
Abstract
Insulin and insulin-like growth factor 1 (IGF-1) signaling regulate cellular growth and glucose metabolism in the myocardium. However, their physiological role in the cells of the cardiac conduction system has never been explored. Therefore, we sought to determine the spatiotemporal function of insulin/IGF-1 receptors in the sinoatrial node (SAN). We generated cardiac conduction cell-specific inducible IGF-1 receptor (IGF-1R) knockout (KO) (CSIGF1RKO), insulin receptor (IR) KO (CSIRKO), and IR/IGF-1R double-KO (CSDIRKO) mice and evaluated their phenotypes. Telemetric electrocardiography revealed regular sinus rhythm in CSIGF1RKO mice, indicating that IGF-1R is dispensable for normal pacemaking. In contrast, CSIRKO and CSDIRKO mice exhibited profound sinus bradycardia. CSDIRKO mice showed typical sinus node dysfunction characterized by junctional rhythm and sinus pauses on electrocardiography. Interestingly, the lack of an insulin receptor in the SAN cells of CSIRKO and CSDIRKO mice caused sinus nodal fibrosis. Mechanistically, hyperpolarization-activated cyclic nucleotide-gated channel 4 (HCN4) protein expression significantly decreased in the CSIRKO and CSDIRKO mice relative to the controls. A patch-clamp study of the SAN cells of CSIRKO mice revealed a significant decrease in the funny current, which is responsible for spontaneous diastolic depolarization in the SAN. This result suggested that insulin receptor loss reduces the heart rate via downregulation of the HCN4 channel. Additionally, HCN1 expression was decreased in CSDIRKO mice, explaining their sinus node dysfunction. Our results reveal a previously unrecognized role of insulin/IGF-1 signaling in sinus node structural maintenance and pacemaker function.
Similar content being viewed by others

Introduction
Insulin signaling acts a key regulator of glucose metabolism through the activation of the insulin receptor (IR), predominantly in the liver, skeletal muscle, and adipose tissue1. Insulin-like growth factor 1 (IGF-1) binds to the highly homologous tyrosine kinase receptor IGF-1 receptor (IGF-1R) and regulates cell growth and glucose homeostasis. Although insulin and IGF-1 preferentially bind to their specific receptors, both ligands can bind with lower affinity to alternate receptors that also transduce their signals1,2. In the heart, deletion of IR in cardiomyocytes induced small heart size and switched cardiac substrate utilization from glucose to fatty acids3. Conversely, excess cardiac insulin signaling exacerbated heart failure induced by pressure overload4. In contrast, deletion of IGF-1R in cardiomyocytes resulted in normal postnatal cardiac size but diminished exercise-induced cardiac hypertrophy with delayed cardiac senescence phenotypes5,6. Strikingly, disruption of IR and IGF-1R signaling induced neonatal cardiac autophagic death7.
Sinus node dysfunction (SND) is a cardiac conduction disturbance characterized by profound sinus bradycardia, pauses, arrest, and sinoatrial node exit block8. Furthermore, SND is associated with increased syncope, heart failure, and atrial fibrillation risks. Clinically, symptomatic SND is referred to as sick sinus syndrome. In a prospective cohort study, aging and several cardiovascular risk factors, such as a high body mass index, slow heart rate, and high prevalence of hypertension, were significantly associated with sick sinus syndrome. In contrast, diabetes was only marginally related to the development of sick sinus syndrome9. Furthermore, individuals who developed sick sinus syndrome faced an increased risk of death and cardiovascular disease in two large community-based cohorts10. Early studies demonstrated that hyperglycemia per se induced SND. In a streptozotocin-induced mouse model of diabetes, the heart rate was significantly reduced, and the sinus node recovery time corrected for the heart rate was prolonged through sinus nodal pacemaker cell apoptosis11. Diabetic mice rescued by insulin administration showed an increased heart rate, suggesting that the absence of insulin signaling may contribute to the mechanism of SND. In addition, SND characterized by prolonged sinus node recovery time and autonomic nerve rarefaction was present in obese db/db diabetic mice12.
How insulin signaling to pacemaker cells affects heart rate regulation remains undefined. To dissect the specific mechanism linking IR-dependent signaling to the sinoatrial node (SAN) conduction system without the systemic effect of hyperglycemia, we generated cardiac conduction cell-restricted IR and/or IGF-1R knockout (KO) mice; this was based on the notion that insulin signaling in the heart is transduced through IR and IGF-1R. The aim was to reveal the role of insulin signaling in the SAN.
Materials and methods
Mice
Homozygous IGF-1Rfl/fl and IRfl/fl mice13,14,15 were bred with Hcn4-CreErt2 or Hcn4- CreErt2;ROSA26TomRed mice16 to generate conduction cell–specific KO mice, including IGF-1Rfl/fl;Hcn4-CreErt2;ROSA26TomRed (CSIGF1RKO), IRfl/fl;CreErt2;ROSA26TomRed (CSIRKO), and IGF-1Rfl/fl;IRfl/fl; CreErt2;ROSA26TomRed (CSDIRKO) mice. To delete the locus of X-over P1 (loxP) site by activating CreErt2 in mice, 40 mg/kg tamoxifen was administered by intraperitoneal injection to the mice for 4 days. Non-floxed CreErt2;ROSA26TomRed mice were also injected with tamoxifen and served as the wild-type (WT) group. Animals were fed standard chow and autoclaved water ad libitum and housed in temperature-controlled, pathogen-free facilities with a 12/12 h light/dark cycle. All animals were maintained on a C57BL/6J background, and male mice were used for all experiments. All animal experiments were conducted according to the guidelines approved by the institutional animal care and use committee of Chung-Ang University, Seoul, Korea.
Telemetric ECG
An HD-X11 heart telemetry probe (Data Sciences International, Inc., New Brighton, MN, USA) was subcutaneously implanted into the abdomen (transmitter), chest (electrode), and carotid artery (catheter) of all animals 7 days before recording an ECG to assess adaptation and recovery. After the recovery period, ECG recordings were performed on each mouse for 3 days for cardiac rhythm analysis 3 weeks after the tamoxifen injections. Eight receiver channels were used for monitoring simultaneously.
ECG
Mice were anesthetized with isoflurane (3% for induction and 1.5% for maintenance) via inhalation using a laboratory animal anesthesia machine (Vet Equipment Inc., Pleasanton, CA, USA). Following anesthesia, electrodes were inserted subcutaneously in the left and right forelimbs and left hindlimb in all animals 7 weeks after the administration of the tamoxifen injections. ECG recordings were performed using a PowerLab 4/20T with an animal BioAmp (ADInstruments, Sydney, Australia).
RNA isolation and semiquantitative or quantitative reverse transcription polymerase chain reaction analysis
Total RNA was obtained from the right atrium (RA) and SAN tissues or cells using the RNeasy® Fibrous Tissue Mini Kit (Qiagen GmbH, Hilden, Germany) and the RiboEx™ Total RNA Solution (Geneall Biotechnology Co. Ltd., Seoul, Korea). First-strand deoxyribonucleic acid was synthesized from 20 ng total RNA with random primers using a SuperScript™ III First-Strand Synthesis System (#18080051; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Polymerase chain reaction (PCR) analysis was performed using PCR Master Mix (#K0171; Thermo Fisher Scientific, Inc.) for semiquantitative reverse transcription PCR (RT‒PCR) and SsoFast™ EvaGreen® Supermix (#1725201AP; Bio-Rad Laboratories, Inc., Hercules, CA, USA) for quantitative RT‒PCR (qRT‒PCR). 18S rRNA was used as an internal control for normalization. Primers are listed in Supplementary Table 3. Postamplification melting curve analysis was performed to assess the specificity of the amplified PCR products, and relative quantities were calculated using the comparative cycle threshold method.
Western blot analysis
Western blot analysis was performed as described previously15. RA and SAN tissues or cells were lysed with lysis buffer (20 mM Tris-HCl, pH 7.4; 1% Triton X-100; 1 mM EDTA; 30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES); 50 mM Na4P2O7; 100 mM NaF) with Protease Inhibitor Cocktail (Roche Life Science, Basel, Switzerland) and phosphatase inhibitors. Lysates were incubated on ice for 15 min and then centrifuged at 13,000 rpm at 4 °C for 10 min. The supernatants were used for protein quantification by the bicinchoninic acid assay, and protein samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis on a 10% gel. Protein samples were transferred to a nitrocellulose membrane with a 0.45 µm pore size and blocked with 5% skim milk. Primary antibodies were diluted in blocking solution (5% bovine serum albumin (BSA) in tris-buffered saline with 0.1% Tween® 20 [anti-HCN4; #sc-58622; Santa Cruz Biotechnology, Inc., Dallas, TX, USA] dilution 1:500); anti-HCN1 (#APC-056; Alomone Labs, Jerusalem, Israel [dilution 1:1000]); anti-phospho-Akt (Ser473) ([#4060; Cell Signaling Technology Inc., Danvers, MA, USA [dilution 1:1000]); anti-Akt (#9272; Cell Signaling Technology Inc. [dilution 1:1000]), and anti-glyceraldehyde 3-phosphate dehydrogenase (anti-GAPDH) (#2118; Cell Signaling Technology Inc. [dilution 1:2000]) primary antibodies were used according to the manufacturer’s protocols. The nitrocellulose membranes were then incubated with secondary antibodies in 5% skim milk for 1 h at room temperature, and protein bands were visualized using EzWestLumi ECL solution. Densitometric quantification was performed using Bio-Rad Image Lab Software (Bio-Rad Laboratories, Inc). GAPDH was used as a loading control.
Histological analysis
Mice were sacrificed by cervical dislocation. Tissue preparation and staining were performed as described previously15,17. Isolated SAN tissues were fixed in 4% paraformaldehyde and sectioned at a thickness of 7 μm using a cryostat (Leica Biosystems, Wetzlar, Germany). Frozen SAN slides were used for hematoxylin and eosin staining, Sirius red staining, TUNEL, and immunostaining. Sirius red staining was performed by incubating the tissues in 0.1% Fast Green solution for 15 min at room temperature and then in 0.1% Fast Green solution and 0.1% Sirius red in saturated picric acid for 45 min at room temperature.
For immunohistochemical analysis of HCN4 and LC3II, peroxidase activity was blocked with 3% H2O2 in MeOH for 15 min at room temperature, blocked with 5% BSA for 1 h and treated with primary antibodies. Primary antibodies were anti-HCN4 (dilution 1:100) and anti-LC3II (#L7543; Merck KGaA, Darmstadt, Germany [dilution 1:200]). In addition, slides were incubated with biotinylated secondary antibodies and avidin–biotin complex reagents (#PK6100; Vector Laboratories, Inc., Newark, CA, USA) for 45 min at room temperature. Chromogenic detection was performed using a DAB Substrate Kit. For immunofluorescence staining of HCN4, IGF-1R, and IR, sections were blocked with 5% BSA for 1 h at room temperature, followed by overnight incubation with primary antibodies (anti-HCN4, dilution 1:50; anti-IGF-1 receptor [#sc-713; Santa Cruz Biotechnology, Inc.], dilution 1:50; and anti-insulin receptor [#AHR0271; Thermo Fisher Scientific Inc.], dilution 1:100) at 4 °C. After being washed in phosphate-buffered saline (PBS) with 0.05% Triton X-100 (PBST) for 10 min each, the slides were incubated with secondary antibodies (Alexa Fluor™ 647-conjugated goat anti-rat IgG, Alexa Fluor™ 488-conjugated goat anti-rabbit IgG, Alexa Fluor™ 488-conjugated goat anti-mouse IgG (Thermo Fisher Scientific, Inc.) for 1 h at room temperature. After being washed with PBS, the slides were mounted with diamidino-2-phenylindole (DAPI) solution. Images were obtained using a confocal microscope (LSM 700; Carl Zeiss AG, Jena, Germany). Apoptosis was determined using a colorimetric DeadEnd™ TUNEL Assay Kit (#G7130; Promega Corp., Madison, WI, USA) following the manufacturer’s instructions.
Isolation of SAN pacemaker cells and immunocytochemistry
Mice were anesthetized with isoflurane to extract SAN tissues, and then pacemaker cells were isolated as previously described18. The SAN tissues were chopped into 4–5 pieces and incubated in a low-Ca2+ solution at 36 °C for 5 min. The low-Ca2+ solution contained 140 mM NaCl, 5 mM HEPES, 5.5 mM glucose, 5.4 mM KCl, 0.2 mM CaCl2, 0.5 mM MgCl2, 1.2 mM KH2PO4, 50 mM taurine, and 1% BSA; the pH was adjusted to 6.9 with NaOH. The SAN tissues were then incubated in 3 ml of enzyme solution containing collagenase type II (Worthington Biochemical Corp., Lakewood, NJ, USA), elastase (Worthington Biochemical Corp.), and protease type XIV (Sigma‒Aldrich®, Merck KGaA, Darmstadt, Germany) in a low-Ca2+ solution for 15–20 min. The tissue was then exposed to Kraft-Brule solution at room temperature and slowly dissociated using a wide-diameter glass pipette for 3–5 min. The dissociated cells were plated on laminin-coated coverslips (Supplementary Fig. 4). Subsequently, the samples were immunocytochemically stained or stored at 4 °C to record the If. For immunocytochemical analysis, attached SAN pacemaker cells were fixed with 4% paraformaldehyde at room temperature for 10 min, permeabilized with 0.1% PBST for 10 min, blocked with 5% BSA, and incubated overnight at 4 °C with anti-HCN4 antibody (dilution 1:50). After being washed three times in PBS for 10 min each, the coverslips were treated with secondary antibodies (Alexa Fluor™ 488-conjugated goat anti-rat IgG) for 1 h at room temperature and then mounted with DAPI solution. Images were obtained using a confocal microscope.
Patch-clamp electrophysiology
The If was recorded as previously described19,20. Whole-cell voltage-clamp recordings were carried out in isolated pacemaker cells from the SAN. The extracellular solution contained the following: 140 mM NaCl, 5.4 mM KCl, 1.2 mM KH2PO4, 5 mM HEPES, 5.5 mM glucose, 1 mM MgCl2, and 1.8 mM CaCl2. The pH was adjusted to 7.4 with NaOH. The current was elicited with a holding potential at −50 mV and a hyperpolarizing step to −110 mV for 1.0 s. The patch pipettes were filled with intracellular solution containing 5 mM NaCl, 135 mM KCl, 10 mM HEPES, 1 mM MgCl2, 0.1 mM CaCl2, 10 mM EGTA, 4 mM MgATP, and 5 mM pCr; the pH was adjusted to 7.2 with KOH. After the initial current recording, 0.5 mM BaCl2 was added to the extracellular solution to eliminate the Ba2+-sensitive K+ current. The current density was calculated by dividing pA by pF.
Isolation of neonatal rat cardiomyocytes
Neonatal hearts were obtained from three-day-old Sprague Dawley rats and digested for isolation of cardiomyocytes as previously described21. Neonatal rat cardiomyocytes (NRCMs) were incubated in DMEM/M199 (1:1) containing 10% horse serum, 5% fetal bovine serum, 1% L-glutamine, 1% bromodeoxyuridine, and 1% penicillin/streptomycin. To confirm the relationship between insulin and HCN channel expression, NRCMs were treated with 200 nM insulin (#I9278; Merck KGaA, Darmstadt, Germany) with or without the PI3K inhibitor LY294002 (#L9908; Merck KGaA, Darmstadt, Germany) for 6–24 h and then analyzed by western blotting and qRT‒PCR.
Echocardiography
Mice were anesthetized in an induction chamber using 3% isoflurane, and anesthesia was maintained with 1.5% isoflurane on a heating pad at 36 °C. Echocardiography was performed using the Vevo 770 System (FUJIFILM VisualSonics Inc., Toronto, ON, Canada) with a 30-MHz transducer in the 2-dimensional and motion-mode images22.
Statistical analysis
Data are presented as the mean ± SEM. For intergroup comparisons, the data distribution was first evaluated for normality using the Shapiro‒Wilk test, a further Shapiro‒Wilk test after log-transformation, and a quantile‒quantile plot. The normally distributed data were compared using a t test or two-way analysis of variance. The nonnormally distributed data were analyzed using either an unadjusted Mann‒Whitney U-test or a Mann‒Whitney U-test with a Bonferroni-corrected α value. P < 0.05 was considered statistically significant. All analyses were conducted using the Statistical Package for the Social Sciences (SPSS®, Version 23, IBM Corp., Armonk, NY, USA).
Results
Genetic deletion of IGF-1R and IR in cardiac conduction cells
To elucidate the role of insulin signaling in SAN pacemaker cells, IGF-1Rfl/fl, IRfl/fl, or IGF-1Rfl/fl; IRfl/fl mice13,14,15 were bred with hyperpolarization-activated cyclic nucleotide-gated channel 4-cyclization (Cre) recombinase-estrogen receptor T2 (Hcn4-CreErt2)23 or Hcn4-CreErt2;reverse orientation splice acceptor 26 (ROSA26TomRed) mice16 to generate IGF-1Rfl/fl;Hcn4-CreErt2;ROSA26TomRed (conduction cell-specific IGF-1 receptor [IGF-1R] knockout [KO] [CSIGF1RKO]), IRfl/fl;Hcn4-CreErt2;ROSA26TomRed (conduction cell-specific IR KO [CSIRKO]), and IGF-1Rfl/fl;IRfl/fl;Hcn4-CreErt2;ROSA26TomRed (conduction cell-specific double IR/IGF-1R KO [CSDIRKO]) mice. ROSA26 reporter tandem dimer Tomato (tdTomato) mice produce uniform red fluorescence when exposed to Cre recombinase. We injected tamoxifen for 4 consecutive days to activate Hcn4-CreErt2 and sacrificed the mice 14 days after the tamoxifen injections (Fig. 1A).

A Schematic representation of tamoxifen treatment in conduction cell-specific inducible IGF-1R knockout (KO) (CSIGF1RKO), conduction cell-specific IR KO (CSIRKO), conduction cell-specific double IR/IGF-1R KO (CSDIRKO), and control mice. Eight-week-old mice were injected intraperitoneally with 40 mg/kg tamoxifen for 4 consecutive days. Two weeks after the final tamoxifen injection, the mice were sacrificed and analyzed. B Sinoatrial node (SAN) localization in a fluorescent image of a heart prepared from 8-week-old Hcn4-CreErt2;ROSA26TomRed mice. SVC, superior vena cava; RA, right atrium. C Immunohistochemical staining of hyperpolarization-activated cyclic nucleotide-gated channel 4 (HCN4) in the SAN region. D Western blot analysis of HCN4 in the RA and SAN from Hcn4-CreErt2;ROSA26TomRed mice; α-tubulin was used as a loading control. E Representative semiquantitative RT‒PCR analysis of Igf-1r and ir mRNA expression in the SAN of wild-type (WT), CSIGF1RKO, CSIRKO, and CSDIRKO mice. F, G Relative levels of mRNA expression were normalized to the levels of 18 S rRNA; the expression level in WT mouse hearts was arbitrarily set to 1. Group sizes: WT (n = 9), CSIGF1RKO (n = 8), CSIRKO (n = 10), CSDIRKO (n = 9). H, I Representative immunofluorescence images of IGF-1R (green), IR (green), tandem dimer Tomato (tdTomato) (red), and DAPI (blue) signal in the SAN of WT, CSIGF1RKO, CSIRKO, and CSDIRKO mice. Magnification ×20; scale bar, 50 μm. Data are presented as the mean ± SEM. **P < 0.01; ***P < 0.001; ****P < 0.0001.
Figure 1B shows the anatomic boundaries of the mouse SAN. We dissected the HCN4-expressing fluorescent SAN and the surrounding nonfluorescent right atrial tissues under a fluorescence microscope. The hyperpolarization-activated cyclic nucleotide-gated channels (HCNs) comprise a depolarizing funny current (If) that contributes to SAN pacemaking24. Although HCN isoforms HCN1–4 are present in human SAN cells, mouse SAN tissues express HCN1 and HCN4 proteins exclusively. However, neither HCN1 nor HCN4 protein was found in the right atrium25,26. Therefore, we confirmed that tdTomato successfully marked SAN tissue, as verified by immunohistochemistry (Fig. 1C) and western blotting (Fig. 1D). Messenger ribonucleic acid (mRNA) expression of IGF-1R in the SAN was reduced to 39% (P < 0.01) and 67% (P < 0.001) of the level of WT in CSIGF1RKO and CSDIRKO mice, respectively (Fig. 1E, F). In addition, IR mRNA levels were reduced to ~65% in CSIRKO and CSDIRKO mice (P < 0.0001) (Fig. 1E, G). The residual mRNA expression was likely from non-pacemaker cells within SAN tissue, such as adipocytes and fibroblasts.
We further investigated whole-mount immunostained SAN tissues and confirmed diminished expression of IGF-1R in CSIGF1RKO and CSDIRKO and decreased IR expression in the SAN of CSIRKO and CSDIRKO mice (Fig. 1H, I). Surprisingly, the abundance of the tdTomato reporter was attenuated in the SAN of CSIRKO and CSDIRKO but not CSIGF1RKO mice, suggesting that insulin signaling, but not IGF-1 signaling, may affect HCN4 expression (Fig. 1H, I and Supplementary Fig. 1).
Deletion of IR induces bradycardia, and the combined deletion of IGF-1R and IR causes sinus node dysfunction
To determine whether impaired insulin signaling induces SND, we recorded electrocardiograms (ECGs) from freely moving mice by telemetry 3 weeks after the tamoxifen injections. ECG analysis of the CSIGF1RKO mice showed normal sinus rhythm (Fig. 2A, B). However, compared with those of the WT mice, the ECG of CSIRKO mice displayed slight interbeat (R-R) interval prolongation (Fig. 2C, D). Furthermore, a long R-R interval was statistically significant in CSDIRKO mice compared to WT mice (Fig. 2E, F).

Representative telemetric recordings of the electrocardiogram (ECG) (A) Quantitation of interbeat (R-R) intervals; B wild-type (WT) and conduction cell-specific inducible insulin-like growth factor 1 receptor (IGF-1R) knockout (KO) CSIGF1RKO mice, n = 3 per group. C Representative telemetric ECG recording; D quantitation of R-R intervals; WT and conduction cell-specific insulin receptor (IR) KO (CSIRKO) mice, group sizes: WT (n = 6) and CSIRKO (n = 8); E representative telemetric ECG recording of conduction cell-specific double IR/IGF-1R KO (CSDIRKO) mice showing sinus arrhythmia; F quantification of R-R intervals of CSDIRKO compared to WT mice, group sizes: WT (n = 6) and CSDIRKO (n = 7). Data are presented as the mean ± SEM. ***P < 0.001.
Next, we recorded heart rates for 3 days by telemetry. Baseline heart rates were similar during the light and dark phases in WT (light, 551 ± 2.6 bpm; dark, 556 ± 3.6 bpm; total 554 ± 6.1 bpm) and CSIGF1RKO (light, 526 ± 9.2 bpm; dark, 539 ± 15.1 bpm; total, 532 ± 10.3 bpm) mice (Fig. 3A, B). When the heart rates were sorted into 200 bpm intervals, histograms showed that the most common bpm range, accounting for >60% of WT and CSGF1RKO mice, was 400–600 bpm (WT, 68.3%; CSIGF1RKO, 65.6%). Moreover, 600–800 bpm accounted for 31% of WT and 28% of CSGF1RKO mice, with the least common interval being 200–400 bpm (WT, 0.7%; CSIGF1RKO, 6.0%) (Fig. 3C and Supplementary Table 1). However, CSIRKO and CSDIRKO mice displayed lower heart rates than WT mice at baseline. In particular, the mean heart rate of CSDIRKO mice was decreased compared to that of CSIRKO mice in both the light and dark phases. The heart rate of CSIRKO mice were 512 ± 8.7 bpm (light, 506 ± 8.7 bpm; dark, 517 ± 10.4 bpm) (Fig. 3D, E), while that of CSDIRKO mice was 438 ± 14 bpm (light, 452 ± 13.5 bpm; dark, 425 ± 15.4 bpm) (Fig. 3G, H). The histogram for the bpm intervals more clearly demonstrated this difference. Figure 3F, I shows that the most frequent interval in CSIRKO mice was 200–600 bpm (81.6%); for CSDIRKO mice, it was 200–400 bpm (53.5%) (Supplementary Table 1). Surprisingly, CSDIRKO, but not CSIGF1RKO or CSIRKO mice, demonstrated sinus arrest with junctional escape rhythm in the unconscious state ECG, which is characteristic of SND (Supplementary Fig. 2). Motion-mode (M-mode) echocardiography showed that CSDIRKO mice displayed preserved cardiac contractile function; this was demonstrated by fractional shortening or ejection fraction measurement (Supplementary Table 2). Therefore, CSDIRKO mice exhibit an arrhythmic phenotype with preserved cardiac contraction.

A, B Mean heart rate with a 12 h/12 h light/dark cycle (8:00 AM to 8:00 PM) in wild-type (WT) and conduction cell-specific inducible insulin-like growth factor 1 receptor (IGF-1R) knockout (KO) (CSIGF1RKO); D, E conduction cell-specific insulin receptor (IR) KO (CSIRKO); G, H conduction cell–specific double IR/IGF-1R KO (CSDIRKO); electrocardiogram (ECG) recording over 72 h; heart rate values were analyzed at 10-s intervals. C Mean heart rate histograms of the WT and CSIGF1RKO mice: F CSIRKO; I CSDIRKO mice. Group sizes: WT (n = 3), CSIGF1RKO (n = 4); WT (n = 6), CSIRKO (n = 8); WT (n = 6), CSDIRKO (n = 7). Data are presented as the mean ± SEM. **P < 0.01; ***P < 0.001.
Deletion of IR causes sinus nodal fibrosis
Next, we examined whether the deletion of IR induces morphological changes in the SAN. As previously reported27, WT mouse SAN tissue comprises a compact nodal structure with densely packed nodal cells (Fig. 4A). Notably, the SAN tissue of CSIRKO and CSDIRKO mice showed a diffuse, loosely packed structure. We hypothesized that the reduced SAN density may have resulted from apoptosis or autophagy. However, there were no positive signals from terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) or microtubule-associated protein 1A/1B-light chain 3-II (LC3II) immunostaining (Supplementary Fig. 3a, b). Furthermore, the mRNA expression levels of bax, p62, vps34, and gabalapl1 were unchanged (Supplementary Fig. 3c–f).

Staining of the sinoatrial node (SAN) of wild-type (WT), conduction cell–specific inducible insulin-like growth Factor 1 receptor (IGF-1R) knockout (KO) (CSIGF1RKO), conduction cell–specific insulin receptor (IR) KO (CSIRKO), and conduction cell–specific double IR/IGF-1R KO (CSDIRKO) mice at 2 weeks after four consecutive days of tamoxifen injections. A Hematoxylin and eosin staining (magnification ×40; scale bar, 20 μm). B Sirius red stain (magnification ×63; scale bar, 20 μm). Arrows indicate the sinoatrial node artery. C Quantification of the fibrosis area. Group sizes: WT (n = 11), CSIGF1RKO (n = 9), CSIRKO (n = 11), and CSDIRKO (n = 14). D–H mRNA quantification of collagen I (D); collagen III (E); mmp2 (F); ctnt (G); and actc1 (H). The results were normalized to 18S rRNA; the expression level in the WT group was arbitrarily set to 1. Group sizes: WT (n = 6–11), CSIGF1RKO (n = 6–11), CSIRKO (n = 6–10), CSDIRKO (n = 6–11). Data are presented as the mean ± SEM. **P < 0.01; ***P < 0.001; ****P < 0.0001.
We then analyzed whether fibrotic tissue replaced the myocyte composition. In CSIRKO and CSDIRKO mice, dense collagen was interspersed within the SAN (Fig. 4B, C). In addition, collagen I, collagen III, and mmp2 mRNA levels were significantly higher in the SAN of CSIRKO and CSDIRKO mice than in that of WT mice (Fig. 4D–F). However, the myocyte marker genes ctnt and actc1 showed decreased expression in the SAN tissue of CSIRKO and CSDIRKO but not CSIGF1RKO mice (Fig. 4G, H). Together, these results suggest that SAN fibrosis may contribute to the development of SND in CSIRKO and CSDIRKO mice.
Insulin signaling regulates HCN expression
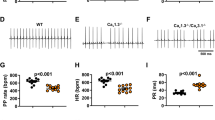
To further explore the mechanism of profound sinus bradycardia in CSIRKO and CSDIRKO mice, we evaluated the HCN channels based on our observation that tdTomato red fluorescence was attenuated in the SAN of CSIRKO and CSDIRKO mice. As expected, tdTomato positivity colocalized with HCN4-positive cells, and the fluorescence intensity was decreased in pacemaker cells isolated from the SAN tissues of CSIRKO and CSDIRKO mice (Fig. 5A). Next, we performed patch-clamp recordings of If from isolated pacemaker cells. As shown in Fig. 5B, C, the amplitude of If was significantly reduced in CSIRKO mice compared to WT mice (mean current density, current amplitude divided by membrane capacitance [pA/pF]; 29.9 in WT and 16.8 in CSIRKO mice, P < 0.05). To examine whether the remaining If in CSIRKO mice could be explained by HCN1, SAN tissue sections were immunostained for HCN1 and HCN4. Expression of the HCN4 protein was decreased in the SAN of CSIRKO and CSDIRKO mice; HCN1 expression was reduced only in the SAN of CSDIRKO mice (Fig. 5D). In addition, the mRNA levels of HCN4 were decreased by 46% in CSIRKO mice and 72% in CSDIRKO mice. However, no difference was observed in CSIGF1RKO mice compared to WT mice (Fig. 5e). Furthermore, HCN1 mRNA expression was reduced by 53% in CSDIRKO mice compared to WT mice (Fig. 5F). Together, these results suggest that insulin signaling is critical for the regulation of HCN genes in SAN tissues.

A Representative images of HCN4 immunocytochemistry sinoatrial node (SAN) pacemaker cells isolated from wild-type (WT), conduction cell–specific inducible insulin-like growth factor 1 receptor (IGF-1R) knockout (KO) (CSIGF1RKO), conduction cell–specific insulin receptor (IR) KO (CSIRKO), and conduction cell–specific double IR/IGF-1R KO (CSDIRKO) mouse SAN tissue labeled with tandem dimer Tomato (tdTomato) red fluorescence. Magnification ×40; scale bar, 50 μm. B, C The mean current density (pA/pF) from the CSIRKO mouse SAN cells (16.8 ± 3.61, n = 10) was significantly lower than the current density recorded from wild-type (WT) pacemaker cells (29.9 ± 3.54, n = 11). D Representative images of HCN1 and HCN4 immunostaining of WT, CSIGF1RKO, CSIRKO, and CSDIRKO mouse SAN. Arrows indicate the sinoatrial node artery. (HCN4, magnification ×20; scale bar, 50 μm; HCN1, magnification 20×; scale bar, 100 μm). E, F mRNA quantification of hcn4 (E) and hcn1 (F). The results were normalized to 18S rRNA; the expression level in the WT group was arbitrarily set to 1. Group sizes: WT (n = 11), CSIGF1RKO (n = 11), CSIRKO (n = 10), and CSDIRKO (n = 11). Data are presented as the mean ± SEM. *P < 0.05; ***P < 0.001; ****P < 0.0001.
To obtain direct evidence that insulin signaling regulates HCN expression, we performed an additional in vitro study using neonatal rat cardiomyocytes, which are known to express If28,29. Cells were treated with 200 nM insulin for up to 24 h. Insulin upregulated HCN4 mRNA expression in a time-dependent manner. However, insulin increased HCN1 mRNA expression only at 6 h (Fig. 6A, B). Cotreatment with the phosphoinositide 3-kinase (PI3K) inhibitor LY294002 completely suppressed insulin-induced Akt phosphorylation and HCN4 protein expression (Fig. 6C–E). However, HCN1 expression was not affected by LY294002 treatment (Fig. 6F). Collectively, these data further indicated that insulin directly regulates HCN4 expression through an Akt-dependent pathway.

Cells were treated with 200 nM insulin for 24 hr. mRNA quantification of hcn4 (A) and hcn1 (B), n = 7–8 per group. The results were normalized to 18S rRNA. Western blot analysis (C) and densitometric ratios of phosphorylated Akt at Ser473 to total Akt; (D), HCN4 (E), and HCN1 (F). GAPDH was used as a loading control. n = 3–4 per group. Data are presented as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Extensive studies of the role of cardiac IR/IGF-1R using cardiomyocyte-specific Cre recombinase in mice have revealed that insulin signaling is critical for myocardial growth and substrate metabolism under physiological and pathological conditions3,5,6,7,30. However, cardiac electrical phenotypes have not been reported in mice. Although several cardiac-specific inducible Cre mouse lines target the myocardium, these lines do not specifically target the cardiac conduction system31. In this study, we demonstrated that conduction cell-specific KO of IR or IR/IGF-1R in adult mice leads to SAN structural remodeling through the development of nodal fibrosis. Furthermore, insulin signaling regulates sinus rhythm and rate by maintaining the If.
The SAN is a spindle-shaped structure located in the right atrium near the opening of the superior vena cava. The SAN consists of a fibrous matrix with a densely packed population of specialized contractile cardiomyocytes. Pacemaker cells in the SAN generate an electrical impulse, propagated through an electrical conduction system, to the atrioventricular node and the ventricular myocardium for cardiac contraction. The intrinsic properties of the pacemaker cells are tightly controlled to maintain normal heart rhythm and allow adaptation to changes in physiological and pathological demands32. In addition, sympathetic and parasympathetic innervation to the SAN determines the heart rate. Diabetes is associated with an increased risk of arrhythmias, and sudden cardiac death is more common33.
Tachycardia is one manifestation of cardiac autonomic neuropathy associated with damage to the vagus nerve innervating the heart, including the SAN34. In db/db diabetic mice, reduced nerve density within the pacemaker tissue has been reported, as have abnormal heart rate fluctuations12. However, few studies have investigated sinus arrhythmia in the context of intrinsic SAN dysfunction in a diabetes model. Krishnaswamy et al. reported a baseline reduction in heart rates and SAN function in diabetic Akita mice35. While there is a connection between hyperglycemia and SND, a relationship between impaired insulin signaling in the SAN and normoglycemia has never been reported. Our data clearly showed that insulin signaling regulates the heart rate independently of the autonomic nervous system or hyperglycemia.
Recently, it has been reported that a streptozotocin-induced mouse model of diabetes exhibited SND, including macrophage infiltration, fibrosis, and oxidative stress, in the SAN36. Additionally, in the context of myocardial infarction, it has been shown that oxidized calcium/calmodulin-dependent protein kinase II mediates SAN cell apoptosis in mice with streptozotocin-induced diabetes11. These studies highlight the potential contribution of high-glucose-induced oxidative stress to SND. In this study, attenuated insulin signaling in the SAN did not affect apoptosis or autophagy. Notably, attenuation of insulin signaling per se resulted in SAN fibrosis. Mechanistically, it is possible that activated fibroblasts within the SAN of CSIRKO mice promoted collagen deposition. Several studies have suggested that crosstalk between pacemaker cells and fibroblasts within the SAN driven by increased expression of the secreted protein transforming growth factor (TGF)β can promote fibroblast proliferation and differentiation into myofibroblasts37,38. Therefore, our findings encourage further investigation into the inverse relationship between the amount of fibrosis within the SAN and heart rate.
Next, our major finding was the alteration in HCN4 expression in CSIRKO and CSDIRKO mice. Furthermore, reduced expression of HCN1 in the SAN of CSDIRKO mice indicated a more severe phenotype, such as sinus pause. Deletion of HCN4 in adult mice eliminated most of the SAN If current, causing cardiac arrhythmia characterized by recurrent sinus pauses and bradycardia; this suggested that HCN4 channels are essential for normal heart impulse generation and conduction39,40. Moreover, in a recent study, HCN1−/− mice displayed congenital SND26. Given the SAN dysfunction following both IGF-1R and IR deletion, we speculate that both channels have compensatory effects on pacemaking activity. Although our data unveiled the critical role of insulin signaling on If current, the contribution of ion channels, such as Na+, Ca2+, and K+ channels, to the bradycardia phenotype in CSIRKO mice remains to be determined. Therefore, we cannot exclude the involvement of changes in other pacemaker ion channels.
In summary, this study provided compelling evidence that insulin signaling is critical for maintaining a physiological heart rate. Therefore, restoring insulin signaling is a promising therapeutic strategy to prevent or improve the treatment of SND.
References
Boucher, J., Kleinridders, A. & Kahn, C. R. Insulin receptor signaling in normal and insulin- resistant states. Cold Spring Harb Perspect. Biol. 6, a009191 (2014).
Belfiore, A., Frasca, F., Pandini, G., Sciacca, L. & Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 30, 586–623 (2009).
Belke, D. D. et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Investig. 109, 629–639 (2002).
Shimizu, I. et al. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J. Clin. Investig. 120, 1506–1514 (2010).
Kim, J. et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 22, 2531–2543 (2008).
Ock, S. et al. Deletion of IGF-1 receptors in cardiomyocytes attenuates cardiac aging in male mice. Endocrinology 157, 336–345 (2016).
Riehle, C. et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J. Clin. Investig. 123, 5319–5333 (2013).
John, R. M. & Kumar, S. Sinus node and atrial arrhythmias. Circulation 133, 1892–1900 (2016).
Jensen, P. N. et al. Incidence of and risk factors for sick sinus syndrome in the general population. J. Am. Coll. Cardiol. 64, 531–538 (2014).
Alonso, A. et al. Association of sick sinus syndrome with incident cardiovascular disease and mortality: the Atherosclerosis Risk in Communities study and Cardiovascular Health Study. PLoS ONE 9, e109662 (2014).
Luo, M. et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Investig. 123, 1262–1274 (2013).
Soltysinska, E., Speerschneider, T., Winther, S. V. & Thomsen, M. B. Sinoatrial node dysfunction induces cardiac arrhythmias in diabetic mice. Cardiovasc. Diabetol. 13, 122 (2014).
Ock, S. et al. Conditional deletion of insulin receptor in thyrocytes does not affect thyroid structure and function. Endocr. J. 58, 1013–1019 (2011).
Ock, S. et al. IGF-1 receptor deficiency in thyrocytes impairs thyroid hormone secretion and completely inhibits TSH-stimulated goiter. FASEB J. 27, 4899–4908 (2013).
Ock, S. et al. IGF-1 protects against angiotensin II-induced cardiac fibrosis by targeting αSMA. Cell Death Dis. 12, 688 (2021).
Mezzano, V. et al. Desmosomal junctions are necessary for adult sinus node function. Cardiovasc. Res. 111, 274–286 (2016).
Ock, S. et al. Connexin43 and zonula occludens-1 are targets of Akt in cardiomyocytes that correlate with cardiac contractile dysfunction in Akt deficient hearts. Biochim. Biophys. Acta. Mol. Basis Dis. 1864, 1183–1191 (2018).
Gao, Z. et al. I(f) and SR Ca(2+) release both contribute to pacemaker activity in canine sinoatrial node cells. J. Mol. Cell. Cardiol. 49, 33–40 (2010).
Yeh, Y. H. et al. Funny current downregulation and sinus node dysfunction associated with atrial tachyarrhythmia: a molecular basis for tachycardia-bradycardia syndrome. Circulation 119, 1576–1585 (2009).
Mangoni, M. E. & Nargeot, J. Properties of the hyperpolarization-activated current (I(f)) in isolated mouse sino-atrial cells. Cardiovasc. Res. 52, 51–64 (2001).
O’Neill, B. T. et al. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 6, 294–306 (2007).
Ock, S. et al. Receptor activator of nuclear factor-κB ligand is a novel inducer of myocardial inflammation. Cardiovasc. Res. 94, 105–114 (2012).
Hoesl, E. et al. Tamoxifen-inducible gene deletion in the cardiac conduction system. J. Mol. Cell. Cardiol. 45, 62–69 (2008).
Mesirca, P. et al. Pharmacologic approach to sinoatrial node dysfunction. Annu. Rev. Pharmacol. Toxicol. 61, 757–778 (2021).
Marionneau, C. et al. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J. Physiol. 562, 223–234 (2005).
Fenske, S. et al. Sick sinus syndrome in HCN1-deficient mice. Circulation 128, 2585–2594 (2013).
Liu, J., Dobrzynski, H., Yanni, J., Boyett, M. R. & Lei, M. Organisation of the mouse sinoatrial node: structure and expression of HCN channels. Cardiovasc. Res. 73, 729–738 (2007).
Kuratomi, S. et al. The cardiac pacemaker-specific channel Hcn4 is a direct transcriptional target of MEF2. Cardiovasc. Res. 83, 682–687 (2009).
Scavone, A. et al. Embryonic stem cell-derived CD166+ precursors develop into fully functional sinoatrial-like cells. Circ. Res. 113, 389–398 (2013).
Abel, E. D. Insulin signaling in the heart. Am. J. Physiol. Endocrinol. Metab. 321, E130–E145 (2021).
Doetschman, T. & Azhar, M. Cardiac-specific inducible and conditional gene targeting in mice. Circ. Res. 110, 1498–1512 (2012).
MacDonald, E. A., Rose, R. A. & Quinn, T. A. Neurohumoral control of sinoatrial node activity and heart rate: insight from experimental models and findings from humans. Front. Physiol. 11, 170 (2020).
Cosentino, F. et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 41, 255–323 (2020).
Pop-Busui, R. et al. Effects of cardiac autonomic dysfunction on mortality risk in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Diabetes Care 33, 1578–1584 (2010).
Krishnaswamy, P. S. et al. Altered parasympathetic nervous system regulation of the sinoatrial node in Akita diabetic mice. J. Mol. Cell. Cardiol. 82, 125–135 (2015).
Kondo, H. et al. Interleukin-10 treatment attenuates sinus node dysfunction caused by streptozotocin-induced hyperglycaemia in mice. Cardiovasc. Res. 115, 57–70 (2019).
Nakajima, H. et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ. Res. 86, 571–579 (2000).
Zheng, M. et al. Hippo-Yap signaling maintains sinoatrial node homeostasis. Circulation 146, 1694–1711 (2022).
Baruscotti, M. et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. USA 108, 1705–1710 (2011).
Herrmann, S., Stieber, J., Stöckl, G., Hofmann, F. & Ludwig, A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 26, 4423–4432 (2007).
Acknowledgements
We thank Dr. Stefan Herrmann, Friedrich-Alexander Universität (Bavaria, Germany) for the HCN4- Ert2 Cre mouse line, Dr. Martin Holzenberger at INSERM (Paris, France) for the IGF-1Rfl/fl mouse line, and Dr. Farah Sheikh at the University of California-San Diego (San Diego, CA, USA) for the HCN4- Ert2;ROSA26TomRed mouse line. We also thank Dr. E. Dale Abel and Dr. Leonid V. Zingman at the University of Iowa (Iowa City, IA, USA) for technical assistance.
Funding
This work was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (2018R1D1A1B07046712 to S.O., 2020R1A2C1009647 to W.-S.L., and 2020R1A2C1010217 to J.K.).
Author information
Authors and Affiliations
Contributions
Conception and design: W.-S.L. and J.K. Acquisition of data: S.O., S.W.C., and S.H.C. Interpretation of data: S.O., S.J.P., W.-S.L., and J.K. Statistical analysis: H.K. Writing and review of the manuscript: W.-S.L. and J.K. Study supervision: W.-S.L. and J.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ock, S., Choi, S.W., Choi, S.H. et al. Insulin signaling is critical for sinoatrial node maintenance and function. Exp Mol Med 55, 965–973 (2023). https://doi.org/10.1038/s12276-023-00988-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-00988-0
