
Abstract
The coronavirus (COVID-19) pandemic, caused by severe acute respiratory system coronavirus 2 (SARS-CoV-2), has created significant challenges for scientists seeking to understand the pathogenic mechanisms of SARS-CoV-2 infection and to identify the best therapies for infected patients. Although ACE2 is a known receptor for the virus and has been shown to mediate viral entry into the lungs, accumulating reports highlight the presence of neurological symptoms resulting from infection. As ACE2 expression is low in the central nervous system (CNS), these neurological symptoms are unlikely to be caused by ACE2-virus binding. In this review, we will discuss a proposed interaction between SARS-CoV-2 and Toll-like receptor 2 (TLR2) in the CNS. TLR2 is an innate immune receptor that recognizes exogenous microbial components but has also been shown to interact with multiple viral components, including the envelope (E) protein of SARS-CoV-2. In addition, TLR2 plays an important role in the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). Based on these observations, we hypothesize that TLR2 may play a critical role in the response to SARS-CoV-2 infiltration in the CNS, thereby resulting in the induction or acceleration of AD and PD pathologies in patients.
Similar content being viewed by others
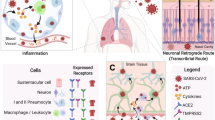

Introduction
The coronavirus pandemic, which is caused by severe acute respiratory system coronavirus 2 (SARS-CoV-2), has resulted in the infection of over 326 million people, with more than 5.5 million deaths due to the disease1. The disease is considered to be primarily respiratory, with the most common symptoms including cough, fatigue, headache, muscle aches, and a loss of taste and/or smell, among others2. The symptoms that are centralized in the lungs present due to the damage to the alveolar tissue caused by the virus, specifically, the resulting pneumonia, which coincides with inflammation3. These symptoms are associated with the expression of angiotensin-converting enzyme 2 (ACE2) in the lungs, which is known as a receptor for SARS-CoV-2 and modulates the cellular entry of the virus. The spike (S) proteins of coronaviruses are known to bind to ACE2, and SARS-CoV-2 has been found to be more infectious than earlier coronaviruses since its binding affinity for ACE2 is higher4.
As the pandemic continues, increasing evidence has drawn attention to the various local and systemic inflammatory effects of the virus, such as cytokine storms5. These inflammatory effects allow the transition of focus from localized damage in the lungs to systemic damage in the body, specifically in the central nervous system (CNS), where the virus was shown to produce pathologies resembling various “classic” forms of neurodegeneration6. It has been noted that patients with dementia have an increased risk of contracting the virus, and it has also been suggested that those who contracted the virus had pathological symptoms that resembled those of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD)7,8,9.
AD and PD are the most common neurodegenerative disorders, with ~6 and 1 million people, respectively, having the conditions in the United States alone in 2021, with both values expected to increase in the future as the average age increases10,11. In AD, the cognitive deficits are caused by abnormal accumulations of amyloid-β peptide (Aβ) and tau protein, called Aβ plaques and neurofibrillary tangles, respectively, which are considered the pathological hallmarks12. PD is a progressive neurological movement disorder13. Common symptoms of PD include tremor, rigidity, and bradykinesia, but nonmotor symptoms, such as depression and anxiety, also occur13. PD is characterized by the loss of dopaminergic neurons in the substantia nigra, and the pathological hallmarks are the abnormal deposition of misfolded proteins called Lewy bodies (LBs) and Lewy neurites (LNs), which are primarily composed of α-synuclein (α-syn)13,14.
Toll-like receptor 2 (TLR2) belongs to a family of pattern recognition receptors (PRRs) and is expressed on the surface of numerous cells, including innate immune cells15. TLR2 recognizes a variety of microbial components, such as lipopeptides and peptidoglycan15. While TLR2 plays an important role in the innate immune system, it has been demonstrated that TLR2 also plays an important role in the pathogenesis of neurodegenerative diseases, including AD and PD16,17,18,19,20. Therefore, TLR2 has been proposed as a new therapeutic target for these diseases18,19,21.
In addition to being a systemic respiratory disease, infection with SARS-CoV-2 induces neuropathology in patients22,23,24,25,26. Although recent studies have reported the presence of SARS-CoV-2 infection in the CNS26,27, it is still under debate whether SARS-CoV-2 can infect cells in the CNS28. Despite the controversy, accumulating evidence supports the invasion of SARS-CoV-2 into the CNS in patients, which could possibly induce further delayed neurological complications28. Therefore, it is reasonable to assume that there could be a receptor that might recognize a component of SARS-CoV-2 in the brain, such as TLR2. TLR2 has been known to interact with bacterial pathogens, and recent studies have demonstrated that TLR2 can also detect various viruses, including SARS-CoV-229,30,31,32. In addition, TLR2 is widely expressed in brain resident cells, such as neurons and glial cells33,34. Therefore, in this review, we speculate that a pathogenic interaction between SARS-CoV-2 and TLR2 occurs in the CNS, and we will examine its potential effects on AD and PD pathology.
Materials and methods
Human brain immunohistochemical analysis
The paraffin-embedded brain sections of control and SARS-CoV-2-infected patients were kindly provided by Dr. Avindra Nath (National Institute of Neurological Disorders and Stroke). The procedure for immunochemical analysis has been described elsewhere35,36. Briefly, blinded brain sections were incubated with anti-ionized calcium-binding adapter molecule 1 (IBA-1, citrate buffer treatment, 1:2000, Wako Chemicals, Richmond, VA), anti-transmembrane protein 119 (TMEM119, citrate buffer treatment, 1:500, Abcam, Cambridge, UK), anti-cluster of differentiation 3 (CD3, citrate buffer treatment, 1:2000, Thermo Fisher Scientific, Waltham, MA), or anti-phospho-α-synuclein (S129) (81 A, citrate buffer treatment, 1:10,000, gift from Drs. Virginia Lee and John Trojanowski, University of Pennsylvania, PA) at 4 °C overnight. The next day, the sections were incubated with a biotinylated secondary antibody and detected with an avidin D-HRP detection system (ABC elite, Vector Laboratories, Burlingame, CA). The immunostained sections were imaged by an Olympus BX41 microscope (Olympus, Tokyo, Japan).
SARS-CoV-2 and neurodegenerative disorders
Neuropathology of SARS-CoV-2-infected patients
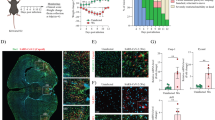
There is increasing evidence that patients infected with SARS-CoV-2 have neurological symptoms along with respiratory symptoms37,38. Approximately 36% of SARS-CoV-2-infected patients have neurological symptoms39. The common neurological symptoms of patients include headaches and nausea, but patients can also present with more severe neurological disorders, such as meningo-encephalitis and acute cerebrovascular disease22,23,24,25,26. Neuropathologies of SARS-CoV-2-infected patients are varied, but common neuroinflammatory findings have been reported, including astrogliosis, microgliosis, ischemia, hemorrhage, and microvascular lesions in the CNS of patients40. Similar to previous studies, our postmortem analysis revealed the activation of microglia in the patients’ brains (Fig. 1). Whether T cells infiltrate the CNS is controversial;41,42 however, a recent study suggested that subpopulations of CD3+ and CD4+ T cells infiltrate the CNS and interact with resident microglial cells in SARS-CoV-2-infected patients43. Our postmortem study also detected a small number of infiltrated CD3+ T cells in the cortex of SARS-CoV-2-infected patients (Fig. 1).

White matter sections obtained from one control and two SARS-CoV-2-infected patients were immunostained with anti-IBA-1 and anti-TMEM119 for microglia and anti-CD3 for T cells. Scale bars represent 50 μm.
How does SARS-CoV-2 enter the CNS?
Although the expression of ACE2 is very limited in the CNS and the amount of SARS-CoV-2 present in the CNS after infection is still disputed14,26, postmortem studies have identified the existence of SARS-CoV-2 in the CNS of patients44. The presence of SARS-CoV-2 in the CNS was initially hypothesized due to anosmia that presented as a common symptom of the infection45. This led to the speculation of the olfactory bulb as a potential route of entry for the virus into the brain46. Meinhardt et al. suggested that the neural-mucosal interface could be a potential route for SARS-CoV-2 neuroinvasion (Fig. 2)46. However, the study also demonstrated the presence of the virus in other brain regions that had no direct connection to this interface, leading them to suggest the existence of other routes for SARS-CoV-2 neuroinvasion46. There are four other potential CNS entry mechanisms of SARS-CoV-2, although none have been proven (Fig. 2). Armocida and colleagues proposed that the virus could infect neurons in the peripheral nervous system and then take advantage of axonal transport to gain access to the CNS47. McQuaid et al. suggested the lateral ventricles and choroid plexus as a CNS entry mechanism for SARS-CoV-248. Since these regions contain epithelial cells, which express ACE2, it has been suggested that the virus could cross the blood-cerebrospinal fluid barrier and enter the choroid plexus and ventricular system. Recent studies demonstrated that the expression of ACE2 is relatively high in the corneal epithelium and suggested that ocular conjunctival inoculation was enough to cause COVID-1949,50. In addition, various sampling studies identified the presence of SARS-CoV-2 RNA within regions of the visual system, such as the retina, optic nerve, conjunctiva, and vitreous body, in patients with confirmed SARS-CoV-2 diagnoses51,52. A recent study revealed that the S protein of the novel coronavirus can cross the blood-brain barrier (BBB)53. Therefore, it has also been suggested that SARS-CoV-2 could impair the functional integrity of the BBB54,55.

SARS-CoV-2 can infiltrate the CNS via the penetration of the blood-brain barrier (BBB), the blood-cerebrospinal fluid barrier in the epithelium of the choroid plexus, the corneal epithelium of the eye, the olfactory epithelium of the olfactory bulb (nasal route), and the peripheral nervous system, including the mucosal epithelium, enterocytes, and smooth muscle cells.
Alzheimer’s disease and SARS-CoV-2
A recent clinical study found that the risk of SARS-CoV-2 infection for patients with dementia was increased 2–3-fold compared with cognitively healthy individuals8. In addition, the levels of total tau, phosphorylated tau181, and glial fibrillary acidic protein, all biomarkers for AD, were elevated in SARS-CoV-2-infected patients with severe symptoms, suggesting a potential correlation between AD and SARS-CoV-2 infection severity56. Transcriptomic and interactomic data also showed a relationship between SARS-CoV-2 and β-amyloid production and clearance, leading to the conclusion that SARS-CoV-2 infection may exacerbate AD neuropathology57. In addition, patients with the homozygous allele apolipoprotein E4 (APOE4), an AD-associated gene, showed an increased risk for SARS-CoV-2 infection, and APOE4 may also affect the severity of the host response to infection58,59. Furthermore, it was found that SARS-CoV-2-infected patients with AD had a higher rate of death due to the disease than SARS-CoV-2-infected patients without AD60.
In addition to viral infection, the characteristic behaviors of AD patients may increase the risk for SARS-CoV-2 infection and severity. First, patients may not be able to follow the recommendations from public health providers to reduce the spread of the virus61. Second, the lack of social interaction due to the pandemic may increase mental and psychological stress in AD patients62. Increased psychological stress further accelerates the deterioration of cognitive function in AD patients62.
ACE2 is a known receptor for SARS-CoV-2, and its role in AD has been extensively studied during the SARS-CoV-2 pandemic; however, its role in AD is controversial63. A recent study found an inverse correlation between ACE2 activity and AD patient neuropathology, such as the accumulation of Aβ and phosphorylated tau64. In addition, Kehoe and colleagues reported a reduction in ACE2 activity in the brain homogenate of AD patients carrying the APOE4 allele64. However, Lim and colleagues showed an increased level of ACE2 in the brain tissue of AD patients65. Zhao and colleagues found that ACE2 expression was upregulated in the occipital and temporal lobes and the hippocampal CA1 region in AD patients compared to healthy controls66. Therefore, further studies are required to evaluate the role of ACE2 in AD pathogenesis.
Parkinson’s disease and SARS-CoV-2
To date, the risk factor associating PD with SARS-CoV-2 infection has not been clearly identified67. However, it has been suggested that SARS-CoV-2 infection may trigger parkinsonism symptoms in healthy individuals68. A case study by Lee and colleagues suggested the potential effects of SARS-CoV-2 infection on the dopaminergic mechanisms that led to the development of dysphagia in PD patients69. In addition, a recent study reported that infection with SARS-CoV-2 may worsen the symptoms of PD70. It was demonstrated that the nucleocapsid (N) protein of SARS-CoV-2 sped up the process of α-syn aggregation in vitro71.
The SARS-CoV-2 pandemic has also increased the level of psychological stress in PD patients72 which may worsen the symptoms of PD. For example, the accumulation of psychological stress has been shown to cause the temporary aggravation of motor symptoms73,74. One observational study found that 40% of PD patients showed an exacerbation of their motor symptoms during the pandemic75. In addition, the limitation of physical exercise increases psychological stress72.
TLR2 as a potential SARS-CoV-2 receptor in the CNS
TLR2
Toll-like receptors (TLRs) belong to a family of innate immune receptors known as PRRs15. To date, 10 human (TLR1-10) and 13 murine (1-13) subtypes of TLRs have been identified76. TLRs are type I transmembrane proteins and have a leucine-rich repeat (LRR) motif, transmembrane domain, and cytoplasmic Toll/IL-1 receptor (TIR) domain77. TLRs are abundantly expressed in multiple peripheral organs but are also expressed in the neuronal and nonneuronal cells of the CNS78. TLRs can recognize both exogenous pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPs)79. TLRs form a homo-/heterodimer to recognize different shapes of pathogens80. For example, the TLR4 homodimer recognizes the lipopolysaccharide of gram-negative bacteria, and the TLR3 homodimer recognizes double-stranded RNA81. Once activated, TLRs trigger intracellular signaling cascades via myeloid differentiation primary-response protein 88 (MyD88), except for TLR3, which initiates signaling via Toll/interleukin 1 receptor domain-containing adaptor interferon-β (TRIF), thereby resulting in the induction of inflammatory cytokines and chemokines15.
The role of TLR2 in AD
TLR2 is an innate immune receptor, but increasing evidence demonstrates its role in neurodegenerative diseases, including AD and PD18,82. Recent genetic studies identified TLR2 as a risk factor for late-onset AD (LOAD) in Han Chinese and Azeri Turk ancestry populations83,84. While genetic association studies of TLR2 with AD are still open to debate76, accumulating in vitro and in vivo studies provide evidence for the pathogenic role of TLR2 in AD.
In microglia, the brain resident innate immune cells, TLR2 mediates fibrillar Aβ-induced immune responses85. In addition, the activation of TLR2 enhances pathogenic Aβ uptake in microglia86. On the other hand, genetic depletion of TLR2 reduces the Aβ42-induced immune response and enhances Aβ clearance in cultured microglia16,82. In an animal model of AD (APP/PS1 mice), functional inhibition of TLR2 decreases microgliosis, astrogliosis, Aβ plaque deposition, and phosphorylated tau accumulation in the brain regions, thereby improving cognitive function19,87,88. In addition, genetic depletion of TLR2 shows protective effects against memory and cognitive impairments in an AD mouse model89,90. The expression of TLR2 is increased in AD patients and animal models91,92. Furthermore, immunohistochemical analysis demonstrates that the localization of microglial TLR2 is associated with Aβ plaques in the brains of AD patients and aged mouse models82,85,93.
TLR2 in PD
Although the pathogenic role of TLR2 in PD was demonstrated a few years later than that in AD, it has also been extensively studied for this short period18. Genetic associations of TLR2 polymorphisms with PD were identified in northeastern Han Chinese and Greek populations94,95. In 2013, we first demonstrated that neuron-released oligomeric forms of α-syn activated microglial TLR2, thereby inducing neurotoxic inflammation through the activation of nuclear factor kappa B (NF-κB)17,96. This finding was supported by subsequent in vitro and in vivo studies. Roodveldt et al. demonstrated that pretreatment with a TLR2 agonist, but not other TLR agonists, increased microglial susceptibility against α-syn97. Qiao et al. showed that functional and genetic inhibition of TLR2 prevented microglial responses against neuron-released α-syn98. Daniele et al. reported that α-syn treatment induced microglial inflammatory responses by forming a TLR1/2 heterodimer complex99. We also demonstrated that leucine-rich repeat kinase 2 (LRRK2), a PD-associated gene, and nuclear factor of activated T cell 1 (NFAT1) are downstream signaling molecules of TLR2 in microglia, thereby modulating neurotoxic microglial activation100. In a mouse model of PD, Drouin-Ouellet et al. reported that the overexpression of α-syn increased the expression of TLR2101. La Viola et al. demonstrated that oligomeric forms of α-syn induced memory impairment through TLR2102. Interestingly, exercise showed a protective effect in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse model via the downregulation of TLR2 and downstream signaling molecules, including MyD88103. In PD patients, the level of TLR2 was elevated in the blood compared to healthy controls101. Furthermore, the expression of TLR2 was also increased in the specific brain regions of PD patients and aged animal models in accordance with disease stages91,101.
Although TLR2 is primarily expressed in innate immune cells, recent studies have demonstrated that neurons also express TLR281,104, which has been demonstrated to play an important role in PD. We demonstrated that the activation of neuronal TLR2 impaired autophagy through the AKT/mammalian target of rapamycin signaling cascade, thereby inducing the accumulation of neurotoxic α-syn aggregates105. These findings were supported by Dzamko and colleagues, who also demonstrated the elevation of neuronal TLR2 in the brains of PD patients106.
The neuron-to-neuron and neuron-to-glial transmission of α-syn has been proposed to play a central role in PD pathogenesis and disease progression107. Although the primary role of TLR2 is recognizing pathogens, TLR2 also modulates pathogen phagocytosis in cells108. Genetic or pharmacological activation of TLR2 increased extracellular α-syn uptake by neuronal and glial cells, while it was inhibited by genetic and functional depletion of TLR217,21,105. Specifically, the α-syn transmission assay indicated that the activation of TLR2 not only increased α-syn transmission in neurons but also increased its propagation21. In microglia, the internalization of monomeric α-syn was not affected by TLR2, but the uptake of α-syn oligomer was significantly decreased by TLR2 inhibition in the cells17. In addition, the deposition of α-syn increased in astrocytes that did not express α-syn in either PD patients or mouse models21. These observations were reproduced in PD models in which functional inhibition of TLR2 significantly reduced astroglial α-syn deposition in both a PD mouse model and a neuron-to-astrocyte α-syn monitoring system21.
Given that TLR2 plays an important role in PD, targeting TLR2 has been proposed as a promising immunotherapeutic option for the disease18. Indeed, the administration of a TLR2 functional blocking antibody improved α-syn neuropathology, neuroinflammation, and motor behavioral deficits in a PD mouse model21.
TLR2 and SARS-CoV-2
Lipopeptides and gram-positive bacteria-derived lipoprotein are considered the traditional ligands of TLR215. However, increasing evidence also supports the interaction of TLR2 with viruses. Varicella-zoster virus activates the inflammatory response in monocytes via TLR2109. Furthermore, various viral proteins, such as the structural proteins (p17, p24, and gp41) of human immunodeficiency virus 1, the core protein of hepatitis C virus, and glycoprotein B of herpes simplex virus 1, have been known to interact with TLR2, thereby inducing cytokine gene expression110. In addition, it was found that the S protein of SARS-CoV-1 activated TLR2 in peripheral leukocytes, which resulted in the induction of proinflammatory cytokine and chemokine gene expression, including interleukin-6 (IL-6) and TNF-α111.
The exact pathogenic mechanism of SARS-CoV-2 is still largely unknown. However, increasing evidence supports that TLRs might play a role during SARS-CoV-2 pathogenesis112. It has been shown that the surface proteins of SARS-CoV-2 could behave as a PAMP, thereby inducing the upregulation of inflammatory factors in the rodent model through TLR2 and TLR4113. Prophylactic administration of a TLR2 agonist showed a protective effect against SARS-CoV-2 infection and decreased virus transmission through the activation of the innate immune system114. More importantly, Zheng et al. demonstrated that TLR2 can sense the envelope (E) protein of SARS-CoV-2, thereby inducing an inflammatory response30. Signaling molecules downstream from TLR2, including MyD88 and TRIF, were significantly increased in SARS-CoV-2-infected patients with severe/critical conditions compared to healthy controls. In addition, a pharmacological inhibitor of TLR2 inhibited the inflammatory responses of human leukocytes against SARS-CoV-2 infection. Furthermore, genetic depletion of TLR2 prevented inflammation and tissue damage in the lungs of mice expressing human ACE2. These findings suggest that the surface proteins of SARS-CoV-2 induce the activation of TLR2, leading to inflammatory responses.
Prospective: Does SARS-CoV-2 affect AD and PD patients through TLR2?
ACE2 is a primary receptor for SARS-CoV-2. After infection, the number of SARS-CoV-2-positive cells rapidly increases in the host peripheral system115. However, the viral load in the CNS is lower than that in the periphery26. Based on these observations, we speculate that SARS-CoV-2 infection is limited in the CNS during the early infection period for two reasons: the inhibition of SARS-CoV-2 CNS infiltration by the existence of a physical barrier (the BBB) and the low level of SARS-CoV-2 high-affinity receptors in the CNS116. However, persistent infection with SARS-CoV-2 leads to excessive peripheral immune responses, which could induce damage to the BBB117. Therefore, a greater number of viruses can infiltrate the CNS via the damaged barriers, which may increase the chance that a viral component will meet a responding receptor, such as TLR2, in the CNS (Fig. 3).

E protein of brain-infiltrated SARS-CoV-2 induces microglial TLR2, thereby increasing the susceptibility of TLR2 to α-syn and Aβ oligomers. The activation of microglial TLR2 by the E protein of SARS-CoV-2, α-syn, or Aβ increases the release of neurotoxic, proinflammatory cytokines, which may also induce vascular degeneration in the brain, thereby increasing SARS-CoV-2 infiltration into the CNS. In neurons, the E protein of SARS-CoV-2 may activate neuronal TLR2, which impairs neuronal autophagic processes, resulting in an accumulation of neurotoxic α-syn aggregates.
Many of the severe and critical conditions of SARS-CoV-2-infected patients result in death115. However, vaccination against SARS-CoV-2 will help reduce the numbers of patients who become severely or critically ill. In addition, developing a medication for COVID-19 would reduce the death rate of patients in the future. To date, two treatments have been approved by the FDA: an antibody cocktail targeting SARS-CoV-2 (REGN-COV2, REGENERON) and an oral antiviral medicine (Molnupiravir, Merck). On the other hand, CNS-infiltrated viruses might survive longer than those in the peripheral system due to the lack of an adaptive immune system and the high selectivity of the BBB against drugs117. Therefore, this prolonged presence of SARS-CoV-2 in the CNS may cause further problems in the brain that might not present until much later. Specifically, SARS-CoV-2 viral components may directly affect patients with neurodegenerative diseases. According to Zheng et al., the E protein of SARS-CoV-2 is activated and induces TLR2 expression in innate immune cells30. Microglia, brain resident innate immune cells, express TLR2, which plays a critical role in the neuroinflammation of AD and PD patients17,85. Therefore, we speculate that the viral components, especially the E protein, of brain-infiltrated SARS-CoV-2 induces the activation of microglial TLR2, thereby increasing the susceptibility of TLR2 to Aβ and α-syn deposition in patients (Fig. 3). The chronic activation of TLR2 can induce chronic neuroinflammation, which will accelerate disease pathogenesis118. TLR2 is also expressed in neurons, and prolonged infection with SARS-CoV-2 may induce neuronal TLR2 activation in the brain. The induction of neuronal TLR2 is associated with pathological α-syn neuron-to-neuron transmission and propagation, which is known to be associated with disease progression18. In addition, the activation of neuronal TLR2 has been shown to impair neuronal autophagy, thereby increasing abnormal accumulation of neurotoxic misfolded proteins, such as α-syn105. Therefore, the induction of neuronal TLR2 susceptibility by the E protein of SARS-CoV-2 may lead to the deposition of abnormal protein in the cells, thereby affecting the disease onset and/or accelerating the disease progression of proteinopathy-associated neurodegenerative diseases. Indeed, our preliminary postmortem analysis revealed that the accumulation of phosphorylated α-syn, one of the pathogenic forms of α-syn, was increased in the brains of SARS-CoV-2-infected patients (Fig. 4). For these reasons, further detailed studies are required to understand the pathogenic interaction between SARS-CoV-2 and TLR2 and the potential of TLR2 as target for COVID-19 treatment.

White matter sections obtained from one control and two SARS-CoV-2-infected patients were immunostained with anti-phospho-α-syn. The arrowhead represents the LN-like accumulation of phospho-α-syn (p-S129). Scale bar represents 50 μm.
References
World Health Organization WHO Coronavirus Dashboard, https://covid19.who.int (2021).
Centers for Disease Control and Prevention COVID-19, https://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.html (2021).
Seyed Hosseini, E. et al. The novel coronavirus Disease-2019 (COVID-19): mechanism of action, detection and recent therapeutic strategies. Virology 551, 1–9 (2020).
Wong, S. K., Li, W., Moore, M. J., Choe, H. & Farzan, M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 279, 3197–3201 (2004).
Tufan, A., Avanoglu Guler, A. & Matucci-Cerinic, M. COVID-19, immune system response, hyperinflammation and repurposing antirheumatic drugs. Turk. J. Med. Sci. 50, 620–632 (2020).
Ferini-Strambi, L. & Salsone, M. COVID-19 and neurological disorders: are neurodegenerative or neuroimmunological diseases more vulnerable? J. Neurol. 268, 409–419 (2021).
Zhou, Y. et al. Network medicine links SARS-CoV-2/COVID-19 infection to brain microvascular injury and neuroinflammation in dementia-like cognitive impairment. Alzheimer’s Res. Ther. 13, 110 (2021).
Wang, Q., Davis, P. B., Gurney, M. E. & Xu, R. COVID-19 and dementia: analyses of risk, disparity, and outcomes from electronic health records in the US. Alzheimer’s Dement. 17, 1297–1306 (2021).
Krey, L., Huber, M. K., Hoglinger, G. U. & Wegner, F. Can SARS-CoV-2 Infection Lead to Neurodegeneration and Parkinson’s Disease? Brain Sci. 11, 1654 (2021).
Matthews, K. A. et al. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015-2060) in adults aged >/=65 years. Alzheimer’s Dement. 15, 17–24 (2019).
Marras, C. et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinson’s Dis. 4, 21 (2018).
John, A. & Reddy, P. H. Synaptic basis of Alzheimer’s disease: focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 65, 101208 (2021).
Beitz, J. M. Parkinson’s disease: a review. Front. Biosci. Scholar Ed. 6, 65–74 (2014).
Gomez-Benito, M. et al. Modeling Parkinson’s Disease With the Alpha-Synuclein Protein. Front. Pharmacol. 11, 356 (2020).
Oliveira-Nascimento, L., Massari, P. & Wetzler, L. M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 3, 79 (2012).
Liu, S. et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J. Immunol. 188, 1098–1107 (2012).
Kim, C. et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562 (2013).
Kwon, S., Iba, M., Masliah, E. & Kim, C. Targeting Microglial and Neuronal Toll-like Receptor 2 in Synucleinopathies. Exp. Neurobiol. 28, 547–553 (2019).
Rangasamy, S. B. et al. Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J. Clin. Investig. 128, 4297–4312 (2018).
Lax, N., Fainstein, N., Nishri, Y., Ben-Zvi, A. & Ben-Hur, T. Systemic microbial TLR2 agonists induce neurodegeneration in Alzheimer’s disease mice. J. Neuroinflammation 17, 55 (2020).
Kim, C. et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating alpha-synuclein transmission and neuroinflammation. Mol. Neurodegener. 13, 43 (2018).
Wang, D. et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
McAbee, G. N., Brosgol, Y., Pavlakis, S., Agha, R. & Gaffoor, M. Encephalitis Associated with COVID-19 Infection in an 11-Year-Old Child. Pediatr. Neurol. 109, 94 (2020).
Moriguchi, T. et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int. J. Infect. Dis. 94, 55–58 (2020).
Lodigiani, C. et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 191, 9–14 (2020).
Al-Sarraj, S. et al. Invited review: the spectrum of neuropathology in COVID-19. Neuropathol. Appl. Neurobiol. 47, 3–16 (2021).
Song, E. et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 218, e20202135 (2021).
Norouzi, M., Miar, P., Norouzi, S. & Nikpour, P. Nervous System Involvement in COVID-19: a review of the current knowledge. Mol. Neurobiol. 58, 3561–3574 (2021).
Dosch, S. F., Mahajan, S. D. & Collins, A. R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 142, 19–27 (2009).
Zheng, M. et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 22, 829–838 (2021).
Khan, S. et al. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. bioRxiv https://doi.org/10.1101/2021.03.16.435700 (2021).
Qian, Y. et al. Direct activation of endothelial cells by SARS-CoV-2 nucleocapsid protein is blocked by Simvastatin. J. Virol. https://doi.org/10.1128/JVI.01396-21 (2021).
Xia, Y. et al. Reactive microglia enhance the transmission of exosomal alpha-synuclein via toll-like receptor 2. Brain 144, 2024–2037 (2021).
Caplan, I. F. & Maguire-Zeiss, K. A. Toll-Like Receptor 2 Signaling and Current Approaches for Therapeutic Modulation in Synucleinopathies. Front. Pharmacol. 9, 417 (2018).
Kim, C. et al. LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci. Transl. Med. 12, eaay0399 (2020).
Kim, C. et al. Effects of innate immune receptor stimulation on extracellular alpha-synuclein uptake and degradation by brain resident cells. Exp. Mol. Med. 53, 281–290 (2021).
Harapan, B. N. & Yoo, H. J. Neurological symptoms, manifestations, and complications associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease 19 (COVID-19). J. Neurol. 268, 3059–3071 (2021).
Maiese, A. et al. SARS-CoV-2 and the brain: a review of the current knowledge on neuropathology in COVID-19. Brain Pathol. 31, e13013 (2021).
Heneka, M. T., Golenbock, D., Latz, E., Morgan, D. & Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res. Ther. 12, 69 (2020).
Matschke, J. et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 19, 919–929 (2020).
Liu, R. et al. Decreased T cell populations contribute to the increased severity of COVID-19. Clin. Chim. Acta 508, 110–114 (2020).
Du, R. H. et al. Predictors of mortality for patients with COVID-19 pneumonia caused by SARS-CoV-2. Eur. Respir. J. 56, 2000524 (2020).
Schwabenland, M. et al. Deep spatial profiling of human COVID-19 brains reveals neuroinflammation with distinct microanatomical microglia-T-cell interactions. Immunity 54, 1594–1610 e1511 (2021).
Solomon, I. H. et al. Neuropathological Features of Covid-19. N. Engl. J. Med. 383, 989–992 (2020).
Butowt, R. & von Bartheld, C. S. Anosmia in COVID-19: Underlying Mechanisms and Assessment of an Olfactory Route to Brain Infection. Neuroscientist 27, 582–603 (2020).
Meinhardt, J. et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 24, 168–175 (2021).
Armocida, D., Palmieri, M., Frati, A., Santoro, A. & Pesce, A. How SARS-Cov-2 can involve the central nervous system. A systematic analysis of literature of the department of human neurosciences of Sapienza University, Italy. J. Clin. Neurosci. 79, 231–236 (2020).
McQuaid, C., Brady, M. & Deane, R. SARS-CoV-2: is there neuroinvasion? Fluids Barriers CNS 18, 32 (2021).
Deng, W. et al. Ocular conjunctival inoculation of SARS-CoV-2 can cause mild COVID-19 in rhesus macaques. Nat. Commun. 11, 4400 (2020).
Sun, K., Gu, L., Ma, L. & Duan, Y. Atlas of ACE2 gene expression reveals novel insights into transmission of SARS-CoV-2. Heliyon 7, e05850 (2021).
Casagrande, M. et al. Detection of SARS-CoV-2 genomic and subgenomic RNA in retina and optic nerve of patients with COVID-19. Br. J. Ophthalmol. https://doi.org/10.1136/bjophthalmol-2020-318618 (2021).
Penkava, J. et al. Detection of SARS-CoV-2-RNA in post-mortem samples of human eyes. Graefes Arch. Clin. Exp. Ophthalmol. https://doi.org/10.1007/s00417-021-05529-x (2021).
Rhea, E. M. et al. The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat. Neurosci. 24, 368–378 (2021).
Erickson, M. A., Rhea, E. M., Knopp, R. C. & Banks, W. A. Interactions of SARS-CoV-2 with the Blood-Brain Barrier. Int. J. Mol. Sci. 22, 2681 (2021).
Alexopoulos, H. et al. Anti-SARS-CoV-2 antibodies in the CSF, blood-brain barrier dysfunction, and neurological outcome: studies in 8 stuporous and comatose patients. Neurol. Neuroimmunol. Neuroinflamm. 7, e893 (2020).
Frontera, J. A. et al. Comparison of serum neurodegenerative biomarkers among hospitalized COVID-19 patients versus non-COVID subjects with normal cognition, mild cognitive impairment, or Alzheimer’s dementia. Alzheimers Dement. https://doi.org/10.1002/alz.12556 (2022).
Chiricosta, L., Gugliandolo, A. & Mazzon, E. SARS-CoV-2 Exacerbates Beta-Amyloid Neurotoxicity, Inflammation and Oxidative Stress in Alzheimer’s Disease Patients. Int. J. Mol. Sci. 22, 13603 (2021).
Kasparian, K., Graykowski, D. & Cudaback, E. Commentary: APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. Front. Immunol. 11, 1939 (2020).
Safieh, M., Korczyn, A. D. & Michaelson, D. M. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med. 17, 64 (2019).
Zhang, Q. et al. COVID-19 Case Fatality and Alzheimer’s Disease. J. Alzheimer’s Dis. 84, 1447–1452 (2021).
Brown, E. E., Kumar, S., Rajji, T. K., Pollock, B. G. & Mulsant, B. H. Anticipating and Mitigating the Impact of the COVID-19 Pandemic on Alzheimer’s Disease and Related Dementias. Am. J. Geriatr. Psychiatry 28, 712–721 (2020).
Boutoleau-Bretonniere, C. et al. The Effects of Confinement on Neuropsychiatric Symptoms in Alzheimer’s Disease During the COVID-19 Crisis. J. Alzheimer’s Dis. 76, 41–47 (2020).
Ni, W. et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 24, 422 (2020).
Kehoe, P. G., Wong, S., Al Mulhim, N., Palmer, L. E. & Miners, J. S. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-beta and tau pathology. Alzheimer’s Res. Ther. 8, 50 (2016).
Lim, K. H., Yang, S., Kim, S. H. & Joo, J. Y. Elevation of ACE2 as a SARS-CoV-2 entry receptor gene expression in Alzheimer’s disease. J. Infect. 81, e33–e34 (2020).
Zhao, Y., Li, W. & Lukiw, W. Ubiquity of the SARS-CoV-2 receptor ACE2 and upregulation in limbic regions of Alzheimer’s disease brain. Folia Neuropathol. 59, 232–238 (2021).
Fearon, C. & Fasano, A. Parkinson’s Disease and the COVID-19 Pandemic. J. Parkinson’s Dis. 11, 431–444 (2021).
Merello, M., Bhatia, K. P. & Obeso, J. A. SARS-CoV-2 and the risk of Parkinson’s disease: facts and fantasy. Lancet Neurol. 20, 94–95 (2021).
Lee, M. Y., Oh, B. M. & Seo, H. G. Prolonged Dysphagia After a COVID-19 Infection in a Patient With Parkinson Disease. Am. J. Phys. Med. Rehabil. 100, 837–839 (2021).
Jaiswal, V. et al. The Influence of Coronavirus Disease-2019 (COVID-19) On Parkinson’s Disease: an updated systematic review. J. Prim. Care Community Health 12, 21501327211039709 (2021).
Semerdzhiev, S. A., Fakhree, M. A. A., Segers-Nolten, I., Blum, C. & Claessens, M. Interactions between SARS-CoV-2 N-Protein and alpha-Synuclein Accelerate Amyloid Formation. ACS Chem. Neurosci. 13, 143–150 (2022).
Helmich, R. C. & Bloem, B. R. The Impact of the COVID-19 Pandemic on Parkinson’s Disease: Hidden Sorrows and Emerging Opportunities. J. Parkinson’s Dis. 10, 351–354 (2020).
Ellis, T. & Rochester, L. Mobilizing Parkinson’s disease: the future of exercise. J. Parkinson’s Dis. 8, S95–S100 (2018).
Xu, X., Fu, Z. & Le, W. Exercise and Parkinson’s disease. Int. Rev. Neurobiol. 147, 45–74 (2019).
Kainaga, M., Shirota, Y., Kodama, S., Toda, T. & Hamada, M. Effects of the Coronavirus Disease 2019 Pandemic on Motor Symptoms in Parkinson’s Disease: an observational study. Mov. Disord. 36, 2461–2463 (2021).
Wang, Y., Song, E., Bai, B. & Vanhoutte, P. M. Toll-like receptors mediating vascular malfunction: lessons from receptor subtypes. Pharmacol. Ther. 158, 91–100 (2016).
Botos, I., Segal, D. M. & Davies, D. R. The structural biology of Toll-like receptors. Structure 19, 447–459 (2011).
Kawasaki, T. & Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 5, 461 (2014).
Piccinini, A. M. & Midwood, K. S. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010, 672395 (2010).
Jin, M. S. & Lee, J. O. Structures of the toll-like receptor family and its ligand complexes. Immunity 29, 182–191 (2008).
Okun, E., Griffioen, K. J. & Mattson, M. P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 34, 269–281 (2011).
Jana, M., Palencia, C. A. & Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J. Immunol. 181, 7254–7262 (2008).
Yu, J. T., Mou, S. M., Wang, L. Z., Mao, C. X. & Tan, L. Toll-like receptor 2 -196 to -174 del polymorphism influences the susceptibility of Han Chinese people to Alzheimer’s disease. J. Neuroinflammation 8, 136 (2011).
Rezazadeh, M. et al. Genetic Factors Affecting Late-Onset Alzheimer’s Disease Susceptibility. Neuromol. Med. 18, 37–49 (2016).
Reed-Geaghan, E. G., Savage, J. C., Hise, A. G. & Landreth, G. E. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 29, 11982–11992 (2009).
Chen, K. et al. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J. Biol. Chem. 281, 3651–3659 (2006).
McDonald, C. L. et al. Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer’s disease. Brain Behav. Immun. 58, 191–200 (2016).
Pourbadie, H. G. et al. Early minor stimulation of microglial TLR2 and TLR4 receptors attenuates Alzheimer’s disease-related cognitive deficit in rats: behavioral, molecular, and electrophysiological evidence. Neurobiol. Aging 70, 203–216 (2018).
Richard, K. L., Filali, M., Prefontaine, P. & Rivest, S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J. Neurosci. 28, 5784–5793 (2008).
Vollmar, P. et al. Active immunization with amyloid-beta 1-42 impairs memory performance through TLR2/4-dependent activation of the innate immune system. J. Immunol. 185, 6338–6347 (2010).
Doorn, K. J. et al. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2, 90 (2014).
Frank, S., Copanaki, E., Burbach, G. J., Muller, U. C. & Deller, T. Differential regulation of toll-like receptor mRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neurosci. Lett. 453, 41–44 (2009).
Udan, M. L., Ajit, D., Crouse, N. R. & Nichols, M. R. Toll-like receptors 2 and 4 mediate Abeta(1-42) activation of the innate immune response in a human monocytic cell line. J. Neurochem. 104, 524–533 (2008).
Li, X., Xue, L., Sun, J., Sun, Y. & Xie, A. Single nucleotide polymorphisms in the toll-like receptor 2 (TLR2) gene are associated with sporadic Parkinson’s disease in the North-eastern Han Chinese population. Neurosci. Lett. 656, 72–76 (2017).
Kalinderi, K., Bostantjopoulou, S., Katsarou, Z. & Fidani, L. TLR9 -1237 T/C and TLR2 -194 to -174 del polymorphisms and the risk of Parkinson’s disease in the Greek population: a pilot study. Neurol. Sci. 34, 679–682 (2013).
Kim, C., Lee, H. J., Masliah, E. & Lee, S. J. Non-cell-autonomous Neurotoxicity of alpha-synuclein Through Microglial Toll-like Receptor 2. Exp. Neurobiol. 25, 113–119 (2016).
Roodveldt, C. et al. Preconditioning of microglia by alpha-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS ONE 8, e79160 (2013).
Qiao, H. et al. Elevated neuronal alpha-synuclein promotes microglia activation after spinal cord ischemic/reperfused injury. NeuroReport 26, 656–661 (2015).
Daniele, S. G. et al. Activation of MyD88-dependent TLR1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal 8, ra45 (2015).
Kim, N. H. et al. Toll-like receptor 2 downregulation and cytokine dysregulation predict mortality in patients with Staphylococcus aureus bacteremia. BMC Infect. Dis. 20, 901 (2020).
Drouin-Ouellet, J. et al. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 18, pyu103 (2014).
La Vitola, P. et al. Alpha-synuclein oligomers impair memory through glial cell activation and via Toll-like receptor 2. Brain Behav. Immun. 69, 591–602 (2018).
Koo, J. H. et al. Treadmill exercise produces neuroprotective effects in a murine model of Parkinson’s disease by regulating the TLR2/MyD88/NF-kappaB signaling pathway. Neuroscience 356, 102–113 (2017).
Liu, H. Y., Chen, C. Y. & Hsueh, Y. P. Innate immune responses regulate morphogenesis and degeneration: roles of Toll-like receptors and Sarm1 in neurons. Neurosci. Bull. 30, 645–654 (2014).
Kim, C. et al. Antagonizing Neuronal Toll-like Receptor 2 Prevents Synucleinopathy by Activating Autophagy. Cell Rep. 13, 771–782 (2015).
Dzamko, N. et al. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 133, 303–319 (2017).
Stefanis, L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a009399 (2012).
Fang, L., Wu, H. M., Ding, P. S. & Liu, R. Y. TLR2 mediates phagocytosis and autophagy through JNK signaling pathway in Staphylococcus aureus-stimulated RAW264.7 cells. Cell. Signal. 26, 806–814 (2014).
Wang, J. P. et al. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J. Virol. 79, 12658–12666 (2005).
Cai, M. et al. The herpes simplex virus 1-encoded envelope glycoprotein B activates NF-kappaB through the Toll-like receptor 2 and MyD88/TRAF6-dependent signaling pathway. PLoS ONE 8, e54586 (2013).
Wang, W. et al. Up-regulation of IL-6 and TNF-alpha induced by SARS-coronavirus spike protein in murine macrophages via NF-kappaB pathway. Virus Res. 128, 1–8 (2007).
Khanmohammadi, S. & Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 93, 2735–2739 (2021).
Frank, M. G. et al. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav. Immun. 100, 267–277 (2021).
Proud, P. C. et al. Prophylactic intranasal administration of a TLR2/6 agonist reduces upper respiratory tract viral shedding in a SARS-CoV-2 challenge ferret model. EBioMedicine 63, 103153 (2021).
Koh, H. K., Geller, A. C. & VanderWeele, T. J. Deaths From COVID-19. JAMA 325, 133–134 (2021).
Tremblay, M. E., Madore, C., Bordeleau, M., Tian, L. & Verkhratsky, A. Neuropathobiology of COVID-19: the role for Glia. Front. Cell. Neurosci. 14, 592214 (2020).
Varatharaj, A. & Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 60, 1–12 (2017).
Poewe, W. et al. Parkinson disease. Nat. Rev. Dis. Prim. 3, 17013 (2017).
Acknowledgements
We thank the editor for the invitation and contribution. This research was supported entirely by the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
Author information
Authors and Affiliations
Contributions
C.K. conceived the idea and supervised manuscript preparation. M.I., A.N., and E.M. performed the brain tissue analysis. M.P.S. and C.K. prepared draft and M.I., A.N., and E.M. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szabo, M.P., Iba, M., Nath, A. et al. Does SARS-CoV-2 affect neurodegenerative disorders? TLR2, a potential receptor for SARS-CoV-2 in the CNS. Exp Mol Med 54, 447–454 (2022). https://doi.org/10.1038/s12276-022-00755-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00755-7
This article is cited by
-
Virus-induced brain pathology and the neuroinflammation-inflammation continuum: the neurochemists view
Journal of Neural Transmission (2024)
