BIOCLAIMS Cohort
The BIOCLAIMS study was a randomised controlled clinical trial of male and female volunteers aged 18–65 years recruited in Southampton UK 2012–2013 (http://bioclaims.uib.eu). The study was approved by the National Research Ethics Service Committee South Central Berkshire. Written informed consent was obtained from all subjects. The study was registered at www.isrctn.com as ISRCTN96712688. Subcutaneous adipose tissue biopsies were collected from 81 volunteers at baseline (lean n = 37, obese n = 44). Participants were grouped as lean or obese according to their BMI: Lean participants were determined as those with a BMI 18.5–25 kg/m2, whilst obese participants had a BMI 30–40 kg/m2 with waist circumference > 94 cm for men and >80 cm for women (Table 1).
DNA extraction
For SWS offspring, genomic DNA was prepared by a high-salt method from a 5 to 10 cm segment cut from the mid portion of the umbilical cord [14]. Genomic DNA was extracted from peripheral blood (RAINE cohort) using the Puregene DNA isolation kit (Qiagen, Germany) and from adipose tissue (BIOCLAIMS cohort) using the QIAamp DNA mini kit (Qiagen, Germany).
Pyrosequencing
Bisulphite conversion and pyrosequencing reactions were carried out [24] using primers listed in Table S1, which were designed across the 5 CpG sites within the SLC6A4 DMR (TSS + 1450 bp, Hg19 Chr:17(−) 28561505-28561468) identified previously [14] (Fig. 1a). Summary statistics for SLC6A4 methylation shown in Table S2. To identify potential transcription factor binding sites across the CpG sites within the SLC6A4 DMR Matinspector analysis was carried out (omictools.com/matinspector-tool).

Location of the SLC6A4 DMR. Location of the SLC6A4 DMR and known genetic variants is shown in relation to the SLC6A4 gene. a The location of the SLC6A4 DMR (GRCh37/hg19 Chr17:2856138128561680negative strand) is shown in relation to the first exon of SLC6A4. b The location of the CpG’s measured by pyrosequencing and nearby characterised genetic variants is shown. The DMR is shown in grey (reverse strand Chr17:28561381-28561680). The underlined region indicates the region sequenced by Pyrosequencing (either SNP or methylation assay)
Genotyping analysis
Genotyping PCR reactions (Fig. 1b) on SWS and BIOCIAIMS participants used 50 ng genomic DNA extracted from UC and adipose tissue, respectively (Table S1), and were analysed using PyroMark MD 1.0 software (Biotage). Genotyping of the RAINE participants used 250 ng DNA on the Illumina Human 660-W Quad Array (San Diego, CA) at the Centre for Applied Genomics (Toronto, Canada). Additional imputation using Mach 1.0.18.c [25] for pre-phasing and MiniMac [26] for imputation against reference panel 1000G Phase3 data. A total of 97,718 SNPs were removed at genotyping QC due to a call rate < 0.95%, 919 SNPs were removed due to Hardy–Weinberg equilibrium < 5.7 × 10E−7, and 35,502 SNP with a MAF < 0.05 were removed.
RT-PCR
Adipose tissue was processed using the RNeasy Lipid Tissue kit (Qiagen, Germany) according to manufacturer’s instructions. A total of 500 ng RNA was incubated with 10 µM random nonamers, 0.5 µM dNTP’s and 200 U M-MLV RT, followed by cDNA amplification using the QuantiTect SLC6A4 primers (Qiagen, Germany).
Statistical methods
Statistical analysis was carried out using Stata (Statacorp) versions 13.1/14.0/14.2. Distributions were examined prior to analysis, and post analysis diagnostic checks were performed where necessary to ensure assumptions were not violated. All outcome and predictor variables were standardised to a mean of zero and standard deviation (SD) of one, for ease of interpretation. Adiposity was measured as % fat and total fat, and methylation was measured as % methylation, with regression coefficients (β) representing the SD change in % fat (or total fat) for each SD change in methylation (Table S2). The skinfold thickness measurements used at each age were calculated using the mean of three skinfold measures, with regression coefficients (β) representing the standard deviation change in skinfold thickness for each SD change in methylation. Where appropriate, measures of adiposity and skinfold thickness were transformed to satisfy the assumption of normality. Regression models were built using child’s fat mass, % fat mass or skinfold thickness as outcomes, and CpG methylation as the predictor. All results are presented as regression coefficients (β) with their associated p-values and 95% confidence intervals. Statistical comparisons of gene expression, or CpG methylation with the lean and obese groups from the BIOCLAIMS cohort was by logistic regression. Analyses were adjusted for sex, and for age where appropriate.
Two of the SLC6A4 SNPs (rs2020934 and rs2020935) were analysed to assess their impact on results. Where possible, both additive and co-dominant models were considered. Mann–Whitney/Kruskal–Wallis tests were performed on CpG 1-5 for rs2020934 and rs2020935, respectively. DXA total fat (at birth, 4 and 6 years of age) was the outcome used as the primary measure of child’s fat mass across all ages in the following regressions; our regression models were adjusted (i) for SNPs individually and (ii) for both rs2020934 and rs2020935 together.
Results
SWS cohort characteristics
Genomic DNA was extracted from the umbilical cord of SWS infants (n = 680) who had measurements of adiposity using DXA or triceps skinfold thickness during infancy and childhood. The infants had a median birth weight of 3.48 kg and gestational age of 40.1 weeks; 50% were female. Median maternal age at birth was 31.5 years, pre-pregnancy median body mass index (BMI) 24.3 kg/m2 and pregnancy weight gain 0.35 kg/week; 48% were in their first pregnancy and 12% smoked in late pregnancy. Table 1 shows additional characteristics and median (5th–95th percentile) cord tissue SLC6A4 methylation values for CpGs 1-5.
Umbilical cord SLC6A4 CpG methylation and percent and total fat mass in infancy/childhood
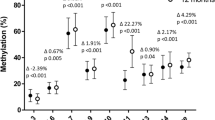
Examining the relationship between cord tissue SLC6A4 methylation at birth and infant/child adiposity, lower CpG1 (Hg19:28561601) and CpG2 (Hg19:28561578) methylation were associated with lower % fat mass age 6–7 years (CpG1, β = 0.118 (95% CI = 0.015, 0.221), p = 0.025; CpG2, β = 0.091 (0.0001, 0.183), p = 0.05, respectively) (Table 2), but were not associated with % fat mass at birth or 4 years or with total fat mass at birth, 4 or 6–7 years. In contrast, lower CpG5 (Hg19:28561468) methylation was associated with higher % fat mass at 6–7 years (β = −0.159 (−0.267, −0.052), p = 0.004). Lower CpG5 methylation was also associated with higher total fat mass at birth (β = −0.102 (−0.213, 0.009), p = 0.072), 4 years (β = −0.108 (−0.206, −0.010), p = 0.031) and 6–7 years (β = −0.219 (−0.325, −0.113), p = 0.0001) (Table 2, Fig. 2a). There were no associations between the methylation of CpG3 (Hg19:28561505) or CpG4 (Hg19:28561490) and % or total fat mass at any age.

Lower methylation of SLC6A4 CpG5 is associated with increased fat mass in the SWS and BIOCLAIMS cohorts. a Association between the methylation of SLC6A4 CpG5 total and % fat mass at 6 years of age. Means are plotted for each individual. b Associations between the methylation of SLC6A4 CpG5 at birth and triceps skinfold thickness at birth, 6 months, 1, 2, 3 and 6 years of age. Means are plotted for each individual. c Trajectory of triceps skinfold thickness from 6 months to 6 years with respect to quarters of SLC6A4 CpG5 methylation. Values are age and sex adjusted (n = 244). Means ± SEM. d Adipose tissue SLC6A4 CpG5 methylation (n = 51) and expression (n = 61) is shown for lean and obese individuals from the BIOCLAIMS cohort. Whiskers show min to max. *p = 0.01–0.05, **p ≤ 0.01
Multivariate analysis of DXA % fat and total fat mass at birth, 4 and 6 years of age adjusted for sex and age showed that methylation of CpG5 was associated with total fat mass at birth (β = −0.1261 (−0.250, −0.002), p = 0.046), 4 (β = −0.1104 (−0.215, −0.006), p = 0.039) and 6 years (β = −0.2039 (−0.314, −0.094), p = 0.0003) of age and with % fat mass (β = −0.1447 (−0.257, −0.033), p = 0.011) at 6 years; there was no association however between CpG2 methylation and % or total fat mass at any time point (Table S3). The effect size was such that for each standard deviation increase in methylation of SLC6A4 CpG5, there was an average decrease of 0.219 standard deviations in total fat mass at 6–7 years of age, accounting for sex, age and batch effect. Additional adjustments for smoking in pregnancy, parity, IOM pregnancy weight gain and mother’s pre-pregnancy BMI did not substantially alter the association between CpG5 methylation and total fat mass at 4 and 6 years but did weaken the association with total fat at birth (adjusted β = 0.0099 (−0.124,0.144), p = 0.884) and % fat mass at age 6 years (adjusted β = −0.0986 (−0.214,0.017), p = 0.0946).
Umbilical cord SLC6A4 methylation and triceps skinfold thickness from birth to age 6–7 years
To determine whether cord SLC6A4 methylation was associated with additional measures of adiposity in childhood, we investigated the relationship between SLC6A4 methylation and triceps skinfold thickness in the SWS children from birth through to 6–7 years. Lower methylation of CpG5 was associated with higher triceps skinfold thickness at birth (β = −0.0993 (−0.177, −0.021), p = 0.013,), 6 months (β = −0.090 (−0.174, −0.005), p = 0.038,), 12 months (β = −0.0781 (−0.160, 0.004), p = 0.062), 2 years (β = −0.155 (−0.238, −0.072), p = 0.0003), 3 years (β = −0.168, (−0.248, −0.088), p = 0.00004) and 6 years (β = −0.104 (−0.186, −0.022), p = 0.013) (Table S4, Fig. 2b). Tracking the association of SLC6A4 CpG5 methylation with skinfold thickness from birth to age 6–7 years, infants with the lowest quarter of SLC6A4 methylation at birth had, on average, the highest triceps skinfold thickness at each of the ages from birth to 6–7 years, while individuals with the highest quarter of SLC6A4 methylation had, on average, the lowest skinfold thickness from birth to 6–7 years (Fig. 2c). Lower CpG4 methylation was associated with higher triceps skinfold thickness at birth (β = −0.137 (−0.254, −0.019), p = 0.023) but there were no associations at later ages. There were no associations between the methylation of CpGs 1, 2 or 3 and triceps skinfold thickness at any age.
Effects of genetic variation within the SLC6A4 DMR on methylation of SLC6A4 DMCpGs in the SWS cohort
Genotyping analysis in all participants excluded the presence of genetic variation within this sample set at the five CpGs sites measured, including the known SNP sites rs141303113 and rs56105746, which overlap with CpG2 and CpG4, respectively. SNPs rs56377481, rs2020934 and rs2020935, which lie within the 300 bp SLC6A4 DMR were also measured (Fig. 1b). Genotyping analysis excluded the presence of variation at rs56377481 (Table S5), but the influence of the higher frequency SNPs rs2020934 and rs2020935 on SLC6A4 methylation was examined further (Table S6). There were differences in methylation at CpG1 (p = 0.0004) and CpG3 (p = 0.005) with respect to genotype at rs2020934, and in the methylation at CpG4 (p = 0.034) and CpG5 (p = 0.024) with respect to genotype at rs2020935. However, for both SNPs the median difference in methylation measurements at CpG sites was <2% (Table S7). The association between SLC6A4 CpG5 methylation and total fat mass remained after adjusting the regression models for SNPs rs2020934 and rs2020935 (Table S8). Genotype alone was not significantly associated with any of the adiposity outcomes measured (Table S9).
Maternal weight gain and parity is associated with altered infant SLC6A4 methylation
To determine the potential maternal drivers of altered infant SLC6A4 methylation, we examined the relationship between SLC6A4 CpG5 methylation and maternal BMI, gestational weight gain, parity, smoking and Vitamin D status, five modifiable factors previously associated with increased infant adiposity [27,28,29,30,31,32]. Higher maternal pregnancy weight gain and lower parity were both associated with lower SLC6A4 CpG5 methylation (p = 0.046; p = 0.029, respectively). There were no associations with pre-pregnancy maternal BMI, smoking or maternal vitamin D status (Table 3).
SLC6A4 CpG5 methylation is associated with adiposity in adolescents at 17 years of age from the Raine Study
The methylation of SLC6A4 CpG5 was measured in DNA from peripheral blood from adolescents aged 17 years from the RAINE cohort (n = 1200) (Table 4). Adjusting for age and sex, lower methylation of CpG5 was associated with greater BMI (β = −0.167 (−0.246, −0.088), p ≤ 0.001) waist circumference (β = −0.102 (−0.182, 0.0234), p = 0.011), subcutaneous fat (β = −0.136 (−0.216, −0.057), p = 0.001), subscapular skinfold thickness (β = −0.123 (−0.2015, −0.045), p = 0.002), abdominal skinfold thickness (β = −0.099 (−0.174, −0.025), p = 0.008), and suprailiac skinfold thickness (β = −0.1096 (−0.185, −0.034), p = 0.004) at age 17 years. Genotyping analysis showed no significant effects of rs2020934 and rs2020935 on CpG5 methylation in the RAINE cohort (Table S6). Blood cell counts were measured in these samples but strong collinearity (Table S10) between blood cell counts and BMI (variance inflation factors: 10.6–112.9) was observed, increasing the variance of the regression coefficients and making them unstable (Table S11). It was therefore not possible to adjust the association between methylation and BMI for cell type.
Lower SLC6A4 methylation is associated with obesity in adipose tissue from adults in the BIOCLAIMS cohort
We next investigated whether SLC6A4 CpG5 was differentially methylated in subcutaneous adipose tissue of obese and lean adults from the BIOCLAIMS cohort (n = 65) (Table 1). Controlling for age and sex, obese individuals had lower SLC6A4 CpG5 methylation (p = 0.019) and SLC6A4 mRNA expression (p = 0.008) in adipose tissue compared to lean individuals (Fig. 2d, Table S12). Genotyping analysis in all participants excluded the presence of SNPs at the cytosine of CpG5.
In silco analysis of putative transcription factor binding sites across the DMR
CpG sites in close proximity are often co-regulated, however here, the CpG sites within the identified DMR of SLC6A4 show different and opposite directions of associations with measures of adiposity. We therefore carried out in silco analysis of the SLC6A4 DMR to identify the transcription factors that may bind across the CpGs of interest and potentially lead to their differential regulation. Matinspector analysis showed that the CpG sites of interest lay within the binding sites of a number of transcription factors, with CpG2 lying within a potential vitamin D-Retinoid X receptor (VDR-RXR) response element and CpG5 within a potential ZNF300 binding site (Table S13).
Discussion
Our findings show that lower DNA methylation in umbilical cord and peripheral blood of a specific CpG site within the SLC6A4 gene is associated with multiple measures of adiposity in infancy, childhood and adolescence. Moreover, in obese adults lower SLC6A4 methylation in adipose tissue was accompanied by a change in SLC6A4 mRNA expression, suggesting that altered SLC6A4 methylation may be of functional relevance in obesity.
The SLC6A4 gene encodes the serotonin transporter, which has an important role in the regulation of emotion, behaviour, energy balance and appetite control. Alterations in SLC6A4 expression have been associated with food intake and obesity in animals and humans; transgenic mice overexpressing SLC6A4 are lighter and shorter than controls [33], whereas SLC6A4 knockout mice develop late onset obesity, hepatic steatosis, glucose intolerance, and insulin resistance [34,35,36]. In humans, a promoter polymorphism within the SLC6A4 gene results in the formation of either a long or a short allele [37]. The short allele reduces transcription compared to the long allele [38], and is associated with obesity in children [18], adult males [19] and with type-2 diabetes [38, 39]. Furthermore, in humans long-term use of SSRI (selective serotonin reuptake inhibitors), is associated with increased obesity and dyslipidaemia [40, 41].
Recent studies have suggested that serotonin has an important role in regulating metabolism in peripheral tissues. Serotonin receptors have been identified in virtually all organs. Adipocytes express a functional system for serotonin synthesis, reuptake and receptor activation, suggesting that serotonin may directly regulate adipocyte function [42]. Consistent with this, mice with a conditional adipose-specific knockout of tryptophan hydroxylase 1, the enzyme that catalyses the rate limiting step in serotonin biosynthesis, exhibit increased energy expenditure, reduced weight gain and decreased lipogenesis in white adipose tissue, suggesting that adipocyte-derived serotonin is important in energy homoeostasis [43].
In cord tissue from infants from the SWS cohort, methylation of SLC6A4 CpG5 showed the strongest and most consistent association with measures of adiposity from birth through to 6 years of age. Two developmental pathways to obesity have been proposed; first, when foetal undernutrition is followed by rapid weight gain, and secondly, when foetal overnutrition is linked to greater adiposity at birth and beyond [22, 27]. Our findings suggest that lower SLC6A4 CpG5 methylation, associated with higher adiposity from birth to 6–7 years, is a marker of the latter pathway. Three of these results passed a strict Bonferroni correction (p < 0.00083) in the SWS discovery cohort, notwithstanding that Bonferroni is likely overly conservative as the different measures of adiposity were highly correlated. Interestingly the associations between CpG5 methylation and total and % fat mass strengthened with age, suggesting an altered response to exposure to risk factors of obesity in later life, consistent with the paradigm of the developmental programming of obesity. Understanding how early life alters later life responses to an obesogenic environment, potentially through alterations in energy homoeostasis, appetite control or food choice will be critical for the development of effective intervention strategies. Weaker positive associations were also found between the methylation of SLC6A4 CpGs 1 and 2 and % fat mass at 6–7 years. Developmentally induced changes in methylation are often CpG site specific [44], so this difference in the strength and direction of the association observed between the methylation of the CpG’s within the SLC6A4 DMR may reflect different regulatory roles of the CpG sites within this region. SLC6A4 CpGs 1 and CpG5 are 135 bp apart and although CpG’s in close proximity are often co-regulated and methylation levels correlated, this is not what is observed for these sites. However, in silico analysis does show that these CpG sites are located within the response elements of different regulatory transcription factors, suggesting that they may be differentially regulated (Table S13).
In an independent cohort, lower methylation of SLC6A4 CpG5 in peripheral blood was associated with multiple measures of adiposity at age 17 years, suggesting that SLC6A4 methylation may be a robust marker of adiposity in peripheral tissue types across a range of ages from infancy to adolescence. Interestingly Zhou et al. [20] have reported that higher SLC6A4 promoter methylation (−69 to –213 from the TSS) in peripheral blood leucocytes was associated with obesity in an adult monozygotic twin study. In our study using cord tissue, peripheral blood and adipose tissue, methylation of CpG5, located 1450 bp downstream of the TSS, was inversely associated with adiposity. This difference in the direction of the association between SLC6A4 methylation and adiposity between the two studies may reflect the different location of the CpGs measured. Recent studies have shown that promoter and gene body methylation are inversely related, with high promoter methylation being associated with reduced transcription, and high gene body methylation positively associated with transcription [45]. Thus, the different direction of associations in the two studies may both reflect a lower level of SLC6A4 transcription associated with increased adiposity. Consistent with this, we found lower SLC6A4 CpG5 methylation in adipose tissue was accompanied by lower SLC6A4 mRNA expression.
To date, the functional significance of altered SLC6A4 methylation in peripheral blood cells or cord tissue is not known, although a number of studies have found that the methylation status of CpGs in peripheral tissues such as blood correlates with that of internal tissues [46, 47]. Interestingly, the methylation of specific CpGs in the promoter of SLC6A4 in T cells and monocytes has been associated with in vivo measures of brain serotonin synthesis [48]. In agreement with this, we showed that differential methylation of SLC6A4 in adipose tissue from a separate population was also associated with obesity. Although the downstream effects of reduced adipose tissue SLC6A4 mRNA expression in our study are not known, this change would suggest an increase in peripheral serotonin availability in the obese state. Consistent with this, increased serum serotonin levels have been associated with obesity in mice [49].
The driving forces for the altered methylation of the SLC6A4 DMR are currently unknown. Lower SLC6A4 methylation was, however, associated with both higher maternal gestational weight gain and lower parity, maternal factors previously linked to increased offspring adiposity [28, 31]. Whether altered SLC6A4 methylation lies on a causal pathway is not known. A substantial contribution to variation in methylation levels between individuals can also be a direct result of local genetic polymorphisms, so-called methylation quantitative trait loci (methQTL); the peak enrichment for distance across cis methQTLs has been experimentally determined as 45 bp [50]. In our study, the SNPs rs2020934 and rs2020935 were statistically associated with the methylation of several of the CpGs within the DMR of SLC6A4, but effect sizes were small and did not significantly affect the association between SLC6A4 CpG methylation and measures of adiposity.
Our study has several limitations. First, we analysed DNA methylation in umbilical cord, blood and adipose tissue, which represent different cell populations with distinct epigenetic profiles. We measured blood cellular heterogeneity in the RAINE cohort, but here the cellular proportions strongly associated with measurements of adiposity. There was a particularly strong association between BMI and the peripheral blood neutrophil count, and previous studies have also reported a linear relationship between BMI and neutrophil number [51, 52]. Many studies have also shown increased neutrophil activation in obese subjects [53]. Multicolinearity was seen between all cell types and BMI for CpG5, overinflating the standard errors of the coefficients and making them unstable. Therefore adjusting the association between methylation and BMI for cellular proportions would violate the assumption of non-colinearity, which is a prerequisite for regression analysis. Consequently, we were unable to assess the dependence on cellular heterogeneity of the association of SLC6A4 methylation in blood and BMI. We cannot rule out the possibility that the association between SLC6A4 methylation in peripheral blood with adiposity in adolescence is dependent upon differences in cellular composition in blood. However, the existence of this association in different tissues, which are (in the case of adipose and cord) more homogeneous in their cellular composition, suggests that the relationship is not completely explained by cell type. In any case, our data showing that methylation status of SLC6A4 CpG5 can distinguish levels of adiposity across a range of tissue types, suggests that altered methylation of this CpG maybe a valuable prognostic biomarker to identify individuals at risk and the efficacy of therapeutic interventions, especially as such biomarkers are likely to be measured in DNA from whole tissue samples rather than isolated specific cell types.
A second limitation of the study is that the number of participants in the SWS cohort with DEXA measurements at birth, 4 and 6 years and with sufficient DNA for methylation analysis did not completely overlap, so there are different numbers of participants at each time point. Moreover, there was also a limited availability of tissues in each cohort and in the SWS cohort, there were no measures of maternal glycemia, which has been suggested to be a driver of offspring epigenetic changes, so we could not determine whether infant’s SLC6A4 methylation was associated with maternal dysglycemia. Comparison of SLC6A4 methylation across tissue types within the same individual would permit a greater understanding of tissue-specific differences in DNA methylation and the utility of SLC6A4 as a marker of future obesity. Thirdly, we did not have longitudinal methylation data, so we cannot ascertain whether methylation of SLC6A4 changes during the life course or identify the factors that modify its methylation. However, this is the first study to define biomarkers of fat mass in perinatal tissue and replicate this result in the adipose tissue of adults from a separate cohort with associated alterations in gene expression. Therefore, not only is this epigenetic mark a potential biomarker of trajectory towards obesity, but it is also possible that its altered methylation may have functional consequences on energy balance, making it a potential target for intervention strategies to optimise health over the life course, or possibly to reverse obesity.
References
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94.
Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–75.
Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24.
Hanson MA. Developmental origins of obesity and non-communicable disease. Endocrinol Nutr. 2013;60(Suppl 1):10–1.
Wellcome Trust Case Control C, Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–20.
Godfrey KM, Barker DJ. Fetal programming and adult health. Public Health Nutr. 2001;4:611–24.
Bertram CE, Hanson MA. Animal models and programming of the metabolic syndrome. Br Med Bull. 2001;60:103–21.
Catalano PM, Kirwan JP. Maternal factors that determine neonatal size and body fat. Curr Diab Rep. 2001;1:71–7.
Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007;97:1064–73.
Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ Res. 2007;100:520–6.
Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008;118:2316–24.
Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60:1528–34.
Clarke-Harris R, Wilkin TJ, Hosking J, Pinkney J, Jeffery AN, Metcalf BS, et al. PGC1alpha Promoter Methylation in Blood at 5-7 Years Predicts Adiposity From 9 to 14 Years (EarlyBird 50). Diabetes. 2014;63:2528–37.
Lillycrop K, Murray R, Cheong C, Teh AL, Clarke-Harris R, Barton S, et al. ANRIL promoter DNA methylation: a perinatal marker for later adiposity. EBioMedicine. 2017;19:60–72.
Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, et al. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci USA. 1993;90:2542–6.
Burke LK, Heisler LK. 5-hydroxytryptamine medications for the treatment of obesity. J Neuroendocrinol. 2015;27:389–98.
Marston OJ, Garfield AS, Heisler LK. Role of central serotonin and melanocortin systems in the control of energy balance. Eur J Pharmacol. 2011;660:70–9.
Sookoian S, Gemma C, Garcia SI, Gianotti TF, Dieuzeide G, Roussos A, et al. Short allele of serotonin transporter gene promoter is a risk factor for obesity in adolescents. Obesity. 2007;15:271–6.
Sookoian S, Gianotti TF, Gemma C, Burgueno A, Pirola CJ. Contribution of the functional 5-HTTLPR variant of the SLC6A4 gene to obesity risk in male adults. Obesity. 2008;16:488–91.
Zhao J, Goldberg J, Vaccarino V. Promoter methylation of serotonin transporter gene is associated with obesity measures: a monozygotic twin study. Int J Obes. 2013;37:140–5.
Inskip HM, Godfrey KM, Robinson SM, Law CM, Barker DJ, Cooper C, et al. Cohort profile: The Southampton Women’s Survey. Int J Epidemiol. 2006;35:42–8.
Crozier SR, Inskip HM, Godfrey KM, Cooper C, Harvey NC, Cole ZA, et al. Weight gain in pregnancy and childhood body composition: findings from the Southampton Women’s Survey. Am J Clin Nutr. 2010;91:1745–51.
Huang RC, Burrows S, Mori TA, Oddy WH, Beilin LJ. Lifecourse adiposity and blood pressure between birth and 17 years old. Am J Hypertens. 2015;28:1056–63.
Lillycrop KA, Costello PM, Teh AL, Murray RJ, Clarke-Harris R, Barton SJ, et al. Association between perinatal methylation of the neuronal differentiation regulator HES1 and later childhood neurocognitive function and behaviour. Int J Epidemiol. 2015;44:1263–76.
Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34.
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–9.
Crozier SR, Harvey NC, Inskip HM, Godfrey KM, Cooper C, Robinson SM, et al. Maternal vitamin D status in pregnancy is associated with adiposity in the offspring: findings from the Southampton Women’s Survey. Am J Clin Nutr. 2012;96:57–63.
Starling AP, Brinton JT, Glueck DH, Shapiro AL, Harrod CS, Lynch AM, et al. Associations of maternal BMI and gestational weight gain with neonatal adiposity in the Healthy Start study. Am J Clin Nutr. 2015;101:302–9.
Oken E, Huh SY, Taveras EM, Rich-Edwards JW, Gillman MW. Associations of maternal prenatal smoking with child adiposity and blood pressure. Obes Res. 2005;13:2021–8.
Oken E, Taveras EM, Kleinman KP, Rich-Edwards JW, Gillman MW. Gestational weight gain and child adiposity at age 3 years. Am J Obstet Gynecol. 2007;196:322–8.
Joshi NP, Kulkarni SR, Yajnik CS, Joglekar CV, Rao S, Coyaji KJ, et al. Increasing maternal parity predicts neonatal adiposity: Pune Maternal Nutrition Study. Am J Obstet Gynecol. 2005;193(3 Pt 1):783–9.
Robinson SM, Crozier SR, Harvey NC, Barton BD, Law CM, Godfrey KM, et al. Modifiable early-life risk factors for childhood adiposity and overweight: an analysis of their combined impact and potential for prevention. Am J Clin Nutr. 2015;101:368–75.
Pringle A, Jennings KA, Line S, Bannerman DM, Higgs S, Sharp T. Mice overexpressing the 5-hydroxytryptamine transporter show no alterations in feeding behaviour and increased non-feeding responses to fenfluramine. Psychopharmacology. 2008;200:291–300.
Murphy DL, Lesch KP. Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci. 2008;9:85–96.
Homberg JR, la Fleur SE, Cuppen E. Serotonin transporter deficiency increases abdominal fat in female, but not male rats. Obesity. 2010;18:137–45.
Chen X, Margolis KJ, Gershon MD, Schwartz GJ, Sze JY. Reduced serotonin reuptake transporter (SERT) function causes insulin resistance and hepatic steatosis independent of food intake. PLoS ONE. 2012;7:e32511.
Heils A, Teufel A, Petri S, Seemann M, Bengel D, Balling U, et al. Functional promoter and polyadenylation site mapping of the human serotonin (5-HT) transporter gene. J Neural Transm Gen Sect. 1995;102:247–54.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–31.
Iordanidou M, Tavridou A, Petridis I, Arvanitidis KI, Christakidis D, Vargemezis V, et al. The serotonin transporter promoter polymorphism (5-HTTLPR) is associated with type 2 diabetes. Clin Chim Acta. 2010;411:167–71.
Raeder MB, Bjelland I, Emil Vollset S, Steen VM. Obesity, dyslipidemia, and diabetes with selective serotonin reuptake inhibitors: the Hordaland Health Study. J Clin Psychiatry. 2006;67:1974–82.
Fava M, Judge R, Hoog SL, Nilsson ME, Koke SC. Fluoxetine versus sertraline and paroxetine in major depressive disorder: changes in weight with long-term treatment. J Clin Psychiatry. 2000;61:863–7.
Stunes AK, Reseland JE, Hauso O, Kidd M, Tommeras K, Waldum HL, et al. Adipocytes express a functional system for serotonin synthesis, reuptake and receptor activation. Diabetes Obes Metab. 2011;13:551–8.
Oh CM, Namkung J, Go Y, Shong KE, Kim K, Kim H, et al. Regulation of systemic energy homeostasis by serotonin in adipose tissues. Nat Commun. 2015;6:6794.
Lillycrop KA, Phillips ES, Torrens C, Hanson MA, Jackson AA, Burdge GC. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPARalpha promoter of the offspring. Br J Nutr. 2008;100:278–82.
Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–91.
Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43.
Slieker RC, Bos SD, Goeman JJ, Bovee JV, Talens RP, van der Breggen R, et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 2013;6:26.
Wang D, Szyf M, Benkelfat C, Provencal N, Turecki G, Caramaschi D, et al. Peripheral SLC6A4 DNA methylation is associated with in vivo measures of human brain serotonin synthesis and childhood physical aggression. PLoS ONE. 2012;7:e39501.
Kim HJ, Kim JH, Noh S, Hur HJ, Sung MJ, Hwang JT, et al. Metabolomic analysis of livers and serum from high-fat diet induced obese mice. J Proteome Res. 2011;10:722–31.
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952.
Bahadir A, Baltaci D, Turker Y, Turker Y, Iliev D, Ozturk S, et al. Is the neutrophil-to-lymphocyte ratio indicative of inflammatory state in patients with obesity and metabolic syndrome? Anatol J Cardiol. 2015;15:816–22.
Furuncuoglu Y, Tulgar S, Dogan AN, Cakar S, Tulgar YK, Cakiroglu B. How obesity affects the neutrophil/lymphocyte and platelet/lymphocyte ratio, systemic immune-inflammatory index and platelet indices: a retrospective study. Eur Rev Med Pharmacol Sci. 2016;20:1300–6.
Xu X, Su S, Wang X, Barnes V, De Miguel C, Ownby D, et al. Obesity is associated with more activated neutrophils in African American male youth. Int J Obes. 2015;39:26–32.
Acknowledgements
We thank all participants included in the studies, the EpiGen Operational Management Group for their project management and Dr. Irma Silva Zolezzi, Nestlé Research Center, for helpful discussion of the manuscript. This work was supported by funding from the Medical Research Council (MC_UU_12011/4, MC_UP_A620_1017 and MC_ST_U12055), British Heart Foundation (RG/15/17/3174 and RG/07/009), Nestec (BIDG/2013/00456), NIHR Musculoskeletal Biomedical Research Unit, University of Oxford, NIHR Southampton Biomedical Research Centre, University of Southampton and University Hospital Southampton NHS Foundation Trust. KMG is supported by the National Institute for Health Research (NF-SI-0515-10042 and through the NIHR Southampton Biomedical Research Centre), by the European Union’s Seventh Framework Programme (FP7/2007-2013 projects EarlyNutrition and ODIN under Grant Agreement Nos 289346 and 613977), by the US National Institute On Aging of the National Institutes of Health (Award Number U24AG047867) and by the UK Economic and Social Research Council and the Biotechnology and Biological Sciences Research Council (Award Number ES/M00919X/1). The BIOCLAIMS study was supported by the European Commission Seventh Framework Programme (Grant Agreement No. 244995). The RAINE study was supported by The Australian National Health and Medical Research Council (NHMRC) (1059711). In the RAINE cohort, data collection at the 17 year follow-up was funded by the National Health and Medical Research Council (Programme Grant ID 353514 and Project Grant ID 403981). Core funding support is provided by University of Western Australia (UWA), the Raine Medical Research Foundation, the Telethon Kids Institute, the UWA Faculty of Medicine, Dentistry and Health Sciences, Curtin University and Edith Cowan University. We would like to thank Melanie White for DNA extraction.
Members of EpiGen Consortium
Graham Burdge, Chong Yap Seng, Cyrus Cooper, Wayne Cutfield, Keith Godfrey, Nicholas Harvey, Joanna Holbrook, Mary Chong Foong-Fong, Neerja Karnani, Lee Yung Seng, Karen Lillycrop, Walter Stunkel and Allan Shepherd.
Author information
Authors and Affiliations
Consortia
Contributions
ESG, RJSM, EC, RCH and PMC carried out the molecular epigenetic studies. ESG, PT, PEM, SJB, JDH, RCH performed the statistical analysis and prepared the tables/figures. ESG, PT,KAL, JDH, KMG drafted the manuscript. KL, KMG, CC, GCB, HMI, NCH, JCH, CEC, CPC, PCC, RCH, TAM and LB participated in the study design and/or collected the samples/physiological measurements. KL, KMG, CC, MAH, PDG conceived of the study, its design, and its coordination. All authors helped draft the manuscript, participated in manuscript editing and read/approved the final.
Corresponding author
Ethics declarations
Conflict of interest
The following authors have no financial interest related to this work: KAL, ESG, RJSM, PT, PEM, EC, SJB, RCH, PMC, JDH, JCH, CEC, CPC, TAM, LB, GCB, HMI, NCH, MAH, PDG, RCH. Both PCC and KMG have received travel reimbursement for speaking at conferences sponsored by companies selling nutritional products.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the EpiGen Consortium are listed below Acknowledgements.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lillycrop, K.A., Garratt, E.S., Titcombe, P. et al. Differential SLC6A4 methylation: a predictive epigenetic marker of adiposity from birth to adulthood. Int J Obes 43, 974–988 (2019). https://doi.org/10.1038/s41366-018-0254-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-018-0254-3
This article is cited by
-
Methylation of serotonin regulating genes in cord blood cells: association with maternal metabolic parameters and correlation with methylation in peripheral blood cells during childhood and adolescence
Clinical Epigenetics (2024)
-
Comparison of methylation capture sequencing and Infinium MethylationEPIC array in peripheral blood mononuclear cells
Epigenetics & Chromatin (2020)
-
Developmental Programming of Body Composition: Update on Evidence and Mechanisms
Current Diabetes Reports (2019)
