
Abstract
Formyl peptide receptors (FPRs), which are seven-membrane G-protein coupled receptors, recognize chemotactic signals to protect hosts from pathogenic infections and mediate inflammatory responses in the body. There are three isoforms of FPRs in humans—FPR1, FPR2, and FPR3—and they bind to N-formyl peptides, except FPR3, and to various endogenous agonists. Among FPR family members, FPR2 has a lower affinity for N-formyl peptides than FPR1 and binds with a wide range of endogenous or exogenous agonists. Thus, FPR2 is considered the most ambiguous member. Accumulating evidence has shown that FPR2 is involved in the host’s defense against bacterial infection and inflammation in liver diseases, such as nonalcoholic fatty liver disease, liver fibrosis, and liver cancer, suggesting the pathophysiological relevance of FPR2 to the liver. However, FPR2 has been shown to promote or suppress inflammation, depending on the type of FPR2-expressing cell and FPR2-bound ligands in the liver. Therefore, it is important to understand FPR2’s function per se and to elucidate the mechanism underlying immunomodulation initiated by ligand-activated FPR2 before suggesting FPR2 as a novel therapeutic agent for liver diseases. In this review, up-to-date knowledge of FPR2, with general information on the FPR family, is provided. We shed light on the dual action of FPR2 in the liver and discuss the hepatoprotective roles of FPR2 itself and FPR2 agonists in mediating anti-inflammatory responses.
Similar content being viewed by others

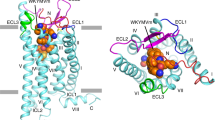

Introduction
Inflammation is a host defense response to the loss of tissue homeostasis1. However, excessive inflammation is injurious to the body and can become a causative factor for a variety of diseases, significantly contributing to their pathophysiology2. The liver is continually exposed to pathogenic antigens from dietary and commensal metabolites delivered from the gastrointestinal tract through the blood3. In the liver, hepatic innate immune cells, such as Kupffer cells, dendritic cells, and natural killer cells, function as the first line of defense by clearing invading pathogens or toxins4. When the liver is exposed to chronic and/or severe damage, injured or dying hepatocytes release different signals to stimulate hepatic immune cells and to recruit neutrophils and monocyte-derived macrophages into the liver5,6. An inflammatory response initially promotes the wound-healing process to recover liver homeostasis, whereas unresolved persistent inflammation aggravates liver damage, leading to chronic liver disease7. This is why chronic inflammation is regarded as a common feature of liver diseases, such as nonalcoholic fatty liver disease (NAFLD), liver fibrosis, hepatitis C virus infection, and liver cancer8. Thus, modulating inflammation is important in preventing the progression of chronic liver disease.
When a host is attacked by invading pathogens or other factors, pattern recognition receptors (PRRs) play an important role in regulating the innate immune response as a nonspecific “sensor” for pathogen-associated molecular patterns or damage-related molecular patterns9. A member of the PRR class, formyl peptide receptors (FPRs), were first identified by Shiffmann et al. as receptors that recognize N-formyl peptides derived from degraded bacteria or damaged mitochondria10. FPRs belong to the seven transmembrane chemoattractant G-protein coupled receptors and consist of three members: FPR1, FPR2, and FPR311. Among these, FPR2 is the most intriguing member because it creates a pro- or anti-inflammatory response according to its diverse ligands12. For instance, several polypeptides, such as serum amyloid A (SAA) and LL-37, elicit the proinflammatory action of Fpr2, while bioactive lipid molecules, including lipoxin A4 (LXA4) and resolvin D1 (RvD1), trigger an anti-inflammatory signaling cascade through FPR213. A double-faceted action of FPR2 has also been shown regarding its impact on disease progression. FPR2 activation promotes the production of proinflammatory cytokines, which aggravate atherosclerosis and diabetic retinopathy14,15. In contrast, FPR2 is involved in suppressing disease progression in several organs/tissues, such as the liver, lung, brain, and kidney, by exerting anti-inflammatory effects. Oldekamp et al.16 reported that Fpr2 deficiency caused severe inflammation and higher mortality in mice with pneumococcal meningitis. LXA4-induced FPR2 activation decreased collagen accumulation and the expression of inflammatory cytokines by blocking TGF-β/Smad signaling in radiation-induced pulmonary fibrosis17. In the liver, FPR2 is related to disease progression. It was shown that FPR2 protected against bacterial hepatic infection by triggering neutrophil recruitment18,19. RvD1 treatment decreased hepatocyte death and mitochondrial dysfunction and increased the engulfment of dead and dying cells by macrophages, alleviating ischemia/reperfusion-induced liver injury20,21. In liver cancer, LXA4 enhanced cancer cell apoptosis and reduced HCC metastasis22. Lee et al.23 also demonstrated that FPR2 protected the liver from lipotoxicity and suppressed NAFLD progression. Contrary to these findings, however, there are also conflicting results. Chen et al.24 reported that Fpr2 deficiency attenuated high-fat diet (HFD)-induced obesity, insulin resistance, and macrophage infiltration into the liver. Therefore, it is necessary to focus on research on FPR2 and the development of therapeutics targeting FPR2. In this review, we summarize the general information on FPRs in human and experimental animal models. In addition, we focus on the pathophysiological role of FPR2 in inflammation-related liver disease and suggest its therapeutic potential.
General information on the formyl peptide receptor family
FPR sequences are highly conserved in mammals, but their genetic characteristics, including their structure, gene number, and sequence, differ between humans and other species, such as rats, rabbits, guinea pigs, and mice25,26. Three FPRs have been identified in humans, namely, FPR1, FPR2 (referred to as formyl peptide receptor-like 1, FPRL1), and FPR3 (or FPRL2), and they are clustered on human chromosome 19q13.327. They exhibit significant homology with each other in their amino acid sequences. FPR1 shares 69% identity with FPR2 and 56% homology with FPR3, while FPR2 and FPR3 share 83% identity in humans13. FPRs also share overlapping physiological roles, such as host defense against pathogens, regulation of inflammation, and clearance of damaged cells28. The murine Fpr family is most widely studied. Compared to human FPRs, the mouse Fpr gene family includes eight identified members: Fpr1, Fpr2 (Fpr-rs2), Fpr-rs1, Fpr-rs3, Fpr-rs4, Fpr-rs5, Fpr-rs6, and Fpr-rs729. The entire mouse Fpr gene family is located within a cluster on mouse chromosome 17A3.229. Among these mouse Fprs, the Fpr-rs5 gene is not encoded as a functional receptor despite the absence of typical pseudogene characteristics. The products of five other mouse Fpr genes, that is, Fpr-rs1, Fpr-rs3, Fpr-rs4, Fpr-rs6, and Fpr-rs7, are known chemosensory vomeronasal receptors in olfactory sensory neurons30. Mouse Fpr1 is considered a counterpart of human FPR1, and mouse Fpr2 functionally corresponds to human FPR231. The mouse counterpart to human FPR3 has not been fully elucidated, but mouse Fpr-rs1 and Fpr2 partially replace the function of human FPR3 by interacting with F2L, a potent agonist of FPR332,33. FPR1 and FPR2 are predominantly expressed in the membranes of myeloid cells, including leukocytes, monocytes, macrophages, natural killer cells, and dendritic cells34,35. They are also identified in nonhematopoietic cells. FPR1 has been detected in smooth muscle cells, lens epithelial cells, fibroblasts, astrocytes, and hepatocytes, and FPR2 has been observed in astrocytoma cells, epithelial cells, hepatocytes, microvascular endothelial cells, and neuroblastoma cells28. While FPR1 and FPR2 are found in diverse nonimmune cell types, FPR3 is more restricted to immune cells, such as monocyte-derived macrophages, mature dendritic cells, and tissue-specific macrophages in the colon, skin, and lung, but not in the liver36.
Although FPRs recognize N-formylate peptides, they have different binding affinities with these peptides according to their composition, origin, and length. N-Formylated peptides containing a methionine residue at the 5′ terminus have at least a 100-fold more potent affinity for FPR1 than identical nonformylated peptides37. Most gram-negative bacteria-derived formylated peptides interact more potently with FPR1 than FPR228. For example, FPR1 binds to Escherichia coli (E. coli)-derived formyl-N-formyl-Met-Leu-Phe (fMLF) with an ~400-fold higher affinity than FPR238. Formylated peptide-bound FPR1 provides a signal to recruit immune cells into the lesions of bacterial infection or injured tissue39. Although most formylated peptides bind to FPR1 with a higher binding affinity, some long formylated peptides, such as alpha-type phenol-soluble modulins (PSMα) and mitocryptide-2 (MCT-2), prefer to bind to FPR2 rather than to FPR1. PSMα produced by Staphylococcus aureus binds to FPR2 and stimulates neutrophil recruitment, the production of proinflammatory cytokines, and bacterial phagocytosis40. MCT-2, which originates from mitochondrial DNA-encoding cytochrome b, increases intracellular Ca2+ flux in neutrophils by interacting with FPR241. In addition, FPR2 shows a more selective response to N-formylated peptides with a positive charge at the C-terminus, such as N-formyl-Met-Leu-Phe-Lys (fMLFK) and N-formyl-Met-Leu-Phe-Ile-Lys (fMLFIK), compared to N-formylated peptides with a negative charge on the C-terminus, such as N-formyl-Met-Leu-Phe-Glu (fMLFE) and fMLF42. FPR3 is nonresponsive to formylated peptides, despite a higher sequence identity between FPR1 and FPR333 (Table 1).
Double-edged action of FPR2 in the inflammatory response
Aside from formyl peptides, FPRs also recognize various types of endogenous lipids, nonformylated peptides, and proteins. Cathepsin G and F2L, which are endogenous ligands, are known to bind to FPR1 and FPR3, respectively, although few endogenous ligands for FPR1 and FPR3 have been identified. Cathepsin G, a neutrophil granule protein, has a weaker binding affinity to FPR1 than fMLF but enhances the chemotaxis of inflammatory cells by activating protein kinase Cζ, without inducing strong Ca influx in phagocytes43. FPR3, which exhibits a high affinity for endogenous F2L, is poorly understood32. Compared with FPR1 and FPR3, FPR2 has a broader range of ligands, including nonformylated peptides and proteins, lipid metabolites, and endogenous ligands28. Specifically, FPR2 could promote or inhibit the inflammatory response, depending on the type of ligand binding to it. For example, amyloidogenic peptide antimicrobial LL-37 stimulates a proinflammatory response44,45, and LXA4, RvD1, and annexin A1 (AnxA1) induce an anti-inflammatory response46,47,48. Herein, we have described the properties of FPR2 ligands and FPR2-mediated intracellular signaling involved in modulating the inflammatory response (Table 2).
Fpr2 as a proinflammatory mediator
FPR2 recognizes amyloidogenic peptides, such as serum amyloid A (SAA), β-amyloid peptide 42 (Aβ42), and prion peptide fragment 106-126 (PrP106-126), which are associated with chronic inflammation. SAA is mainly produced by hepatocytes and macrophages and binds to extracellular loops I and II of FPR249,50. SAA-stimulated FPR2 upregulates the amounts of proinflammatory cytokines and their receptors and promotes the survival of immune cells, enhancing the inflammatory response. He et al.44 reported that SAA bound to FPR2 activated nuclear factor-kappa B (NF-κB) signaling and increased the expression of proinflammatory IL-1, IL-6, IL-8, TNF-α and their receptors in myeloid-lineage phagocytes. It was shown that SAA interacting with FPR2 rapidly increased extracellular signal-regulated kinase (ERK) and AKT phosphorylation to induce myeloid cell leukemia-1 expression, a key regulator of neutrophil apoptosis, thus contributing to suppressed apoptosis in neutrophils51. SAA-activated FPR2 is also involved in facilitating the recruitment of monocytes by triggering ERK phosphorylation and NF-κB activation52. In addition, FPR2 senses neurotoxic Aβ42 and PrP106-126, which act as the main causative factors of Alzheimer’s disease by impacting the proinflammatory response. Aβ42 binds to extracellular loops and helices II, III, V, VI, and VII of FPR2 and is internalized in microglial cells53,54. In these cells, Aβ42 stimulates ERK phosphorylation and elevates the number of various cytokines and chemokines, including IL-1β, IL-6, interferon-γ, chemokine (C-C motif) ligand 2 (CCL2), chemokine (C-X-C motif) ligand (CXCL) 8, and CXCL1055. However, FPR2 also serves as a receptor for neuroprotective polypeptide humanin (HN), which acts as a competitive inhibitor of Aβ42 by occupying a region similar to the extracellular regions of FPR2 to which Aβ42 binds54,56. HN-bound FPR2 attenuates the fibrillary formation and cytotoxicity of Aβ4256. PrP106-126 interacting with FPR2 is internalized into glial cells, which are specialized macrophages in the central nervous system. Internalized PrP106-126 increases Ca2+ mobilization and the production of proinflammatory cytokines, such as IL-1β and IL-6, and induces the activation and migration of monocytic cells57. In addition, LL-37, which is an antimicrobial peptide derived from the breakdown of the neutrophil granule protein cathelicidin, is a proinflammatory ligand for FPR2. LL-37 bound with FPR2 induces the migration of neutrophils and eosinophils and stimulates differentiation of monocytes into the M1 phenotype of macrophages58. LL-37 also inhibits the apoptosis of neutrophils by blocking caspase-3 activity and upregulating anti-apoptotic Bcl-xL45.
Fpr2 as an anti-inflammatory mediator
FPR2 is also capable of inhibiting inflammation by interacting with anti-inflammatory agonists, such as LXA4, RvD1 and AnxA1. LXA4 is a lipid metabolite derived from ω-6 arachidonic acid that retains anti-inflammatory properties59. LXA4 has a high binding affinity for FPR2 at extracellular loop III and the seventh transmembrane domain49,60. LXA4 binding to FPR2 leads to a conformational change in FPR2 to block the further binding of proinflammatory ligands such as Aβ42 and SAA61. The interaction between LXA4 and FPR2 activates various intracellular signaling pathways to relieve inflammation and results in a decrease in excessive neutrophil infiltration and the expression of proinflammatory cytokines and an increase in phagocytosis of apoptotic cells62. LXA4 binding to FPR2 inactivates NF-κB signaling in neutrophils and inhibits the production of proinflammatory cytokines and their proliferation63,64. LXA4 interacting with FPR2 also reduces the activity of p38 mitogen-activated protein kinase (MAPK) and elevates the levels of nuclear factor erythroid 2-related factor 2 and peroxisome proliferator-activated receptor gamma (PPARγ) which are the factors alleviating the expression of proinflammatory genes47. Maderna et al.62 reported that LXA4 stimulated macrophages through FPR2 and increased phagocytosis of apoptotic cells, attenuating the inflammatory response. FPR2-expressing macrophages significantly eliminated apoptotic cells under the condition of LXA4 stimulation, whereas Fpr2-deficient macrophages rarely did, even though LXA4 treatment was given62.
RvD1, a derivative of docosahexaenoic acid, also plays an important role in ameliorating inflammation by interacting with FPR2, although the binding region of RvD1 on FPR2 remains unclear65. RvD1 increases apoptotic cell clearance by macrophages and decreases the migration of inflammatory cells through FPR246,66. Luo et al.46 showed that RvD1 treatment enhanced the phagocytosis of dying cells to stimulate TGF-β expression in macrophages and induced the recruitment of regulatory T cells to promote inflammation resolution in autoimmune neuritis. RvD1 binding to FPR2 inactivates the NF-κB pathway in a PPARγ-dependent manner and reduces the secretion of proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, in chorioamnionitis67. In an experimental animal model of acute lung injury, FPR2 activated by RvD1 suppressed IκBα degradation and NF-κB p65 nuclear translocation and downregulated the proinflammatory cytokines IL-1β, IL-6, and TNF-α68.
In addition to bioactive lipid mediators, AnxA1 and its derived peptide, Ac2-26, bind to FPR2 and are involved in the anti-inflammatory response. Although a high concentration of AnxA1 (100–200 μM) activates FPR1 and has a proinflammatory effect, a low concentration of AnxA1 (10–20 μM) binds to FPR2 and leads to an anti-inflammatory response69. In monocytes, AnxA1 binding to FPR2 stimulates the p38 MAPK/MAPKAPK/HSP27 signaling cascade and increases the production of the anti-inflammatory cytokine IL-1048. Pettetti et al.69 found that both Ac2-26 and AnxA1 interacting directly with FPR2 limited polymorphonuclear neutrophil recruitment to inflammatory loci and suppressed the expression of macrophage inflammatory protein-1α and prostaglandin E2, leading to a decrease in dermal inflammation in an air-pouch model. Ac2-26 was also shown to alleviate granulocyte infiltration and proinflammatory responses during pneumococcal meningitis by interacting with FPR270. Therefore, these ligands bound to FPR2 with anti-inflammatory action have been targeted to develop treatments for diseases with severe inflammation.
Focusing on FPR2 in the liver
Because chronic inflammation is considered a common feature of liver disease, the regulation of inflammation is an important therapeutic strategy for preventing disease progression8. According to the evidence obtained thus far, FPR2 induces opposing inflammatory responses, promoting or inhibiting inflammation, but it is clear that FPR2 is involved in the inflammatory response in the liver. Given the limited information on FPR2 in the liver and the fact that FPR2 ligands causing anti-inflammatory action have therapeutic potential for chronic liver disease, it is necessary to determine and understand the pathogenetic action of FPR2 in liver disease to determine its effective clinical approaches. Herein, we summarized and discussed the accumulated evidence for FPR2’s function in the liver to provide basic knowledge and suggest the research needed on FPR2 in the liver.
FPR2 influences neutrophil recruitment in hepatic infection by bacteria
As one of the FPR members, FPR2 also participates in the host’s immune response against bacterial infection by recognizing pathogen-derived danger signals or antibacterial host responses. Several papers have reported the essential role of FPR2 in host defense in cases of infected liver. When mice were infected with bacteria, Fpr2-deficient mice had a distinctly higher mortality rate than WT mice16,19,71. Liu et al.19 demonstrated that FPR2 activation facilitated H2O2 production in neutrophils to remove bacteria by increasing ERK1/2 phosphorylation. In addition, neutrophils expressing Fpr2 rapidly migrated into the infected liver by reacting with Listeria monocytogenes-released chemotactic signals, whereas neutrophils lacking Fpr2 failed to do so19. FPR2 also senses the E. coli-derived chemotactic peptide fMLF in murine neutrophils71. In addition, when neutrophils were treated with WRW4, an FPR2 antagonist, these cells lost their chemotactic ability for fMLF71. Sun et al.18 have reported that Fpr2-deficient mice had impaired infiltration to the infected site and were more susceptible to Streptococcus agalactiae (also known as group B streptococcus; GBS) infection than WT mice. In their research, FPR2 upregulated CXCL1 and CXCL2 during GBS and indirectly promoted neutrophil recruitment to the liver, rather than directly stimulating the migration of these cells into the liver. These findings clearly present a crucial role for FPR2 in neutrophil recruitment in response to bacterial infection.
FPR2 is associated with the inflammatory response in liver disease
FPR2 has anti-inflammatory properties and attenuates the progression of liver disease, as supported by the results obtained from experiments with Fpr2-depleted mice. Giebeler et al.72 reported that a ubiquitous deficiency of Fpr2 enhanced the infiltration of immune cells with increased levels of proinflammatory genes, such as IL-6, TNF-α, CXCL1, TLR2, and TLR4, in the livers of liposaccharide-injured mice. A higher number of TUNEL-positive apoptotic cells and a lower number of Ki-67-positive proliferating cells were also observed in mice with systemic Fpr2 deletion than in WT mice. Recently, Lee et al.23 demonstrated that FPR2 is involved in preventing the development and/or progression of NAFLD. NAFLD shows a different prevalence based on sex in that men have a higher prevalence than women before age 5073. However, its prevalence increases and becomes higher in women than in men after age 5074. The authors showed significantly higher expression of FPR2 in the hepatocytes and livers of WT female mice than in WT male mice and that this female-specific FPR2 in hepatocytes protected these cells from lipotoxicity, contributing to the resistance to NAFLD development and severity in female mice. These findings suggest that FPR2 is involved in regulating sex-specific responses to nonalcoholic injuries. Contrary to the results of this study, Chen et al.24 reported that ubiquitous Fpr2 deletion attenuated insulin resistance and hepatic steatosis in HFD-fed mice. It was also shown that Fpr2 deficiency reduced macrophage recruitment and the production of serum IL-6, TNF-α, and CCL2 by inhibiting M1 polarization of macrophages. The hepatoprotective effect caused by a lack of Fpr2 disappeared when FPR2 expression was induced in immune cells. In addition, myeloid-specific deletion of Fpr2 alleviated diet-induced liver damage, insulin resistance, and macrophage infiltration. These two papers suggest the contrasting effects of FPR2 in NAFLD and focus on different types of liver cells expressing FPR2: hepatocytes and immune cells. The NAFLD animal models employed in the two groups were also different. Compared to a HFD, a choline-deficient, L-amino acid-defined high-fat diet (CDAHFD) induces more excessive lipid accumulation in the liver by restricting lipid secretion due to choline deficiency, resulting in more severe liver damage, such as inflammation and fibrosis, during the same period of treatment as the HFD75. However, the CDAHFD rarely induces adiposity, body weight gain, or peripheral insulin sensitivity because the diet contains minimal methionine to maintain visceral fat mass, whereas a HFD leads to an increase in body weight and insulin resistance76,77. Although they used different animal models of NAFLD, Lee et al. investigated FPR2 action in a nonalcoholic steatohepatitis (NASH)-like model induced by CDAHFD feeding for a longer time compared to the HFD treatment period, and Chen’s group examined FPR2’s role in NAFL, a milder disease than NASH. Namely, the action of FPR2 seems to have been analyzed at different stages of disease progression. In addition, the expression of hepatic FPR2 hardly changed during either HFD or CDAHFD feeding. According to Lee’s group, hepatic FPR2 expression was significantly lower in males than in females, and FPR2 in hepatocytes was even absent in healthy male mice, whereas it was distinctly present in hepatocytes from healthy female mice. The hepatoprotective effect of FPR2 observed in female mice could occur in male mice by artificially inducing FPR2. These findings indicate that the hepatic function of FPR2 is more influential in female mice than in male mice and suggest the need to assess the presence or absence of Fpr2 expression in the liver before examining its role in the liver. Although Chen’s group found that ubiquitous deletion of Fpr2 reduced the amounts of inflammatory cytokines in serum, the levels of inflammatory markers in the liver itself did not differ significantly between WT and Fpr2-deficient male mice during HFD feeding. Rather, Fpr2 deficiency significantly downregulated inflammatory markers in WAT and muscle, but not liver. Given that crosstalk among liver, fat, and muscle plays an important role in the pathogenesis of NAFLD78, the mitigated inflammation in WAT and muscle caused by Fpr2 removal seems to lead to decreased inflammation in the livers of Fpr2-deficient male mice. However, the functions of FPR2 in hepatocytes and immune cells in the liver remain to be elucidated. Therefore, additional studies are needed to determine the cell-specific function, transcriptional regulation, and intracellular signal cascades impacted by FPR2 in hepatocytes and inflammatory cells.
Ligands binding to FPR2 exert hepatoprotective action
Although FPR2 is known to regulate disease progression as an immune modulator, based on data obtained from Fpr2 knockout animals, its function also depends on which ligands are bound and which cellular signaling systems are activated. Several studies have reported that the anti-inflammatory ligands of FPR2 alleviate liver disease (Fig. 1). Proresolving agonists increased hepatocyte survival in the damaged liver. RvD1 treatment reduced hepatocyte apoptosis and serum levels of inflammatory cytokines, such as TNF-α, IL-6, IL-10, and monocyte chemoattractant protein-1, in a D-galactosamine (D-GalN)-sensitized mouse endotoxin shock model79. Several papers have reported that RvD1 protects the liver against ischemia/reperfusion (IR)-induced injury. Supplementation with RvD1 reduced hepatocyte necrosis in IR-injured livers by activating sphingosine-1-phosphate, which promotes cell growth and survival and inhibits apoptosis20. RvD1 also improved mitochondrial dysfunction by increasing the protein expression associated with mitochondrial biogenesis in IR-damaged liver21. Kang et al.80 revealed that RvD1-activated FPR2 triggered the M2 polarization of macrophages and enhanced efferocytosis, ameliorating IR-induced liver damage. In addition, LXA4 treatment was shown to suppress the activities of caspase-3 and nuclear NF-κB in both hepatocytes and Kupffer cells, leading to a reduction in hepatocyte death in mice with acute liver failure caused by D-GalN/LPS81.

Anti-inflammatory agonists have therapeutic potential for chronic liver disease by activating FPR2. In ischemia/reperfusion (IR)-induced liver, resolvin D1 (RvD1) treatment improves sphingosine-1-phosphate (S-1-P) activity and mitochondrial dysfunction and reduces hepatocyte apoptosis. Lipoxin A4 (LXA4) also inhibits hepatocyte apoptosis by suppressing caspase-3 action in the damaged liver. FPR2 bound with these anti-inflammatory ligands alleviates lipotoxic stress in hepatocytes. Annexin A1 increases insulin receptor substrate 1 signaling and decreases hepatic lipid contents. RvD1 binding to FPR2 lowers hepatic lipid accumulation by downregulating peroxisome proliferator-activated receptor gamma (PPARγ). In immune cells, both LXA4 and RvD1 reduce inflammation by interacting with FPR2. RvD1 treatment decreases the levels of M1 polarization-related genes such as cyclooxygenase-2 (COX-2), interleukin-1 beta (IL-1β), and C-C chemokine receptor type 7 (CXCL7) and increases the expression of the M2 polarization-related gene arginase 1 (ARG1). In liver cancer, RvD1 triggers the FPR2/ROS/FOXM1 signaling pathway and blocks the secretion of cartilage oligomeric matrix protein (COMP) in cancer-associated fibroblasts (CAFs), suppressing epithelial-mesenchymal transition (EMT) and the gain of stemness features in HCC cells. In addition, LXA4 alleviates the levels of integrin-linked kinase (ILK), hypoxia-inducible factor-1α (HIF-1α), and vascular endothelial growth factor (VEGF) and the phosphorylation of Akt and GSK3β and suppresses cancer cell proliferation, EMT and angiogenesis.
Recently, the action of these ligands binding to FPR2 has been proven in NAFLD. Börgeson et al.82 demonstrated that LXA4 attenuated HFD-induced liver injury by reducing hepatic triglyceride accumulation in mice. In obese mice with steatohepatitis, RvD1 treatment decreased the intrahepatic lipid content and insulin resistance by decreasing the expression of PPARγ and increasing the level of circulating adiponectin83. RvD1 also mitigated hepatic inflammation by downregulating M1 polarization markers, such as cyclooxygenase-2, IL-1β, and C-C chemokine receptor type 7, and upregulating the M2 marker arginase 1, thereby reinforcing the effect of calorie restriction83. Li et al.84 demonstrated that RvD1 alleviated NASH progression by decreasing the TLR4-MyD88‐mediated NF‐κB and MAPK signaling pathways and increasing the expression of nuclear factor erythroid-2-related factor 2, a transcription factor for the antioxidant genes heme oxygenase-1, nicotinamide-adenine dinucleotide phosphate, and quinone oxidoreductase-1. AnxA1 treatment attenuated the development of obesity and insulin resistance by improving insulin receptor substrate 1 signaling in HFD-fed mice85. It was also reported that a deficiency of endogenous AnxA1 enhanced the activation of liver macrophages and fibrosis in mice with NASH86. However, because a high concentration of AnxA1 could induce a proinflammatory response via FPR1 rather than FPR2, further study of the AnxA1-stimulated receptor type subsequent to the activated signaling pathway is necessary to determine the action of AnxA1 in NAFLD.
The antitumor action of FPR2 has been reported by a few papers that show that ligand-activated FPR2 inhibits both the proliferation of HCC cells and their acquisition of stemness features. In the tumor microenvironment, hepatic stellate cells (HSCs) serve as cancer-associated fibroblasts and release cartilage oligomeric matrix protein, enhancing the invasion and metastasis of HCC87. Sun et al.87 reported that RvD1 reduced HSC-derived COMP paracrine signaling through the FPR2/ROS/Forkhead Box M1 signaling pathway and inhibited the epithelial-mesenchymal transition (EMT) and stemness of HCC cells. LXA4 treatment downregulated both the expression of integrin-linked kinase expression and the phosphorylation of AKT and GSK3β and interrupted EMT, migration and metastasis of HCC in an in vitro model22. LXA4 treatment also increased the apoptosis of liver cancer cells and blocked their proliferation by effectively blocking proliferation signals from macrophages88. In H22 cells, a mouse hepatocellular carcinoma cell line, exogenous LXA4 exerted an antiangiogenic effect by suppressing the production of vascular endothelial growth factor and reducing hypoxia-inducible factor-1α levels89. FPR2 agonists have consistently been shown to suppress liver cancer progression, suggesting that FPR2 agonists may represent potential candidates for the treatment of liver cancer. However, data demonstrating the action of FPR2 agonists in preventing HCC progression are limited, and further study is required to support their role in HCC before targeting them as therapeutics for liver cancer.
Future perspectives and conclusion
FPR2 modulates inflammation, hepatocellular death, and lipid accumulation and inhibits the invasion and metastasis of liver cancer cells, contributing to controlling the progression of chronic liver disease. In the damaged liver, FPR2 is involved in the inflammatory response and promotes or suppresses disease progression. However, growing evidence indicates that FPR2 functions as an anti-inflammatory modulator to attenuate the progression of liver disease. Hence, effective synthetic agonists and analogs of proresolving FPR2 ligands, such as Trp-Lys-Tyr-Met-Val-D-Met (WKYMVm) and BML-111, have been developed that focus on the anti-inflammatory effect of FPR2. The synthetic peptide WKYMVm is a strong agonist for FPR2, with higher affinity than other FPRs, and has anti-fibrotic and regenerative effects in fibrotic liver90. Jun et al.90 found that WKYMVm treatment decreased the expression of fibrotic markers, α-smooth muscle actin, and type I collagen and increased the levels of angiogenetic factors, vascular endothelial growth factor (VEGF), and VEGF receptors in a bile duct ligation model. WKYMVm also improved hepatocyte proliferation through IL-6/GP130/STAT3 signaling. Recently, the same group reported that WKYMVm improves the proangiogenic, regenerative, and anti-fibrotic abilities of placenta-derived mesenchymal stem cells (PD-MSCs) via FPR2, and combined treatment of WKYMVm with PD-MSCs significantly reduces hepatic damage and improves hepatocyte proliferation in rats with cholestatic liver compared to PD-MSCs alone91. However, WKYMVm also binds to a receptor, namely, the hepatocyte growth factor receptor (HGFR), other than FPR2. Cattaneo et al.92 demonstrated that WKYMVm induced activation of HGFR and that activated HGFR phosphorylated the Y705 and S727 residues of STAT3 to facilitate nuclear translocation of phosphorylated STAT3, which acted as a transcription factor in human prostate epithelial cells. Jun et al. have shown that WKYMVm suppresses HSC activation and liver fibrosis but did not show whether HSCs express FPR2 in response to WKYMVm; this is still unclear. A few reports have shown that FPR2 is rarely expressed by HSCs23,93. It is possible that WKYMVm interacting with other receptors, such as HGFR expressed by HSCs, directly regulates HSC activation or binds to FPR2 expressed by other types of cells, which indirectly impacts HSC activation. Thus, it is necessary to first confirm the expression of FPR2 in HSCs, and further studies are needed to determine how WKYMVm influences HSC activation.
BML-111, an analog of LXA4, has been shown to reduce articular neutrophil accumulation through FPR2. El-Agamy et al.94 revealed that BML-111 treatment lowered the degree of hepatocellular necrosis, oxidative stress, and inflammation in acetaminophen-induced acute liver injury. BML-111 also suppressed HCC progression by inhibiting proliferation, migration, EMT, and metastasis22.
Growing evidence indicates that FPR2 exerts anti-inflammatory action and attenuates liver injury. However, a few studies have demonstrated the pathogenic role of FPR2 in accelerating the disease state. FPR2 was even shown to promote or inhibit NAFLD, depending on the types of cells expressing it. Because FPR2 has shown opposing effects on hepatocytes and inflammatory cells under lipotoxic conditions, it is important to study the cell type-specific role of FPR2 in the liver. In addition, it is first necessary to clarify FPR2 expression before defining its function in the liver. Therefore, an understanding of the sophisticated mechanism of FPR2 activation may help to develop treatments that selectively promote the hepatoprotective effect of FPR2, which may be a promising strategy against liver disease.
References
Chen, L. et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9, 7204–7218 (2018).
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Kanneganti, T. D., Lamkanfi, M. & Amer, A. O. Innate immune pathways in host defense. Mediators Inflamm. 2012, 708972 (2012).
Liaskou, E., Wilson, D. V. & Oo, Y. H. Innate immune cells in liver inflammation. Mediators Inflamm. 2012, 949157 (2012).
Seki, S. et al. The liver as a crucial organ in the first line of host defense: the roles of Kupffer cells, natural killer (NK) cells and NK1.1 Ag+ T cells in T helper 1 immune responses. Immunol. Rev. 174, 35–46 (2000).
Czaja, A. J. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J. Gastroenterol. 20, 2515–2532 (2014).
Seki, E. & Schwabe, R. F. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 61, 1066–1079 (2015).
Del Campo, J. A., Gallego, P. & Grande, L. Role of inflammatory response in liver diseases: therapeutic strategies. World J. Hepatol. 10, 1–7 (2018).
Amarante-Mendes, G. P. et al. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 9, 2379 (2018).
Schiffmann, E., Corcoran, B. A. & Wahl, S. M. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc. Natl Acad. Sci. USA 72, 1059–1062 (1975).
Tian, C. et al. The G-protein coupled formyl peptide receptors and their role in the progression of digestive tract cancer. Technol. Cancer Res. Treat. 19, 1533033820973280 (2020).
Tylek, K. et al. Formyl peptide receptor 2, as an important target for ligands triggering the inflammatory response regulation: a link to brain pathology. Pharmacol. Rep. 73, 1004–1019 (2021).
Ye, R. D. et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 61, 119–161 (2009).
Petri, M. H. et al. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 105, 65–74 (2015).
Yu, Y. et al. The G-protein-coupled chemoattractant receptor Fpr2 exacerbates neuroglial dysfunction and angiogenesis in diabetic retinopathy. FASEB Bioadv. 2, 613–623 (2020).
Oldekamp, S. et al. Lack of formyl peptide receptor 1 and 2 leads to more severe inflammation and higher mortality in mice with of pneumococcal meningitis. Immunology 143, 447–461 (2014).
Kim, H. et al. LXA(4)-FPR2 signaling regulates radiation-induced pulmonary fibrosis via crosstalk with TGF-β/Smad signaling. Cell Death Dis. 11, 653 (2020).
Sun, Z. et al. Fpr2/CXCL1/2 controls rapid neutrophil infiltration to inhibit streptococcus agalactiae infection. Front. Immunol. 12, 786602 (2021).
Liu, M. et al. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2, 786 (2012).
Kang, J. W., Choi, H. S., Shin, J. K. & Lee, S. M. Resolvin D1 activates the sphingosine-1-phosphate signaling pathway in murine livers with ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 514, 1058–1065 (2019).
Kang, J. W., Choi, H. S. & Lee, S. M. Resolvin D1 attenuates liver ischaemia/reperfusion injury through modulating thioredoxin 2-mediated mitochondrial quality control. Br. J. Pharmacol. 175, 2441–2453 (2018).
Xu, F. et al. Lipoxin A(4) and its analog suppress hepatocarcinoma cell epithelial-mesenchymal transition, migration and metastasis via regulating integrin-linked kinase axis. Prostaglandins Other Lipid Mediat. 137, 9–19 (2018).
Lee, C. et al. Formyl peptide receptor 2 determines sex-specific differences in the progression of nonalcoholic fatty liver disease and steatohepatitis. Nat. Commun. 13, 578 (2022).
Chen, X. et al. Fpr2 deficiency alleviates diet-induced insulin resistance through reducing body weight gain and inhibiting inflammation mediated by macrophage chemotaxis and M1 polarization. Diabetes 68, 1130–1142 (2019).
Weiß, E. & Kretschmer, D. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol. 39, 815–829 (2018).
Panaro, M. A. et al. Formyl peptide receptors on immune and nonimmune cells: analysis of sequence conservation in FPR genes. Immunopharmacol. Immunotoxicol. 29, 243–269 (2007).
Migeotte, I., Communi, D. & Parmentier, M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 17, 501–519 (2006).
He, H. Q. & Ye, R. D. The formyl peptide receptors: diversity of ligands and mechanism for recognition. Molecules 22, 455 (2017).
Gao, J. L., Chen, H., Filie, J. D., Kozak, C. A. & Murphy, P. M. Differential expansion of the N-formylpeptide receptor gene cluster in human and mouse. Genomics 51, 270–276 (1998).
Hartt, J. K., Barish, G., Murphy, P. M. & Gao, J. L. N-formylpeptides induce two distinct concentration optima for mouse neutrophil chemotaxis by differential interaction with two N-formylpeptide receptor (FPR) subtypes. Molecular characterization of FPR2, a second mouse neutrophil FPR. J. Exp. Med. 190, 741–747 (1999).
Chen, K. et al. Regulation of inflammation by members of the formyl-peptide receptor family. J. Autoimmun. 85, 64–77 (2017).
Gao, J. L. et al. F2L, a peptide derived from heme-binding protein, chemoattracts mouse neutrophils by specifically activating Fpr2, the low-affinity N-formylpeptide receptor. J. Immunol. 178, 1450–1456 (2007).
Migeotte, I. et al. Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J. Exp. Med. 201, 83–93 (2005).
Yang, D., Chen, Q., Le, Y., Wang, J. M. & Oppenheim, J. J. Differential regulation of formyl peptide receptor-like 1 expression during the differentiation of monocytes to dendritic cells and macrophages. J. Immunol. 166, 4092–4098 (2001).
Kim, S. D. et al. Functional expression of formyl peptide receptor family in human NK cells. J. Immunol. 183, 5511–5517 (2009).
Devosse, T. et al. Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J. Immunol. 182, 4974–4984 (2009).
Prossnitz, E. R. & Ye, R. D. The N-formyl peptide receptor: a model for the study of chemoattractant receptor structure and function. Pharmacol. Ther. 74, 73–102 (1997).
Ye, R. D., Cavanagh, S. L., Quehenberger, O., Prossnitz, E. R. & Cochrane, C. G. Isolation of a cDNA that encodes a novel granulocyte N-formyl peptide receptor. Biochem. Biophys. Res. Commun. 184, 582–589 (1992).
Park, Y. J. et al. Promotion of formyl peptide receptor 1-mediated neutrophil chemotactic migration by antimicrobial peptides isolated from the centipede Scolopendra subspinipes mutilans. BMB Rep. 49, 520–525 (2016).
Kretschmer, D. et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 7, 463–473 (2010).
Seki, T., Fukamizu, A., Kiso, Y. & Mukai, H. Mitocryptide-2, a neutrophil-activating cryptide, is a specific endogenous agonist for formyl-peptide receptor-like 1. Biochem. Biophys. Res. Commun. 404, 482–487 (2011).
He, H. Q., Troksa, E. L., Caltabiano, G., Pardo, L. & Ye, R. D. Structural determinants for the interaction of formyl peptide receptor 2 with peptide ligands. J. Biol. Chem. 289, 2295–2306 (2014).
Sun, R. et al. Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J. Immunol. 173, 428–436 (2004).
He, R., Sang, H. & Ye, R. D. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 101, 1572–1581 (2003).
Nagaoka, I., Tamura, H. & Hirata, M. An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptor-like 1 and P2X7. J. Immunol. 176, 3044–3052 (2006).
Luo, B. et al. Resolvin D1 programs inflammation resolution by increasing TGF-β expression induced by dying cell clearance in experimental autoimmune neuritis. J. Neurosci. 36, 9590–9603 (2016).
Prieto, P. et al. Lipoxin A4 impairment of apoptotic signaling in macrophages: implication of the PI3K/Akt and the ERK/Nrf-2 defense pathways. Cell Death Differ. 17, 1179–1188 (2010).
Cooray, S. N. et al. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl Acad. Sci. USA 110, 18232–18237 (2013).
Bena, S., Brancaleone, V., Wang, J. M., Perretti, M. & Flower, R. J. Annexin A1 interaction with the FPR2/ALX receptor: identification of distinct domains and downstream associated signaling. J. Biol. Chem. 287, 24690–24697 (2012).
Ye, R. D. & Sun, L. Emerging functions of serum amyloid A in inflammation. J. Leukoc. Biol. 98, 923–929 (2015).
El Kebir, D., Gjorstrup, P. & Filep, J. G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl Acad. Sci. USA 109, 14983–14988 (2012).
Lee, H. Y. et al. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem. Biophys. Res. Commun. 330, 989–998 (2005).
Tiffany, H. L. et al. Amyloid-beta induces chemotaxis and oxidant stress by acting at formylpeptide receptor 2, a G protein-coupled receptor expressed in phagocytes and brain. J. Biol. Chem. 276, 23645–23652 (2001).
Zhu, Y. et al. Structural basis of FPR2 in recognition of Aβ(42) and neuroprotection by humanin. Nat. Commun. 13, 1775 (2022).
Gouwens, L. K., Makoni, N. J., Rogers, V. A. & Nichols, M. R. Amyloid-β42 protofibrils are internalized by microglia more extensively than monomers. Brain Res. 1648, 485–495 (2016).
Ying, G. et al. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 172, 7078–7085 (2004).
Le, Y. et al. The neurotoxic prion peptide fragment PrP(106-126) is a chemotactic agonist for the G protein-coupled receptor formyl peptide receptor-like 1. J. Immunol. 166, 1448–1451 (2001).
van der Does, A. M. et al. LL-37 directs macrophage differentiation toward macrophages with a proinflammatory signature. J. Immunol. 185, 1442–1449 (2010).
Serhan, C. N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fat. Acids 73, 141–162 (2005).
Chiang, N., Fierro, I. M., Gronert, K. & Serhan, C. N. Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 191, 1197–1208 (2000).
Dufton, N. & Perretti, M. Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacol. Ther. 127, 175–188 (2010).
Maderna, P. et al. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 24, 4240–4249 (2010).
Serhan, C. N., Chiang, N. & Van Dyke, T. E. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361 (2008).
Serhan, C. N. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 25, 101–137 (2007).
Serhan, C. N. et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037 (2002).
Krishnamoorthy, S. et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl Acad. Sci. USA 107, 1660–1665 (2010).
Li, A. et al. Effect of RvD1/FPR2 on inflammatory response in chorioamnionitis. J. Cell. Mol. Med. 24, 13397–13407 (2020).
Liao, Z. et al. Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARγ/NF-κB pathway. Respir. Res. 13, 110 (2012).
Perretti, M. et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 8, 1296–1302 (2002).
Rüger, M. et al. The formyl peptide receptor agonist Ac2-26 alleviates neuroinflammation in a mouse model of pneumococcal meningitis. J. Neuroinflammation 17, 325 (2020).
Zhang, M. et al. A critical role of formyl peptide receptors in host defense against Escherichia coli. J. Immunol. 204, 2464–2473 (2020).
Giebeler, A. et al. Deficiency of formyl peptide receptor 1 and 2 is associated with increased inflammation and enhanced liver injury after LPS-stimulation. PLoS ONE 9, e100522 (2014).
Vilar-Gomez, E. et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology 155, 443–457.e417 (2018).
Lonardo, A. et al. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology 70, 1457–1469 (2019).
Sugasawa, T. et al. One week of CDAHFD induces steatohepatitis and mitochondrial dysfunction with oxidative stress in liver. Int. J. Mol. Sci. 22, 5851 (2021).
Matsumoto, M. et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 94, 93–103 (2013).
Gupte, A. A. et al. High-fat feeding-induced hyperinsulinemia increases cardiac glucose uptake and mitochondrial function despite peripheral insulin resistance. Endocrinology 154, 2650–2662 (2013).
Zhang, X., Ji, X., Wang, Q. & Li, J. Z. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Protein Cell 9, 164–177 (2018).
Murakami, T., Suzuki, K., Tamura, H. & Nagaoka, I. Suppressive action of resolvin D1 on the production and release of septic mediators in D-galactosamine-sensitized endotoxin shock mice. Exp. Ther. Med. 2, 57–61 (2011).
Kang, J. W. & Lee, S. M. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim. Biophys. Acta 1861, 1025–1035 (2016).
Jiang, X. et al. Lipoxin A4 exerts protective effects against experimental acute liver failure by inhibiting the NF-κB pathway. Int. J. Mol. Med. 37, 773–780 (2016).
Börgeson, E. et al. Lipoxin A4 attenuates obesity-induced adipose inflammation and associated liver and kidney disease. Cell Metab. 22, 125–137 (2015).
Rius, B. et al. Resolvin D1 primes the resolution process initiated by calorie restriction in obesity-induced steatohepatitis. FASEB J. 28, 836–848 (2014).
Li, J. et al. Resolvin D1 mitigates non-alcoholic steatohepatitis by suppressing the TLR4-MyD88-mediated NF-κB and MAPK pathways and activating the Nrf2 pathway in mice. Int. Immunopharmacol. 88, 106961 (2020).
Purvis, G. S. D. et al. Identification of AnnexinA1 as an endogenous regulator of RhoA, and its role in the pathophysiology and experimental therapy of type-2 diabetes. Front. Immunol. 10, 571 (2019).
Locatelli, I. et al. Endogenous annexin A1 is a novel protective determinant in nonalcoholic steatohepatitis in mice. Hepatology 60, 531–544 (2014).
Li, Q. et al. HSCs-derived COMP drives hepatocellular carcinoma progression by activating MEK/ERK and PI3K/AKT signaling pathways. J. Exp. Clin. Cancer Res. 37, 231 (2018).
Hao, H. et al. Lipoxin A4 and its analog suppress hepatocellular carcinoma via remodeling tumor microenvironment. Cancer Lett. 309, 85–94 (2011).
Chen, Y. et al. Lipoxin A4 and its analogue suppress the tumor growth of transplanted H22 in mice: the role of antiangiogenesis. Mol. Cancer Ther. 9, 2164–2174 (2010).
Jun, J. H. et al. Formyl peptide receptor 2 alleviates hepatic fibrosis in liver cirrhosis by vascular remodeling. Int. J. Mol. Sci. 22, 2107 (2021).
Jun, J. H. et al. Combination therapy of placenta-derived mesenchymal stem cells with WKYMVm promotes hepatic function in a rat model with hepatic disease via vascular remodeling. Cells 11, 232 (2022).
Cattaneo, F., Parisi, M. & Ammendola, R. WKYMVm-induced cross-talk between FPR2 and HGF receptor in human prostate epithelial cell line PNT1A. FEBS Lett. 587, 1536–1542 (2013).
Getachew, A. et al. SAA1/TLR2 axis directs chemotactic migration of hepatic stellate cells responding to injury. iScience 24, 102483 (2021).
El-Agamy, D. S., Makled, M. N. & Gamil, N. M. Protective effects of BML-111 against acetaminophen-induced acute liver injury in mice. J. Physiol. Biochem. 70, 141–149 (2014).
Acknowledgements
This research was supported by Y.J. and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) to C.L. (No. 2022R1C1C2008830).
Author information
Authors and Affiliations
Contributions
C.L. and J.H. contributed to this paper with a literature review and drafting the manuscript, and Y.J. contributed to this paper with conception, review, drafting and editing the manuscript, and supervision. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, C., Han, J. & Jung, Y. Formyl peptide receptor 2 is an emerging modulator of inflammation in the liver. Exp Mol Med 55, 325–332 (2023). https://doi.org/10.1038/s12276-023-00941-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-00941-1
