
Abstract
Intervertebral disc degeneration (IDD) is a common degenerative musculoskeletal disorder and is recognized as a major contributor to discogenic lower back pain. However, the molecular mechanisms underlying IDD remain unclear, and therapeutic strategies for IDD are currently limited. Oxidative stress plays pivotal roles in the pathogenesis and progression of many age-related diseases in humans, including IDD. Nuclear factor E2-related factor 2 (Nrf2) is a master antioxidant transcription factor that protects cells against oxidative stress damage. Nrf2 is negatively modulated by Kelch-like ECH-associated protein 1 (Keap1) and exerts important effects on IDD progression. Accumulating evidence has revealed that Nrf2 can facilitate the transcription of downstream antioxidant genes in disc cells by binding to antioxidant response elements (AREs) in promoter regions, including heme oxygenase-1 (HO-1), glutathione (GSH), superoxide dismutase (SOD), catalase (CAT), and NADPH quinone dehydrogenase 1 (NQO1). The Nrf2 antioxidant defense system regulates cell apoptosis, senescence, extracellular matrix (ECM) metabolism, the inflammatory response of the nucleus pulposus (NP), and calcification of the cartilaginous endplates (EP) in IDD. In this review, we aim to discuss the current knowledge on the roles of Nrf2 in IDD systematically.
Similar content being viewed by others

Introduction
Lower back pain (LBP) has become a profoundly debilitating and increasingly prevalent disorder, causing a heavy socioeconomic burden worldwide1. The leading cause of LBP is intervertebral disc degeneration (IDD)2. However, the pathogenesis of IDD is associated with multiple complex factors, including genetic, epigenetic, and environmental factors, and the knowledge about the molecular mechanisms underlying IDD remains elusive3,4. The clinical treatments for IDD are limited to surgery, pharmacological or other nonpharmacological interventions to relieve the symptoms, and more effective therapeutic strategies to address the underlying pathology are needed for this degenerative spine disorder5. Therefore, a better understanding of the molecular signaling involved in IDD has been a research focus, which may help to develop novel therapeutic targets for the successful treatment of IDD6,7,8,9.
Redox homeostasis is crucial for the physiological maintenance of many cellular processes, and dysregulation of redox homeostasis is closely associated with various pathological conditions affecting human health10. Oxidative stress is described as the disruption of redox homeostasis, which occurs when the balance between reactive oxygen species (ROS) production and the scavenging activity of the antioxidant system becomes dysregulated11. Excessive accumulation of ROS induces oxidative stress, which can cause damage to biological macromolecules such as carbohydrates, lipids, nucleic acids, and proteins, impairing the regular functional integrity of cells in the body12. Accumulating evidence has revealed the roles played by oxidative stress in the pathogenesis of various human diseases, especially age-related disorders such as degenerative musculoskeletal diseases13,14,15. Degenerated disks exhibit oxidative stress as well as increased oxidation product levels, contributing to the development of IDD16. Importantly, mounting evidence has revealed that therapies targeting oxidative stress might effectively alleviate or prevent IDD progression17.
Nuclear factor E2-related factor 2 (Nrf2) is a master endogenous antioxidant transcription factor that has been increasingly reported to play crucial roles in protecting cells against oxidative stress18. Physiologically, Nrf2 is critical for the expression of antioxidative genes, cytoprotective enzymes, and export transporters, which constitute an antioxidant defense system that maintains intracellular redox homeostasis19. The activation of Nrf2 signaling is negatively regulated by Kelch-like ECH-associated protein 1 (Keap1), which functions as a redox sensor for ROS and electrophiles20,21. Under resting conditions, the activity of Nrf2 is tightly controlled by Keap1, which mediates ubiquitination-dependent proteasomal degradation of Nrf2 in the cytoplasm. In the presence of oxidative stress, Keap1 undergoes a conformational change and releases Nrf2, which moves to the nucleus, resulting in the initiation of the transcription of multiple antioxidant genes22,23. Nrf2 signaling is considered a central hub that modulates redox homeostasis in cells, and aberrant Keap1-Nrf2 signaling is functionally involved in the pathology of many diseases24,25,26. Interestingly, increasing evidence has revealed the crucial roles played by the Nrf2 signaling pathway in protecting against IDD progression27,28,29. To our knowledge, no systematic review has yet summarized the involvement of Nrf2 in disc degeneration diseases. Therefore, in this review, we synthesize and evaluate the results from the available literature and comprehensively discuss the roles of the Nrf2 antioxidant defense system in IDD.
Pathophysiology of IDD and oxidative stress
Situated between the vertebral bones, each intervertebral disc (IVD) is made of fibro-cartilaginous tissues and is one of the most important structures of the spine. The IVD can distribute the axial compressive load transmitted from the vertebral bodies and enables physiological lateral and rotational flexibility of the spine30. Anatomically, the disc consists of three major parts: the hydrated gel-like nucleus pulposus (NP) in the center, elastic annulus fibrosus (AF) surrounding the NP, and cartilaginous endplates (EP) on the inferior and superior sides31. Oxidative stress, compressive overload, nutrient stress, enhanced inflammation, and other factors can act on these parts and stimulate aberrant cellular responses and progressive structural deficiency, leading to disc degeneration32. IDD is characterized by a loss of centrally situated NP cells, which are replaced with cells with a fibroblast-like phenotype33. Another typical pathological change in disc degeneration is accelerated extracellular matrix (ECM) degradation, such as decreased deposition of type II collagen (Col II) and aggrecan, which is caused by imbalanced anabolism and catabolism34. Additionally, cellular senescence and programmed cell death induced by inflammatory responses or other factors in the disc significantly contribute to the pathological changes during the complicated process of IDD9.
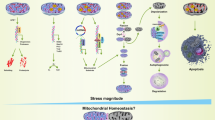
Oxidative stress is a critical mediator in the initiation and progression of IDD. Oxidative stress occurs when the balance between ROS production and the scavenging activity of the antioxidant defense system is disrupted11. Excessive ROS accumulation can induce oxidative stress, which causes damage to the integrity and regular function of cells35. Accumulating evidence has suggested that oxidative stress exerts significant effects on cell fate and function and is closely related to cell viability, senescence, programmed cell death, matrix metabolism, and signaling network transduction of disc cells within the IVD27,36,37,38. Previous studies have reported that aged and degenerated disks exhibit decreased antioxidant activity and elevated concentrations of oxidation products during IDD development16,17. Excessive ROS accumulation and dysfunction of the antioxidant defense system induce cell apoptosis and senescence and trigger inflammatory responses in disc NP cells, accelerating IDD progression39 (Fig. 1). Redox homeostasis in the disc also plays a crucial role in the ECM anabolism and catabolism balance, and oxidative stress has been found to promote ECM degradation by interacting with various important signaling pathways in NP cells, including NF-κB signaling, p38/MAPK signaling, and the Nrf2/ARE signaling pathway17,40,41. Moreover, the annulus fibrosus is a crucial part of the IVD, and oxidative stress is involved in the cell senescence, apoptosis, and ferroptosis of disc AF cells in the pathogenesis of IDD37,42,43,44. Disc EP degeneration is another critical contributor to IDD initiation because it hinders the nutrient supply to the NP and leads to disrupted disc homeostasis. It has been demonstrated that oxidative stress can induce autophagy, apoptosis, and calcification of endplate chondrocytes to modulate the EP degeneration process38,45,46,47. Therefore, elucidating the key molecular mechanisms of oxidative stress in the disc might lead to effective therapeutic strategies for IDD.

Excessive ROS accumulation exerts important effects on the three major types of IVD cells. Oxidative stress induces autophagy, apoptosis, and calcification of EP chondrocytes, while autophagy can act as a protective response to oxidative damage. Oxidative stress promotes cell apoptosis, senescence, ECM degradation, and inflammation response of disc NP cells. Oxidative stress induces cellular senescence, apoptosis, and ferroptosis in disc AF cells.
Nrf2 mediated antioxidant defense in IDD
Nuclear factor E2-related factor 2 (Nrf2), also known as nuclear factor erythroid 2-like 2 (NFE2L2), is a master antioxidant transcription factor encoded by the NFE2L2 gene in humans18. The Nrf2 protein is composed of approximately 605 amino acid residues and possesses seven highly conserved domains, namely, Neh1 to Neh7. Specifically, the Neh2 domain in Nrf2 participates in binding with the Keap1 homodimer and the degradation of Nrf248. The Neh2 domain contains two conserved motifs, DLG and ETGE, with an intervening sequence possessing seven lysine residues that can be ubiquitinated. DLG and ETGE are both associated with the interaction between Nrf2 and Keap1 homodimer. Physiologically, the activation of Nrf2 is regulated by Keap1, which functions as a cysteine-rich oxidative stress sensor. Keap1 is a substrate adaptor protein for the Cullin3 (Cul3)-containing E3 ubiquitin (Ub) ligase complex and is a cytosolic protein that negatively modulates Nrf2 activity49. Structurally, the Keap1 peptide is composed of 624 amino acid residues and possesses five functional regions, namely, the N-terminal region (NTR), intervening region (IVR), Broad complex Tramtrack and Bric-a-Brac (BTB) domain, double glycine repeat (DGR) domain and C-terminal region (CTR). The BTB domain is associated with the formation of the Keap1 homodimer, and the DGR and CTR domains (collectively known as the DC region) are involved in the interaction of Keap1 with Nrf250. The ubiquitin-proteasome system (UPS) is responsible for protein quality control and degradation and plays key roles in the maintenance of intracellular protein homeostasis51. Under unstressed conditions, Keap1 can bind to Nrf2 and target Nrf2 for ubiquitination and subsequent degradation by the proteasome. However, when ROS levels in cells are increased, the cysteine residues of Keap1 are covalently modified, and Keap1 undergoes a conformational change, resulting in blocked ubiquitination of Nrf2 and accumulation of newly synthesized Nrf252. Then, Nrf2 is released into the nucleus, where it forms a heterodimer with small musculoaponeurotic fibrosarcoma (Maf) proteins53. Subsequently, Nrf2-Maf binds to the antioxidant response element (ARE) in DNA to promote the transcription of multiple downstream antioxidant genes, including heme oxygenase-1 (HO-1), glutathione (GSH), superoxide dismutase (SOD), catalase (CAT), and NADPH quinone dehydrogenase 1 (NQO1)48.
IDD is one of the most common age-related degenerative musculoskeletal disorders. As mentioned above, the pathogenesis of IDD is closely associated with oxidative stress. Nrf2 is a crucial transcription factor that can modulate the cellular oxidative stress response. An increasing number of studies have revealed the important roles played by the Nrf2 antioxidant defense system in preventing IDD progression. Multiple antioxidants, including ulinastatin, dimethyl fumarate, and cyanidin-3-glucoside, have been reported to alleviate oxidative stress in disc NP cells by promoting the activity of the Nrf2-mediated HO-1 signaling pathway54,55,56,57. Dimethyl fumarate has also been demonstrated to activate Nrf2 to promote the production of GSH in NP cells, which is one of the most important ROS scavengers55. Another study reported that Nrf2/HO-1 signaling activated by moracin dramatically promoted the expression levels of SOD and CAT in NP cells induced by LPS challenge58. Acacetin and wogonin were also reported to activate the Nrf2 pathway to upregulate the expression of important antioxidant proteins, including HO-1, SOD, and NQO1, to ameliorate IDD progression59,60. Interestingly, it has been reported that activating autophagy promoted Nrf2 signaling to upregulate the expression of antioxidant proteins, including SOD1 and SOD2, and thus protect cartilage endplate stem cells against calcification and ECM degradation during IDD61. In summary, activating Nrf2 signaling facilitated the transcription of downstream antioxidant genes, including HO-1, GSH, SOD, CAT, and NQO1, to defend against oxidative stress in disc cells. The molecular mechanism of the Nrf2 antioxidant pathway is indicated in Fig. 2.

In unstressed conditions, Keap1 binds to the Cul3-containing E3 ubiquitin ligase complex, and two molecules of Keap1 form a homomeric dimer. The Keap1 complex binds to Nrf2 for the ubiquitination and subsequent degradation of Nrf2 by the proteasome. Under conditions of oxidative stress, Keap1 undergoes a conformational change, which leads to blocked ubiquitination of Nrf2 and accumulation of newly synthesized Nrf2. Subsequently, free Nrf2 is translocated to the nucleus, where it forms a heterodimer with small Maf proteins. Then, Nrf2-Maf interacts with the ARE in the promoter regions of DNA to promote the transcription of multiple targeted antioxidant genes, including HO-1, GSH, SOD, CAT, and NQO1. Activating Nrf2 signaling protects against oxidative stress in disc cells to alleviate IDD.
Therapeutic potential of targeting Nrf2 signaling in IDD treatment
Targeting Nrf2 to alleviate apoptosis of NP cells
The highly hydrated NP is the structural and functional center of a disc. Dysregulated NP cell apoptosis causes damage to the normal metabolism in the NP, which disrupts the normal structure and physiological function of the disc and is considered a key contributor to IDD pathogenesis. It has been demonstrated that the apoptosis ratio of NP cells is more than 50% in human degenerative disks, and preventing or alleviating apoptosis of NP cells is a potential effective therapy to treat disc degeneration32,62. Accumulating evidence has reported antiapoptotic roles played by Nrf2 activation in various types of human cells63,64,65. Unexpectedly, targeting Nrf2 signaling regulated the apoptosis of NP cells during IDD progression. Long noncoding RNAs (lncRNAs) constitute a common and diverse class of noncoding RNAs (ncRNAs) without protein-coding capacity66. Recently, Kang et al.27 reported that lncRNA ANPODRT activated Nrf2 signaling to inhibit oxidative stress and apoptosis in human NP cells. Mechanistically, the lncRNA ANPODRT facilitated Nrf2 accumulation and nuclear translocation to activate downstream target genes by disrupting the Keap1-Nrf2 interaction. Moreover, Nrf2 knockdown obliterated the antioxidative and antiapoptotic effects of the lncRNA ANPODRT, indicating that Nrf2 activation is required for the lncRNA ANPODRT to exert protective effects on NP cells. MicroRNAs (miRNAs) comprise another important and large class of short-chain noncoding RNAs that regulate downstream genes by targeting the 3’ untranslated region (3’UTR) posttranscriptionally. A study by Xu et al.67 revealed that a miRNA termed miR-141-3p, which was enriched in platelet-rich plasma (PRP)-derived exosomes, activated Keap1-Nrf2 signaling to reverse the cell apoptosis, pyroptosis, and inflammatory response of NP cells stimulated by H2O2. Mechanistically, miR-141-3p interacted with the 3’UTR of Keap1 mRNA to induce its degradation, thus leading to Nrf2 translocation to the nucleus.
More recently, Hu et al.28 reported the critical roles played the Nrf2 agonist tert-butylhydroquinone (TBHQ) in retarding NP cell apoptosis. The results showed that TBHQ rescued TBHP-induced apoptosis and oxidative stress by promoting Nrf2 expression and translocation to the nucleus. Mechanistically, TBHQ resisted oxidative stress by inducing Nrf2 activity and increasing the Sirt3 expression level to maintain mitochondrial homeostasis and enhance mitochondrial autophagy. Furthermore, the authors validated the therapeutic function and mechanism of TBHQ in a rat tail disc degeneration model in vivo. Mitoquinone (MitoQ) is a known mitochondria‐targeted antioxidant that has shown protective effects in various oxidative damage‐related diseases68. It has been suggested that MitoQ alleviates sustained mitochondrial dysfunction, oxidative stress, and apoptosis of NP cells by stimulating the Nrf2 antioxidant pathway in vitro and ex vivo69. Luo et al.54 found that an anti-inflammatory acidic protein extracted from human urine, ulinastatin, ameliorated the apoptosis of human NP cells by activating the Nrf-2/HO-1 signaling pathway and suppressing the NF-κB signaling pathway. Treatment with ulinastatin reversed the expression of the apoptosis-related proteins Bax and cleaved-caspase 3 and the antiapoptosis molecule Bcl-2. Moreover, increasing evidence has revealed other crucial molecular agents that mitigate excessive apoptosis of NP cells by interacting with Nrf2 signaling; these agents include sinapic acid70, plumbagin71, dimethyl fumarate55, luteoloside72, CDDO-ethyl amide73, cyanidin-3-glucoside57, kinsenoside74, lycopene75, and genistein76. Taken together, these studies revealed that activating Nrf2 signaling is a promising strategy to attenuate the apoptosis of NP cells and treat IDD.
Targeting Nrf2 to inhibit NP cell senescence
Numerous studies have reported that the impairment of NP cell function caused by senescence is a crucial contributor to the dehydration of NP tissue and, more importantly, to the initiation and progression of disc degeneration7,77,78. Senescent disc cells are metabolically active and can secrete various inflammatory cytokines, chemokines and matrix proteases, which collectively are known as the senescence-associated secretory phenotype (SASP)79,80. These inflammatory factors have been found to disrupt the balance between ECM anabolism and catabolism during IDD. Moreover, the SASP of senescent cells can induce senescence in neighboring nonsenescent cells by paracrine effects, which is referred to as paracrine senescence or secondary senescence79,81. The increase in inflammatory factor expression levels as a result of senescence causes a vicious cycle of degeneration and leads to further aggravation of IDD. Obviously, protecting disc NP cells against senescence is conducive to the amelioration of IDD.
In 2019, Cherif et al.82 reported that curcumin and o-vanillin exhibited significant senolytic activity in human degenerative disc NP cells. Curcumin, diferuloylmethane, has wide therapeutic benefits via its antioxidative and anti-inflammatory properties83, and its main metabolite, o-vanillin (2-hydroxy-3-methoxybenzaldehyde), shows similar effects84. This research revealed that curcumin and o-vanillin mediated senolytic effects via Nrf2 signaling and decreased SASP factor secretion by suppressing NF-κB pathway activation. A recent study by Shao et al.85 demonstrated that quercetin, a natural senolytic compound, activated Nrf2 signaling to suppress SASP factor expression and the senescence phenotype acquisition by NP cells. Mechanistically, quercetin suppressed IL-1β-induced activation of NF-κB pathway cascades by directly binding to the Keap1-Nrf2 complex. A previous work reported that kinsenoside activated the AKT-ERK1/2-Nrf2 signaling pathway in NP cells to attenuate IDD both in vitro and in vivo74. Kinsenoside is an active monomer extracted from Anoectochilus roxburghii, a traditional Chinese medicinal herb that exhibits diverse pharmacological actions. Importantly, kinsenoside has been shown to protect NP cells from apoptosis, senescence, and mitochondrial dysfunction in a Nrf2-dependent manner. Polydatin is a resveratrol glucoside that exerts extensive pharmacological antioxidative, anti‐inflammatory, and anti‐aging properties86. It has been reported that polydatin rescued mitochondrial dysfunction, suppressed senescence, and preserved ECM homeostasis in nucleus pulposus cells to attenuate IDD progression by promoting Nrf2 activity87. In summary, triggering Nrf2 activation to inhibit NP cell senescence is a potential therapeutic strategy for IDD.
Targeting Nrf2 to regulate ECM metabolism in NP cells
Physiologically, the ECM endows the IVD with elastic and weight-bearing properties, allowing it to absorb compression loads while maintaining flexibility in the spine88. The ECM is mainly composed of proteoglycans (mainly aggrecan) and Col II in disc NP tissues89. ECM metabolism is generally modulated by degradative enzymes, including matrix metalloproteinases (MMPs) and aggrecanases, and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs)90,91. Degenerative disks are biochemically characterized by an imbalanced ECM metabolism of NP cells, implicating attenuated anabolic activities and enhanced catabolic activities in the disc. In this process, excessive degradation of aggrecan and Col II leads to NP dehydration and resorption and a decline in the ability of the cells to resist mechanical loading, thus contributing to IDD progression92,93.
A recent study reported that lncRNA NEAT1 overexpression accelerated the ECM degradation of NP cells, while the Nrf2 activator TBHQ partially reversed the effects of the lncRNA NEAT1 on ECM metabolism41. These results suggested that the lncRNA NEAT1 ameliorated ECM degradation of NP cells by regulating Nrf2 signaling pathway activation. Dimethyl fumarate is a known agonist of Nrf2-responsive genes and has been applied in certain clinically degenerative diseases94. It has been revealed that dimethyl fumarate helped maintain the ECM metabolic balance of human NP cells, mainly by regulating the Nrf2/HO-1 signaling pathway55. As mentioned above, the anti-inflammatory acidic protein extracted from human urine, ulinastatin, also protected human NP cells from ECM degradation by activating the Nrf-2/HO-1 signaling pathway and suppressing the NF-κB signaling pathway54. Treatment with either curcumin or o-vanillin increased the proteoglycan and type II collagen content and inhibited MMP3 and MMP13 expression in human disc cells. Further experiments suggested that curcumin and o-vanillin promoted ECM synthesis in IVD, which was mediated by the Nrf2 and NF-κB pathways82. Furthermore, it has been reported that some other important biologically active components also regulated ECM metabolism in NP cells by targeting Nrf2 signaling; these compounds included cardamonin95, sinapic acid70, luteoloside72, cyanidin-3-glucoside57, moracin58, acacetin59, wogonin60, lycopene75, genistein76, and polydatin87. Altogether, these results revealed that targeting Nrf2 signaling to alleviate ECM degradation of NP cells is a potential therapeutic strategy for IDD.
Targeting Nrf2 to regulate the inflammatory response in NP cells
In the initiation and progression of IDD, inflammation is widely acknowledged as a major characteristic feature96,97. Accumulating evidence has demonstrated that excessive inflammatory responses can significantly affect the normal function of NP cells and thus contribute to IDD development4,32. Dysregulated expression of proinflammatory cytokines such as interleukin (IL)-1, IL-6, IL-17, and tumor necrosis factor (TNF)-α has been observed in degenerated disc NP tissues and has been involved in the inflammatory response during IDD98. These proinflammatory cytokines also play critical roles in the pathophysiological processes of IDD, including NP cell apoptosis, senescence, ECM remodeling, neovascularization, and oxidative stress4,32,99. Therefore, regulating the inflammatory microenvironment in NP cells is essential for IDD treatment.
Nrf2 is widely involved in the modulation of the inflammatory response in NP cells. As mentioned above, the study by Xu et al.67 revealed that exosomal miR-141-3p activated Keap1-Nrf2 signaling to regulate the inflammatory response of NP cells stimulated by H2O2. Mechanistically, miR-141-3p directly interacted with the 3′UTR of Keap1 mRNA, causing Keap1 degradation, resulting in Nrf2 translocation to the nucleus, and thus inhibiting proinflammatory cytokine (IL-1β, IL-18, TGF-β, and IL-6) production and secretion by NP cells. It has been reported that the anti-inflammatory acidic protein extracted from human urine, ulinastatin, also dramatically suppressed the expression levels of proinflammatory mediators in human NP cells, including IL-6, TNF-α, iNOS, and COX-2, by activating the Nrf-2/HO-1 signaling pathway and suppressing the NF-κB signaling pathway54. Interestingly, studies have shown that cardamonin95, sinapic acid70, and plumbagin71 protected NP cells against inflammation by modulating Nrf2/NF-κB axis activation. The known agonist for the Nrf2-responsive gene dimethyl fumarate has been found to ameliorate NP cell inflammation by promoting the activity of the Nrf2/HO-1 signaling pathway in IDD55,56. Moreover, evidence has suggested that some other biologically active components, including moracin58, acacetin59, and wogonin60, regulated the inflammatory response in NP cells by regulating the Nrf2 signaling pathway. Collectively, these data revealed that targeting Nrf2 signaling to regulate the inflammatory response in NP cells may be a promising therapeutic strategy for IDD.
Targeting Nrf2 to alleviate degeneration and calcification of EP
The human IVD has large vascular channels passing through the cartilaginous endplates at birth. With increasing age, however, these vessels recede, leaving the disc with little direct vascular supply100. The IVD becomes the largest avascular organ of the body in adulthood. The cartilaginous endplates that attach the disc to the adjacent vertebral bodies provide the major portal for the diffusion of nutrients into the disc inner tissues100,101. Therefore, the integrity of the EP structure is of great significance to the maintenance of homeostasis in the IVD. Histology and pathology have revealed that cartilaginous endplate calcification is a major pathological characteristic of disc degeneration100,102. The degeneration and calcification of EP hinder the transport of nutrients and metabolite clearance in IVD and thus impair the survival and functions of disc cells, which is considered a crucial initiating mechanism of IDD103.
Recently, Kang et al.45 revealed the critical roles of oxidative stress and Nrf2 signaling in the cartilaginous endplate homeostasis of IVD. The authors found that H2O2 stimulated oxidative stress, mitochondrial dysfunction, and cell apoptosis of human endplate chondrocytes, which were enhanced by Nrf2 knockdown. Moreover, upregulation of Nrf2 expression by polydatin treatment significantly protected endplate chondrocytes against these detrimental H2O2-induced effects. The study also applied the puncture-induced rat IDD model to validate the beneficial effects of Nrf2 activation on EP and disc degeneration. Interestingly, another study reported that rapamycin, a lipophilic macrolide antibiotic isolated from the actinomycete Streptomyces hygroscopicus, activated autophagy-Nrf2 signaling to protect cartilage endplate stem cells against calcification and ECM degradation61. Tumor necrosis factor-α (TNF-α) treatment induced oxidative stress, cell senescence and the osteogenic differentiation of cartilage endplate stem cells. Mechanistically, rapamycin-induced autophagy to upregulate antioxidant protein expression, scavenge ROS, alleviate cell senescence and promote the chondrogenic differentiation potential of cartilage endplate stem cells. Moreover, the function of rapamycin-activated autophagy in inhibiting TNF-α-induced EP degeneration was realized through the regulated expression and nuclear translocation of Nrf2. Hence, targeting Nrf2 signaling to alleviate degeneration and calcification of EP might be an effective therapeutic means of IDD intervention. A list of the functional mechanisms of Nrf2 activation and related signaling pathways in IDD treatment is presented in Table 1.
Conclusions and perspectives
Oxidative stress has been demonstrated to play pivotal roles in the initiation and progression of a plethora of age-related diseases in humans. IDD is one of the most prevalent degenerative musculoskeletal disorders, and its pathogenesis is closely associated with oxidative stress. Nrf2 is a master antioxidant transcription factor and protects cells against oxidative stress damage, similar to its role in disc cells. As mentioned above, certain noncoding RNAs, including lncRNAs and miRNAs, and important antioxidants, such as bioactive compounds and small molecules from natural products, can activate Nrf2 signaling to alleviate IDD progression. Activating Nrf2 helps maintain the structural and functional integrity of IVD by inhibiting cell apoptosis, senescence, inflammation response, and ECM degradation of NP cells and alleviating degeneration and calcification of EP (Fig. 3). Therefore, targeting the Nrf2 antioxidant defense system is an effective therapeutic strategy for IDD. Although pharmacological Nrf2 activators have proven the benefits of defending against oxidative stress to prevent IDD progression in vitro and in vivo models, further investigations are needed to discover the details of the underlying molecular mechanism. In addition, mitochondria are intimately related to oxidative stress, as they are the main sources of intracellular ROS. Whether and how Nrf2 signaling regulates mitochondrial quality control in IDD might be a difficult but interesting area to address in the future. In addition, crosstalk between Nrf2 and important signaling pathways or cellular protective mechanisms, such as autophagy, is evident. Therefore, there remains a need for further systematic studies to clarify the multiple connected and intertwined mechanisms involved in IDD.

Noncoding RNAs, such as lncRNAs and miRNAs, and other antioxidants can activate Nrf2 to alleviate IDD progression by inhibiting cell apoptosis, senescence, inflammation response, and ECM degradation in NP cells and alleviating the degeneration and calcification of EP.
Data availability
The data used to support this study were included in the article.
References
Buchbinder, R. et al. Low back pain: A call for action. Lancet 391, 2384–2388 (2018).
Binch, A. L. A., Fitzgerald, J. C., Growney, E. A. & Barry, F. Cell-based strategies for IVD repair: Clinical progress and translational obstacles. Nat. Rev. Rheumatol. 17, 158–175 (2021).
Yang, S., Zhang, F., Ma, J. & Ding, W. Intervertebral disc ageing and degeneration: The antiapoptotic effect of oestrogen. Ageing Res. Rev. 57, 100978 (2020).
Francisco, V. et al. A new immunometabolic perspective of intervertebral disc degeneration. Nat. Rev. Rheumatol. 18, 47–60 (2022).
Knezevic, N. N., Mandalia, S., Raasch, J., Knezevic, I. & Candido, K. D. Treatment of chronic low back pain—new approaches on the horizon. J. Pain. Res. 10, 1111–1123 (2017).
Morris, H., Gonçalves, C. F., Dudek, M., Hoyland, J. & Meng, Q. J. Tissue physiology revolving around the clock: circadian rhythms as exemplified by the intervertebral disc. Ann. Rheum. Dis. 80, 828–839 (2021).
Saberi, M., Zhang, X. & Mobasheri, A. Targeting mitochondrial dysfunction with small molecules in intervertebral disc aging and degeneration. Geroscience 43, 517–537 (2021).
Harmon, M. D. et al. Growing a backbone—functional biomaterials and structures for intervertebral disc (IVD) repair and regeneration: challenges, innovations, and future directions. Biomater. Sci. 8, 1216–1239 (2020).
Roh, E. J. et al. Genetic therapy for intervertebral disc degeneration. Int. J. Mol. Sci. 22, 1579 (2021).
Akanji, M. A., Rotimi, D. E., Elebiyo, T. C., Awakan, O. J. & Adeyemi, O. S. Redox homeostasis and prospects for therapeutic targeting in neurodegenerative disorders. Oxid. Med. Cell. Longev. 2021, 9971885 (2021).
Nishimura, Y., Kanda, Y., Sone, H. & Aoyama, H. Oxidative stress as a common key event in developmental neurotoxicity. Oxid. Med. Cell. Longev. 2021, 6685204 (2021).
Venza, M. et al. Cellular mechanisms of oxidative stress and action in melanoma. Oxid. Med. Cell. Longev. 2015, 481782–481782 (2015).
Kimball, J. S., Johnson, J. P. & Carlson, D. A. Oxidative stress and osteoporosis. J. Bone Jt. Surg. Am. 103, 1451–1461 (2021).
Kulkarni, P., Martson, A., Vidya, R., Chitnavis, S. & Harsulkar, A. Pathophysiological landscape of osteoarthritis. Adv. Clin. Chem. 100, 37–90 (2021).
Zhao, M. J. et al. Oxidative stress links aging-associated cardiovascular diseases and prostatic diseases. Oxid. Med. Cell. Longev. 2021, 5896136 (2021).
Seol, D. et al. Targeting oxidative stress with amobarbital to prevent intervertebral disc degeneration: Part I. In vitro and ex vivo studies. Spine J. 21, 1021–1030 (2021).
Feng, C. et al. ROS: Crucial intermediators in the pathogenesis of intervertebral disc degeneration. Oxid. Med. Cell. Longev. 2017, 5601593 (2017).
Tonelli, C., Chio, I. I. C. & Tuveson, D. A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 29, 1727–1745 (2018).
Maldonado, E., Rojas, D. A., Urbina, F. & Solari, A. The use of antioxidants as potential co-adjuvants to treat chronic chagas disease. Antioxidants 10, 1022 (2021).
Itoh, K. et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86 (1999).
Bono, S., Feligioni, M. & Corbo, M. Impaired antioxidant KEAP1-NRF2 system in amyotrophic lateral sclerosis: NRF2 activation as a potential therapeutic strategy. Mol. Neurodegener. 16, 71 (2021).
Tian, L. et al. aPKCι promotes gallbladder cancer tumorigenesis and gemcitabine resistance by competing with Nrf2 for binding to Keap1. Redox Biol. 22, 101149 (2019).
Chen, Q. M. Nrf2 for protection against oxidant generation and mitochondrial damage in cardiac injury. Free Radic. Biol. Med. 179, 133–143 (2021).
Annie-Mathew, A. S. et al. The pivotal role of Nrf2 activators in adipocyte biology. Pharmacol. Res. 173, 105853 (2021).
Yu, C. & Xiao, J. H. The Keap1-Nrf2 system: A mediator between oxidative stress and aging. Oxid. Med. Cell. Longev. 2021, 6635460 (2021).
González-Bosch, C., Boorman, E., Zunszain, P. A. & Mann, G. E. Short-chain fatty acids as modulators of redox signaling in health and disease. Redox Biol. 47, 102165 (2021).
Kang, L., Tian, Y., Guo, X., Chu, X. & Xue, Y. Long noncoding RNA ANPODRT overexpression protects nucleus pulposus cells from oxidative stress and apoptosis by activating Keap1-Nrf2 signaling. Oxid. Med. Cell. Longev. 2021, 6645005 (2021).
Hu, S. et al. Promoting Nrf2/Sirt3-dependent mitophagy suppresses apoptosis in nucleus pulposus cells and protects against intervertebral disc degeneration. Oxid. Med. Cell. Longev. 2021, 6694964 (2021).
Hua, W. et al. Icariin protects human nucleus pulposus cells from hydrogen peroxide-induced mitochondria-mediated apoptosis by activating nuclear factor erythroid 2-related factor 2. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165575 (2020).
Roughley, P. J. Biology of intervertebral disc aging and degeneration: Involvement of the extracellular matrix. Spine 29, 2691–2699 (2004).
Sakai, D. & Grad, S. Advancing the cellular and molecular therapy for intervertebral disc disease. Adv. Drug Deliv. Rev. 84, 159–171 (2015).
Cheng, Z., Xiang, Q., Wang, J. & Zhang, Y. The potential role of melatonin in retarding intervertebral disc ageing and degeneration: A systematic review. Ageing Res. Rev. 70, 101394 (2021).
Sampara, P., Banala, R. R., Vemuri, S. K., Av, G. R. & Gpv, S. Understanding the molecular biology of intervertebral disc degeneration and potential gene therapy strategies for regeneration: A review. Gene Ther. 25, 67–82 (2018).
Vadalà, G., Ambrosio, L., Russo, F., Papalia, R. & Denaro, V. Interaction between mesenchymal stem cells and intervertebral disc microenvironment: From cell therapy to tissue engineering. Stem Cells Int. 2019, 2376172 (2019).
Angelova, P. R. Sources and triggers of oxidative damage in neurodegeneration. Free Radic. Biol. Med. 173, 52–63 (2021).
Trachootham, D., Lu, W., Ogasawara, M. A., Nilsa, R. D. & Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 10, 1343–1374 (2008).
Yang, R. Z. et al. Involvement of oxidative stress-induced annulus fibrosus cell and nucleus pulposus cell ferroptosis in intervertebral disc degeneration pathogenesis. J. Cell. Physiol. 236, 2725–2739 (2021).
Tang, P. et al. Small molecule inhibitor of TAK1 ameliorates rat cartilaginous endplate degeneration induced by oxidative stress in vitro and in vivo. Free Radic. Biol. Med. 148, 140–150 (2020).
Bao, X. et al. HIF-1α-mediated miR-623 regulates apoptosis and inflammatory responses of nucleus pulposus induced by oxidative stress via targeting TXNIP. Oxid. Med. Cell. Longev. 2021, 6389568–6389568 (2021).
Zhang, G. Z. et al. NF-κB signalling pathways in nucleus pulposus cell function and intervertebral disc degeneration. Cell Prolif. 54, e13057 (2021).
Li, C. et al. LncRNA NEAT1 promotes nucleus pulposus cell matrix degradation through regulating Nrf2/ARE axis. Eur. J. Med. Res. 26, 11 (2021).
Park, J. S., Park, J. B., Park, I. J. & Park, E. Y. Accelerated premature stress-induced senescence of young annulus fibrosus cells of rats by high glucose-induced oxidative stress. Int. Orthop. 38, 1311–1320 (2014).
Cai, X. Y. et al. Ropivacaine- and bupivacaine-induced death of rabbit annulus fibrosus cells in vitro: Involvement of the mitochondrial apoptotic pathway. Osteoarthr. Cartil. 23, 1763–1775 (2015).
Xu, W. N. et al. PGC-1α acts as an mediator of Sirtuin2 to protect annulus fibrosus from apoptosis induced by oxidative stress through restraining mitophagy. Int. J. Biol. Macromol. 136, 1007–1017 (2019).
Kang, L. et al. Parkin and Nrf2 prevent oxidative stress-induced apoptosis in intervertebral endplate chondrocytes via inducing mitophagy and anti-oxidant defenses. Life Sci. 243, 117244 (2020).
Zhang, Z. et al. Melatonin protects vertebral endplate chondrocytes against apoptosis and calcification via the Sirt1-autophagy pathway. J. Cell. Mol. Med. 23, 177–193 (2019).
Chen, K. et al. Autophagy is a protective response to the oxidative damage to endplate chondrocytes in intervertebral disc: Implications for the treatment of degenerative lumbar disc. Oxid. Med. Cell. Longev. 2017, 4041768–4041768 (2017).
Zhang, W., Feng, C. & Jiang, H. Novel target for treating Alzheimer’s diseases: Crosstalk between the Nrf2 pathway and autophagy. Ageing Res. Rev. 65, 101207 (2021).
Villeneuve, N. F., Lau, A. & Zhang, D. D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 13, 1699–1712 (2010).
Liu, S., Pi, J. & Zhang, Q. Mathematical modeling reveals quantitative properties of KEAP1-NRF2 signaling. Redox Biol. 47, 102139–102139 (2021).
Vasilopoulou, M., Ioannou, E., Roussis, V. & Chondrogianni, N. Modulation of the ubiquitin-proteasome system by marine natural products. Redox Biol. 41, 101897–101897 (2021).
Telkoparan-Akillilar, P., Suzen, S. & Saso, L. Pharmacological applications of Nrf2 inhibitors as potential antineoplastic drugs. Int. J. Mol. Sci. 20, 2025 (2019).
Otsuki, A. & Yamamoto, M. Cis-element architecture of Nrf2-sMaf heterodimer binding sites and its relation to diseases. Arch. Pharm. Res. 43, 275–285 (2020).
Luo, X. et al. Ulinastatin ameliorates IL-1β-induced cell dysfunction in human nucleus pulposus cells via Nrf2/NF-κB pathway. Oxid. Med. Cell. Longev. 2021, 5558687 (2021).
Wang, R., Luo, D., Li, Z. & Han, H. Dimethyl fumarate ameliorates nucleus pulposus cell dysfunction through activating the Nrf2/HO-1 pathway in intervertebral disc degeneration. Comput. Math. Methods Med. 2021, 6021763 (2021).
Zhu, H. et al. Dimethyl fumarate protects nucleus pulposus cells from inflammation and oxidative stress and delays the intervertebral disc degeneration. Exp. Ther. Med. 20, 269 (2020).
Bai, X. et al. Cyanidin-3-glucoside protects against high glucose-induced injury in human nucleus pulposus cells by regulating the Nrf2/HO-1 signaling. J. Appl. Toxicol. 42, 1137–1145 (2021).
Gu, R. et al. Moracin attenuates LPS-induced inflammation in nucleus pulposus cells via Nrf2/HO-1 and NF-κB/TGF-β pathway. Biosci. Rep. 39, BSR20191673 (2019).
Wang, H., Jiang, Z., Pang, Z., Zhou, T. & Gu, Y. Acacetin alleviates inflammation and matrix degradation in nucleus pulposus cells and ameliorates intervertebral disc degeneration in vivo. Drug Des. Devel. Ther. 14, 4801–4813 (2020).
Fang, W. et al. Wogonin mitigates intervertebral disc degeneration through the Nrf2/ARE and MAPK signaling pathways. Int. Immunopharmacol. 65, 539–549 (2018).
Zuo, R. et al. Rapamycin induced autophagy inhibits inflammation-mediated endplate degeneration by enhancing Nrf2/Keap1 signaling of cartilage endplate stem cells. Stem Cells 37, 828–840 (2019).
Zhao, C. Q., Wang, L. M., Jiang, L. S. & Dai, L. Y. The cell biology of intervertebral disc aging and degeneration. Ageing Res. Rev. 6, 247–261 (2007).
Narasimhan, M. et al. Nrf2 deficiency promotes apoptosis and impairs PAX7/MyoD expression in aging skeletal muscle cells. Free Radic. Biol. Med. 71, 402–414 (2014).
Ala, M. & Eftekhar, S. P. Target sestrin2 to rescue the damaged organ: Mechanistic insight into its function. Oxid. Med. Cell. Longev. 2021, 8790369 (2021).
Jayasuriya, R. et al. Targeting Nrf2/Keap1 signaling pathway by bioactive natural agents: Possible therapeutic strategy to combat liver disease. Phytomedicine 92, 153755 (2021).
Xiang, Q., Zhao, Y., Lin, J., Jiang, S. & Li, W. Epigenetic modifications in spinal ligament aging. Ageing Res. Rev. 77, 101598 (2022).
Xu, J. et al. Platelet-rich plasma attenuates intervertebral disc degeneration via delivering miR-141-3p-containing exosomes. Cell Cycle 20, 1487–1499 (2021).
Rodriguez-Cuenca, S. et al. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic. Biol. Med. 48, 161–172 (2010).
Kang, L. et al. The mitochondria-targeted anti-oxidant MitoQ protects against intervertebral disc degeneration by ameliorating mitochondrial dysfunction and redox imbalance. Cell Prolif. 53, e12779 (2020).
Huang, J. F. et al. Sinapic acid inhibits IL-1β-induced apoptosis and catabolism in nucleus pulposus cells and ameliorates intervertebral disk degeneration. J. Inflamm. Res. 13, 883–895 (2020).
Chu, H., Yu, H., Ren, D., Zhu, K. & Huang, H. Plumbagin exerts protective effects in nucleus pulposus cells by attenuating hydrogen peroxide-induced oxidative stress, inflammation and apoptosis through NF-κB and Nrf-2. Int. J. Mol. Med. 37, 1669–1676 (2016).
Lin, J. et al. Luteoloside inhibits IL-1β-induced apoptosis and catabolism in nucleus pulposus cells and ameliorates intervertebral disk degeneration. Front. Pharmacol. 10, 868 (2019).
Zhang, C. X. et al. Protective effect of CDDO-ethyl amide against high-glucose-induced oxidative injury via the Nrf2/HO-1 pathway. Spine J. 17, 1017–1025 (2017).
Wang, Y. et al. Kinsenoside ameliorates intervertebral disc degeneration through the activation of AKT-ERK1/2-Nrf2 signaling pathway. Aging 11, 7961–7977 (2019).
Lu, Y. et al. Lycopene alleviates disc degeneration under oxidative stress through the Nrf2 signaling pathway. Mol. Cell. Probes 51, 101559 (2020).
Wang, K. et al. Genistein protects intervertebral discs from degeneration via Nrf2-mediated antioxidant defense system: An in vitro and in vivo study. J. Cell. Physiol. 234, 16348–16356 (2019).
Song, Y. et al. Mitochondrial quality control in intervertebral disc degeneration. Exp. Mol. Med. 53, 1124–1133 (2021).
Kang, L. et al. Restoration of autophagic flux rescues oxidative damage and mitochondrial dysfunction to protect against intervertebral disc degeneration. Oxid. Med. Cell. Longev. 2019, 7810320 (2019).
Gorgoulis, V. et al. Cellular senescence: Defining a path forward. Cell 179, 813–827 (2019).
Saito, Y. & Chikenji, T. S. Diverse roles of cellular senescence in skeletal muscle inflammation, regeneration, and therapeutics. Front. Pharmacol. 12, 739510–739510 (2021).
Kaur, J. & Farr, J. N. Cellular senescence in age-related disorders. Transl. Res. 226, 96–104 (2020).
Cherif, H. et al. Curcumin and o-vanillin exhibit evidence of senolytic activity in human IVD cells in vitro. J. Clin. Med. 8, 433 (2019).
Mantzorou, M., Pavlidou, E., Vasios, G., Tsagalioti, E. & Giaginis, C. Effects of curcumin consumption on human chronic diseases: A narrative review of the most recent clinical data. Phytother. Res. 32, 957–975 (2018).
Santosh Kumar, S., Priyadarsini, K. I. & Sainis, K. B. Free radical scavenging activity of vanillin and o-vanillin using 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical. Redox Rep. 7, 35–40 (2002).
Shao, Z. et al. Senolytic agent Quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-κB axis. Osteoarthr. Cartil. 29, 413–422 (2021).
Peng, W., Qin, R., Li, X. & Zhou, H. Botany, phytochemistry, pharmacology, and potential application of Polygonum cuspidatum Sieb.et Zucc.: A review. J. Ethnopharmacol. 148, 729–745 (2013).
Wang, J. et al. Polydatin suppresses nucleus pulposus cell senescence, promotes matrix homeostasis, and attenuates intervertebral disc degeneration in rats. J. Cell. Mol. Med. 22, 5720–5731 (2018).
Rajasekaran, S. et al. Part 1: Profiling extra cellular matrix core proteome of human fetal nucleus pulposus in search for regenerative targets. Sci. Rep. 10, 15684–15684 (2020).
Craddock, R. J. et al. Extracellular matrix fragmentation in young, healthy cartilaginous tissues. Eur. Cells Mater. 35, 34–53 (2018).
Yurube, T. et al. Rat tail static compression model mimics extracellular matrix metabolic imbalances of matrix metalloproteinases, aggrecanases, and tissue inhibitors of metalloproteinases in intervertebral disc degeneration. Arthritis Res. Ther. 14, R51–R51 (2012).
Liang, H. et al. The proteolysis of ECM in intervertebral disc degeneration. Int. J. Mol. Sci. 23, 1715 (2022).
Kepler, C. K., Ponnappan, R. K., Tannoury, C. A., Risbud, M. V. & Anderson, D. G. The molecular basis of intervertebral disc degeneration. Spine J. 13, 318–330 (2013).
Xiang, Q. et al. CircRNA-CIDN mitigated compression loading-induced damage in human nucleus pulposus cells via miR-34a-5p/SIRT1 axis. EBioMedicine 53, 102679 (2020).
Albrecht, P. et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflamm. 9, 163 (2012).
Xie, C. et al. Cardamonin protects nucleus pulposus cells against IL-1β-induced inflammation and catabolism via Nrf2/NF-κB axis. FEBS Lett. 12, 2703–2714 (2021).
Molinos, M. et al. Inflammation in intervertebral disc degeneration and regeneration. J. R. Soc. Interface 12, 20150429 (2015).
Lyu, F. J. et al. Painful intervertebral disc degeneration and inflammation: From laboratory evidence to clinical interventions. Bone Res. 9, 7 (2021).
Risbud, M. V. & Shapiro, I. M. Role of cytokines in intervertebral disc degeneration: Pain and disc content. Nat. Rev. Rheumatol. 10, 44–56 (2014).
Cunha, C. et al. The inflammatory response in the regression of lumbar disc herniation. Arthritis Res. Ther. 20, 251 (2018).
Roberts, S., Evans, H., Trivedi, J. & Menage, J. Histology and pathology of the human intervertebral disc. J. Bone Jt. Surg. Am. 88, 10–14 (2006).
Malandrino, A. et al. The role of endplate poromechanical properties on the nutrient availability in the intervertebral disc. Osteoarthr. Cartil. 22, 1053–1060 (2014).
Rodriguez, A. G. et al. Morphology of the human vertebral endplate. J. Orthop. Res. 30, 280–287 (2012).
Huang, Y. C., Urban, J. P. & Luk, K. D. Intervertebral disc regeneration: Do nutrients lead the way? Nat. Rev. Rheumatol. 10, 561–566 (2014).
Tang, Z. et al. Nrf2 drives oxidative stress-induced autophagy in nucleus pulposus cells via a Keap1/Nrf2/p62 feedback loop to protect intervertebral disc from degeneration. Cell Death Dis. 10, 510 (2019).
Lin, H. et al. Nuclear factor erythroid-2 related factor 2 inhibits human disc nucleus pulpous cells apoptosis induced by excessive hydrogen peroxide. Rev. Assoc. Med. Bras. 66, 986–991 (2020).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (82172480).
Author information
Authors and Affiliations
Contributions
Q.X. conceived and drafted the manuscript. Q.X., Y.Z., J.L., and S.J. collected the references, and Y.Z. and W.L. proofread and revised the manuscript. All authors approved the final manuscript. Q.X. and Y.Z. contributed equally.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiang, Q., Zhao, Y., Lin, J. et al. The Nrf2 antioxidant defense system in intervertebral disc degeneration: Molecular insights. Exp Mol Med 54, 1067–1075 (2022). https://doi.org/10.1038/s12276-022-00829-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00829-6
This article is cited by
-
FSTL1 Accelerates Nucleus Pulposus Cell Senescence and Intervertebral Disc Degeneration Through TLR4/NF-κB Pathway
Inflammation (2024)
-
Adipose-derived stem cell exosomes regulate Nrf2/Keap1 in diabetic nephropathy by targeting FAM129B
Diabetology & Metabolic Syndrome (2023)
-
Programmable DNA hydrogel provides suitable microenvironment for enhancing autophagy-based therapies in intervertebral disc degeneration treatment
Journal of Nanobiotechnology (2023)
-
FSP1 confers ferroptosis resistance in KEAP1 mutant non-small cell lung carcinoma in NRF2-dependent and -independent manner
Cell Death & Disease (2023)
-
An integrated view of lipid metabolism in ferroptosis revisited via lipidomic analysis
Experimental & Molecular Medicine (2023)
