
Abstract
Cancer immune evasion is one of the hallmarks of carcinogenesis. Cancer cells employ multiple mechanisms to avoid immune recognition and suppress antitumor immune responses. Recently, accumulating evidence has indicated that immune-related pathways are epigenetically dysregulated in cancer. Most importantly, the epigenetic footprint of immune-related pathways is associated with the patient outcome, underscoring the crucial need to understand this process. In this review, we summarize the current evidence for epigenetic regulation of immune-related pathways in cancer and describe bioinformatics tools, informative visualization techniques, and resources to help decipher the cancer epigenome.
Similar content being viewed by others
Introduction
Cancer immunotherapy constitutes a major paradigm shift in cancer care. Recent successes with immune checkpoint inhibitors, most notably CTLA41,2 and PD1/PDL-13,4,5,6 inhibitors, have altered the landscape of systemic therapy for cancer. Despite such breakthroughs, the majority of cancer patients remain refractory to existing cancer immunotherapeutic modalities, highlighting the inherent capacity of tumor cells to evade the immune system. Mechanisms that impede immune surveillance during carcinogenesis are required for tumor cells to progress towards the development of macroscopic tumors. Indeed, cancer cells regulate immune-related pathways to suppress the immune system7 via intricate modulation at the transcriptional, translational, and posttranslational levels7,8,9.
Echoing the pivotal importance of cancer immune evasion, immune-related pathways are frequently dysregulated in cancer. However, for heritable changes to impact the entire tumor tissue, the initial cascade of tolerogenic signals must involve genetic or epigenetic changes, and accordingly, the mutational landscape of cancer includes crucial immune pathways regulating tumor immunity in a subset of patients7. However, loss or gain of function via somatic mutations are rare events, whereas every tumor is required to acquire a tolerogenic immune barrier to survive. In this context, there is a growing consensus that the immune-evasive phenotype of cancer cells relies in part on the epigenetic machinery, which is based on changes that in turn make cancer cells more adaptable. Indeed, cancer cells frequently utilize epigenetic dysregulation to silence tumor suppressors or activate oncogenes10; similarly, carcinogenesis may also require epigenetic reprogramming of immune-related pathways to evade immune killing.
Methylation is one of the major epigenetic mechanisms modulating gene transcription in cancer. Furthermore, increased stochastic variations in methylation events are manifested in the cancer epigenome, thus contributing to tumor heterogeneity11. Recent studies have also demonstrated the crucial role of dysregulated methylation in modulating tumor immunity12. Therefore, in this review, we outline the key immune pathways active in cancer cells, the relevance of methylation in regulating these pathways, and the bioinformatic methodologies and resources to decipher the epigenetic manifestations of cancer immune evasion.
The immune synapse
The immune synapse between Antigen-presenting cells (APCs) and cognate T cells relays two signals. Signal 1 refers to the presentation of a specific peptide antigen by major histocompatibility complex (MHC) molecules on APCs to the cognate T cell receptor on tumor-specific T cells. Signal 2 is the “danger signal” that alerts the immune system to elicit functional immune responses via engagement of costimulatory molecules, degradation of immune checkpoints, and release of proinflammatory cytokines, as APCs recognize conserved danger motifs, such as Toll-like receptor ligands, pathogen-associated molecular patterns (PAMPs), or damage-associated molecular patterns (DAMPs). Indeed, the prototypic immune synapse (Fig. 1a) has now been expanded to encompass the complex interaction between APCs and effector T cells (Fig. 1b), and the qualitative and quantitative signal transmitted through the immune synapse determines the ensuing antitumor immune response. Consistent with this observation, many of the molecules comprising the immune synapse, including but not limited to PD1, CTLA4, Tim3, LAG3, and TIGIT, are targeted by the current generation of cancer immunotherapy and new pipeline drugs (Fig. 1b).

a Schematic of the prototypic immune synapse. b Expanded depiction of the immune synapse between APCs/tumor cells and T cells. A comprehensive interaction between costimulatory and immune checkpoint ligand-receptor pairs is visualized. c STING-cGAS signaling pathway. Sensing of double-stranded DNA by cGAS leads to endogenous generation of cGAMP, which stimulates STING tetramerization and downstream signaling through IRF3, TBK1, and NF-κB to elicit interferon signaling.
More recently, STING and cyclic GMP-AMP synthase (cGAS) have gained considerable attention as representing the pivotal pathway that regulates downstream interferon signaling to provide danger signals. The abnormal presence of cytosolic DNA from dying tumor cells leads to cGAS activation and the generation of cyclic GMP-AMP, which, upon binding to STING, activates TANK-binding kinase 1 (TBK1) and IRF3, ultimately triggering type 1 interferon signaling13. The STING-cGAS pathway also induces NF-κB signaling and the transcription of proinflammatory cytokines, including IL-6 and TNFα (Fig. 1c)13.
Methylation in cancer
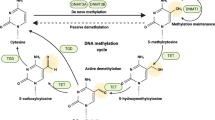
DNA methylation of cytosine residues, primarily on 5’-C-phosphate-G-3 (CpG) dinucleotides, and covalent modifications of histones via acetylation, methylation, phosphorylation, and ubiquitination, confer a heritable epigenetic code that profoundly regulates transcriptional activity in normal tissues and cancer cells. Accumulating evidence shows that aberrant epigenetic reprogramming significantly contributes to tumor initiation and progression14,15. For instance, site-specific promoter hypermethylation of tumor suppressors has been considered the key epigenetic event during carcinogenesis14. More recent studies have shown increased stochastic variations in methylation events in the cancer epigenome11, which may contribute to tumor heterogeneity. Mechanistically, DNA methyltransferase 1 (DNMT1), DNMT3A, and DNMT3B have been shown to be important maintenance and de novo methyltransferases in cancer16,17. By contrast, ten-eleven translocation methylcytosine dioxygenase 1 (TET1), TET2 and TET3 mediate key steps in active DNA demethylation18.
Dysregulated methylation of immune pathways in cancer
In recent years, it has become apparent that immune-related pathways in cancer are regulated by the epigenetic machinery. For instance, mechanisms of human leukocyte antigen (HLA) downregulation include mutations in MHC class I heavy chain genes19,20, mutations in the b2m gene21, mutations in genes encoding JAK/STAT pathway components22 and hypermethylation of MHC class I gene loci23. Furthermore, the expression of costimulatory and immune checkpoint molecules is not limited to immune cells; tumor cells also exploit these interactions to induce a tolerogenic tumor microenvironment7. Indeed, comprehensive profiling of the DNA methylation status of immune synapse genes using level 1 methylation data from 8,186 solid tumors and 745 normal adjacent tissues in 30 solid tumor types from The Cancer Genome Atlas (TCGA) demonstrated hypomethylation of immune checkpoint genes and hypermethylation of costimulatory genes across disease sites in comparison to normal adjacent tissues, thus indicating that these cells adopted an immune-tolerogenic phenotype12. Strikingly, the differential methylation status of the immune synapse genes exhibited prognostic significance in immunogenic cancers, including melanoma, lung cancer, kidney cancer, head and neck cancer, and breast cancer, with associated changes in effector T cell trafficking to the tumor microenvironment12.
Interestingly, the STING-cGAS pathway also appears to be epigenetically regulated in cancer cells24. The methylation status of the STING and cGAS genes in 8426 primary solid tumors and 747 normal adjacent tissues with data deposited in the TCGA database revealed relative hypermethylation of these crucial pathways to suppress cytosolic DNA sensing and interferon signaling24.
Clinical relevance
As dysregulated methylation is deemed critical in modulating antitumor immune responses, translational studies to harness the immunogenic effects of manipulating tumor methylation are underway, with strong evidence from previous preclinical studies demonstrating the efficacy of demethylating agents to augment immunotherapy25,26. Importantly, recent success with demethylating agents in combination with anti-PD1 therapy (nivolumab) in AML suggests the clinical merit of this approach27. However, the negative preliminary findings from the phase II randomized clinical trial of oral 5-azacitidine plus pembrolizumab (a PD1 blocker) vs. pembrolizumab plus placebo in non-small-cell lung carcinoma28 suggest that patient selection may be crucial to harness the therapeutic efficacy of 5-azacitidine. Indeed, given the multifaceted immune tolerance mechanisms employed by cancer cells, the immunogenic effects of 5-azacitidine may be largely limited to patients with unfavorable methylation patterns, suggesting a need for biomarker development to optimize the use of demethylating agents to augment cancer immunotherapy in the clinic.
The Illumina methylation array and bioinformatic pipelines for analysis
The Illumina HumanMethylation450 BeadChip (HM450) proved to be a success, providing an affordable, user-friendly, and high-throughput platform for measuring the DNA methylation status at single-base resolution64. HM450 evaluates over 480,000 CpG sites that predominantly targeting RefSeq genes and CpG islands with 99% and 96% coverage, respectively65.
The MethylationEPIC BeadChip (EPIC) utilizes the same technology and design principles as HM450, but the EPIC chip contains probes for over 850,000 CpG sites, retaining 91.1% percent of the probes on HM45066. EPIC expands coverage of the genome by including 413,745 CpG sites that were not on HM450, 333,265 of which newly target enhancer regions distal to transcription start sites64,66.
Due to the success and popularity of Illumina methylation BeadChips, there is a need for bioinformatics tools that are able to import, preprocess, visualize, and analyze the data produced by these platforms. Specialized software packages that target a specific step in the analysis pipeline are developed and released, often accompanying published research. For example, the implementation of a novel preprocessing method might be made freely available. For many researchers, however, comprehensive software packages that allow the user to easily perform the majority of the steps needed to convert raw data into results are of interest. Table 1 gives an overview of such packages. The majority of the packages are designed for the R programming language and environment67 and are available through the Bioconductor project68,69. Included are three alternatives: a Java-based package, a suite of Python modules, and a web-based application.
Sources of methylation data
Many methylation datasets for different tumor types exist, many of which also include normal samples. Clinical data are frequently included, and some also include matching gene expression data. Below, we describe some of the key sources of methylation datasets, focused on methylation data based on the Illumina Infinium HumanMethylation27 BeadChip (HM27), Illumina Infinium HumanMethylation450 BeadChip (HM450), and Illumina Infinium MethylationEPIC BeadChip (EPIC).
The Cancer Genome Atlas (TCGA, https://www.cancer.gov/tcga) collates molecular data for more than 10,000 tumor and normal tissue samples across 33 different tumor types. The vast majority of the methylation data are from HM450, with some samples from HM27. All data from TCGA are available through the Genomic Data Commons (GDC) data portal70. Normalized methylation data, described in Gao et al.71, are available at https://portal.gdc.cancer.gov/, while the raw idat files can be retrieved through the GDC Legacy Archive (https://portal.gdc.cancer.gov/legacy-archive). Data from GDC can also be downloaded with tools such as TCGAbiolinks72. Moreover, data from individual TCGA studies are available at https://gdc.cancer.gov/about-data/publications, which also includes the complete datasets used in the PanCanAtlas Publications73,74 (https://gdc.cancer.gov/about-data/publications/pancanatlas). This web page provides all types of molecular data, including methylation data, and clinical data for all TCGA samples in simple text files.
The International Cancer Genome Consortium75,76 (ICGC) https://dcc.icgc.org/ is a resource similar to TCGA and also contains TCGA samples. Therapeutically Applicable Research to Generate Effective Treatments (TARGET) is similar to TCGA in the sense that it contains multiple types of molecular data, including methylation data, for several different tumor types. The data are available directly from https://ocg.cancer.gov/programs/target/data-matrix or through the GDC Data Portal.
Gene Expression Omnibus77 (GEO) and ArrayExpress78 are two public molecular data repositories with multiple methylation datasets available. The data can be accessed directly from their corresponding web pages through different application programming interfaces (APIs) and through some of the tools listed in Table 1.
Methylation data are also available from the Cancer Cell Line Encyclopedia79 (CCLE). Multiple types of molecular data, including reduced representation bisulfite sequencing (RRBS) methylation data, are available at https://portals.broadinstitute.org/ccle/data. The publication by Iorio et al.80 provides HM450 methylation data for 1028 CCLE cell lines and is available in GEO (GSE68379) or through ArrayExpress (E-MTAB-3610).
Compiled datasets from GEO, TCGA, and other sources are also available through different methylation databases. EWAS Data Hub81, DiseaseMeth82,83, and PubMeth84 are examples of such databases, but it should be noted that online tools may not always be updated and/or functional, while the source data from GEO, ArrayExpress, and TCGA are always available and up to date.
Visualization of methylation data
Methylation data can be visualized similarly to other types of molecular data, such as gene expression data, e.g., with heatmaps, PCA plots and boxplots, but there are some unique features of methylation data that require some special considerations. The TCGA prostate adenocarcinoma (PRAD) methylation dataset will be used as an example dataset for different types of informative visualizations.
Sample-to-sample density scatter plots
Replicates can be compared using sample-to-sample density scatter plots, as exemplified in Siegel et al.85. In Fig. 2a, four replicates from the TCGA PRAD study are compared by plotting all CpG probes in a scatter plot and assigning colors based on the density of each CpG probe. The density can quickly and easily be calculated using the algorithm described by Eilers and Goeman86. Even if there are several hundred thousand markers in the plot, the density clearly shows how similar each replicate pair is. Most CpG probes are located on or close to the black y=x line, indicating that the CpG probes have the same methylation value across the two replicates. The numbers of CpG probes that have a difference larger than 0.2 and 0.3 (|Δβ| > 0.2, (|Δβ| > 0.3) are listed in the top corner together with the correlation coefficient. It is clear from the density plots and the numbers that replicate 3 is different when compared to the other three replicate pairs.

a Sample-to-sample density scatter plot for the four replicate pairs from the TCGA PRAD methylation dataset. The color indicates the density of the points, and the black line is the y=x line. The Pearson correlation coefficient is listed together with the numbers of probes with |Δβ| > 0.2 and |Δβ| > 0.3. b Histogram of beta-values for the four replicate pairs from the TCGA PRAD methylation dataset. c Scatter plot for the first two principal components, PC1 and PC2, from a PCA model using all the CpG probes and all the samples from the TCGA PRAD methylation dataset. The replicates are indicated by distinct shapes and colors. d Same PCA plot but with the colors and shapes based on the sample type and molecular subtype, respectively.
Beta-value histograms
Histograms of all the beta-values for a sample can be used to find low-quality samples. The beta-value for the eight replicate samples from the TCGA PRAD study is shown in Fig. 2b. A visual inspection of all samples can easily identify samples that do not have a clear bimodal distribution. It is clear from Fig. 2b that replicates 4A and 4B do not show the same bimodal distribution as the other samples, with a much lower right peak and a higher line in the middle. The distribution of beta-values is especially important for samples where there can be a mixed cell population and for FFPE samples where the DNA quality may be poor.
PCA plots
Principal component analysis (PCA)87 score plots can be used to visualize all samples using all CpG probes to find groups, outlier samples, and batch effects. PCA is an unsupervised analysis method that displays the major trends in the data. Figure 2c shows the PCA score plot from a PCA model using the methylation data from the TCGA PRAD samples, including the replicates. The figure highlights the replicates and clearly confirms the findings from the two previous plots. Replicate pairs 1, 2, and 4 are similar to each other, while replicates 3A and 3B are located further from each other. The histogram in Fig. 2b indicates that replicates 4A and 4B are different, which is also confirmed in the PCA plot, since they are located far from the origin.
Figure 2d shows the same PCA score plot as Fig. 2c, but colors are instead assigned by biological subgroup, demonstrating separation based on biology. This plot indicates that the major changes in methylation arise from biological differences and not from batch effects or other technical artifacts. Using distinct colors and shapes improves the visual understanding of the PCA plot. The first principal component, PC1, explains 23.4% of the variation in the data and shows clear separation of the normal samples (green stars) and tumor samples. In other words, the methylation pattern in tumor samples is very different from the methylation pattern in normal samples. The PCA plot suggests that there is less variation in the normal samples (green stars) than in the different types of tumor samples. The second principal component, PC2, explains 12% of the variation and demonstrates separation of tumor samples with TMPRSS2 fusion (blue markers) from those without TMPRSS2 fusion (yellow-red markers). Thus, it becomes clear that the status of TMPRSS2 fusion results in methylation differences between different tumor types. This PCA plot also includes the ~150 samples (gray dots) not used in the TCGA PRAD publication due to RNA quality issues88. The PCA plot indicates that these samples are not different from the original 333 samples, since they are distributed across the whole area and are not separated as a unique group. It is recommended that probes with too many missing values (>50%) are removed before PCA, since they may become overly important to the PCA model. The nonlinear iterative partial least squares (NIPALS) algorithm can handle a moderate number of missing values, obviating the need to completely remove all probes with missing values. There are also available methods to impute missing values, which can be done before the PCA. CpG probes with a narrow range across all samples (e.g., 0.1) can also be removed before the PCA, since they are most likely describing nonbiological changes. It is also recommended to do the analysis on nonscaled data, i.e., without each variable scaled to unit variance. Scaling increases the importance of CpG probes with low variance, which have a lower signal-to-noise ratio. PCA can also be used to summarize the methylation level across multiple CpG probes and genes into one or a few variables12. When summarizing multiple variables using PCA, it is important to confirm that the PCA model describes the expected biology89,90,91.
Gene-level visualization of methylation
Visualizing methylation data for a gene has several challenges. Frequently, there are several CpG probes available for each gene, and the methylation pattern for each CpG probe can be quite different, which makes it problematic to select the CpG probes to include when representing the gene. Using rules such as the position in the gene body (TSS1500, TSS200, 5’UTR, 1st exon, body, or 3’UTR) is not an optimal option, since a CpG probe can be located in different gene regions when the gene has multiple transcripts. Representing the gene by a single CpG probe is not ideal either, since this approach does not use all available data and is more sensitive to missing values and noise. There is also no guarantee that CpG probes within the same gene body region have similar methylation patterns. Displaying the methylation levels across all CpG probes for a gene as boxplots across multiple groups—for example, normal vs. tumor—provides valuable information that can be used when selecting CpG probes. One example of such a plot is the gene structure methylation (GSM) plot92, shown in Fig. 3a. Figure 3a shows the methylation levels of the 15 CpG probes for CD40 in the TCGA PRAD dataset. The methylation level of each CpG probe is shown along the x-axis (ranging from 0 to 1) and is represented by a boxplot for each group, with normal samples shown in green and tumor samples in blue. The genomic location is displayed on the left y-axis, while the right y-axis shows the probe ID with a star (*) indicating a significant difference between the normal and tumor samples. The distance between each probe on the y-axis is logarithmically related to the number of base pairs between the two probes. The leftmost column indicates the presence of CpG islands, and the right column indicates the CpG probes’ positions in the gene structure.

a Gene structure methylation (GSM) plot for CD40 demonstrating the methylation of the 15 CpG probes across the tumor and normal samples from the TCGA PRAD dataset. The x-axis shows the beta-values, which are shown in boxplots for each CpG probe and group, with normal samples shown in green and tumor samples in blue. The left y-axis displays the genomic position, while the right y-axis displays the probe id. The leftmost vertical column indicates CpG islands, and the right vertical column indicates the gene structure location. *q < 0.05 and |Δβ| > 0.1, **q < 0.01 & |Δβ| > 0.2. b The bar plot shows the Pearson correlation coefficient between each CD40 CpG probe and the gene expression level for the TCGA PRAD samples. The heatmap demonstrates the Pearson correlation of the methylation level between each CpG-probe pair. c The CD40 RNAseq gene expression level vs. the average methylation level for the 11 selected CD40 CpG probes using TCGA PRAD samples. Normal samples are shown as green circles, and tumors are shown as blue diamonds.
Twelve of the fifteen CD40 CpG probes show a significant difference in methylation between the normal and tumor samples. Eleven of the fifteen CD40 CpG probes demonstrate hypermethylation in the tumor samples compared to the normal samples, while one CpG probe, cg07222575, shows the opposite trend, with a higher degree of methylation in normal samples. Two of the CpG probes, cg24575067 and cg01149415, demonstrate less variation, with cg24575067 showing a low degree of methylation and cg01149415 showing a high degree of methylation in both normal and tumor samples. This finding clearly exemplifies that CpG probes can show very different methylation patterns for the same gene.
This observation is further confirmed in the correlation heatmap in Fig. 3b, which illustrates the correlation between each probe. The top two probes, cg07222575 and cg06218285, are correlated to each other and to the bottom CpG probe, cg01149415, while showing a negative correlation with all the other CpG probes. Eleven probes, ranging from cg16686951 to cg09053081 and excluding cg4575067, are highly correlated with each other and show a significantly higher methylation level in tumor samples than in normal samples (Fig. 3a). This pattern indicates that the expression of CD40 is suppressed in tumors compared to normal tissues. The bar plot in Fig. 3b clearly demonstrates that these eleven CpG probes show a negative correlation with the gene expression level of CD40. These three figures clearly demonstrate that using the average of these eleven CpG probes provides a good representation of the CD40 methylation level.
The average methylation level of the 11 selected CD40 CpG probes is clearly negatively correlated with the CD40 gene expression level identified by RNAseq, as shown in Fig. 3c. This finding indicates that CD40 is at least partially regulated by methylation in prostate cancer.
Online visualization of methylation data
The figures and graphs described in the previous section can be generated in many programming languages, such as R, MATLAB, Python, and Julia, and through some of the pipelines described in Table 1. Methylation data can also be analyzed and visualized using online tools with preloaded data from TCGA, GEO, and other sources. These tools are great resources for researchers with limited bioinformatics or programming expertise.
DNMIVD (DNA Methylation Interactive Visualization Database) is a web-based tool with many features93. DNMIVD uses TCGA methylation data, RNAseq gene expression data, and survival data together with the cancer hallmark pathways from Zhang et al.94. Both CpG-level and gene-level information are available together with survival information and various quantitative trait loci (QTL) results.
The SMART App is an interactive web application through which TCGA data are also available95. Normal vs. tumor boxplots are provided for specific CpG probes but can also be aggregated for selected CpG probes. Similar boxplots can also be generated to visualize the stage and mutational status. In addition, correlations between methylation and gene expression levels, as well as survival curves, are available.
Wanderer96 provides CpG-probe-level visualization of the methylation status for TCGA data, including both normal and tumor samples. CpG-probe correlation plots for RNAseq data are also available. All results for normal and tumor samples are separated. The data can easily be downloaded.
MEXPRESS97,98 provides a slightly different type of visualization of TCGA methylation data, where many types of clinical data are overlaid with clinical annotations.
MethSurv99 is focused on the survival analysis of patients with 25 types of cancer from TCGA. The analysis is CpG probe specific only, and no gene expression data are available. There are options to create both heatmaps and PCA plots.
Summary
Methylation is one of the main epigenetic mechanisms controlling gene transcription and is thus increasingly recognized as one of the major mechanisms of cancer immune escape. As cancer cells evade the immune system via dysregulated methylation of immune pathway-related genes during carcinogenesis, these alterations evolve into resistance mechanisms against the current immunotherapeutic agents. Epigenetic dysregulation of immune-related pathways in cancers may be mediated by natural selection and clonal expansion of cancer cells that display the immune-evasive phenotype acquired in response to immune surveillance. This evolutionary concept implies the crucial role of tumor-immune crosstalk throughout carcinogenesis and underscores the need for in-depth characterization of the role of methylation in modulating antitumor immune responses, which may reveal actionable targets to overcome immune evasion. There exists a vast amount of methylation data that are ready to be analyzed using well-established bioinformatics pipelines. The results can be visualized in informative figures that clearly show the epigenetic changes in tumor samples.
References
Weber, J. S. et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J. Clin. Oncol. 26, 5950–5956 (2008).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Hamid, O. et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 369, 134–144 (2013).
Weber, J. S. et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 31, 4311–4318 (2013).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812–830.e14 (2018).
Sun, C., Mezzadra, R. & Schumacher, T. N. Regulation and function of the PD-L1 checkpoint. Immunity 48, 434–452 (2018).
Zerdes, I., Matikas, A., Bergh, J., Rassidakis, G. Z. & Foukakis, T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene 37, 4639–4661 (2018).
Ehrlich, M. DNA methylation and cancer-associated genetic instability. Adv. Exp. Med. Biol. 570, 363–392 (2005).
Hansen, K. D. et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 43, 768–775 (2011).
Berglund, A. et al. Methylation of immune synapse genes modulates tumor immunogenicity. J. Clin. Invest. 130, 974–980 (2020).
Barber, G. N. STING: infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770 (2015).
Jones, P. A. & Baylin, S. B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 3, 415–428 (2002).
Jones, P. A. & Baylin, S. B. The epigenomics of cancer. Cell 128, 683–692 (2007).
Sarabi, M. M. & Naghibalhossaini, F. Association of DNA methyltransferases expression with global and gene-specific DNA methylation in colorectal cancer cells. Cell Biochem. Funct. 33, 427–433 (2015).
Zhang, W. & Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 5, 1 (2017).
Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009).
Maleno, I., Lopez-Nevot, M. A., Cabrera, T., Salinero, J. & Garrido, F. Multiple mechanisms generate HLA class I altered phenotypes in laryngeal carcinomas: high frequency of HLA haplotype loss associated with loss of heterozygosity in chromosome region 6p21. Cancer Immunol. Immunother. 51, 389–396 (2002).
Wang, Z., Marincola, F. M., Rivoltini, L., Parmiani, G. & Ferrone, S. Selective histocompatibility leukocyte antigen (HLA)-A2 loss caused by aberrant pre-mRNA splicing in 624MEL28 melanoma cells. J. Exp. Med. 190, 205–215 (1999).
Paschen, A. et al. The coincidence of chromosome 15 aberrations and beta2-microglobulin gene mutations is causative for the total loss of human leukocyte antigen class I expression in melanoma. Clin. Cancer Res. 12, 3297–3305 (2006).
Rodriguez, T. et al. Distinct mechanisms of loss of IFN-gamma mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer 7, 34 (2007).
Serrano, A. et al. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2’-deoxycytidine treatment. Int. J. Cancer 94, 243–251 (2001).
Konno, H. et al. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37, 2037–2051 (2018).
Wrangle, J. et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 4, 2067–2079 (2013).
Chiappinelli, K. B. et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 169, 361 (2017).
Daver, N. et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. 9, 370–383 (2019).
Levy, B. P. et al. Randomised phase 2 study of pembrolizumab plus CC-486 versus pembrolizumab plus placebo in patients with previously treated advanced non-small cell lung cancer. Eur. J. Cancer 108, 120–128 (2019).
Müller, F. et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 20, 55 (2019).
Assenov, Y. et al. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 11, 1138–1140 (2014).
Teschendorff, A. E. et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196 (2013).
Maksimovic, J., Gordon, L. & Oshlack, A. SWAN: Subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44 (2012).
Pidsley, R. et al. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 14, 293 (2013).
Triche, T. J. Jr, Weisenberger, D. J., Van Den Berg, D., Laird, P. W. & Siegmund, K. D. Low-level processing of Illumina Infinium DNA methylation BeadArrays. Nucleic Acids Res. 41, e90–e90 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Smyth, G. K. in Bioinformatics and Computational Biology Solutions Using R and Bioconductor 397–420 (Springer-Verlag, 2005).
Houseman, E. A., Molitor, J. & Marsit, C. J. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 30, 1431–1439 (2014).
Tian, Y. et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33, 3982–3984 (2017).
Morris, T. J. et al. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics 30, 428–430 (2014).
Fortin, J.-P. et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503 (2014).
Butcher, L. M. & Beck, S. Probe Lasso: a novel method to rope in differentially methylated regions with 450K DNA methylation data. Methods 72, 21–28 (2015).
Jaffe, A. E. et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 41, 200–209 (2012).
Peters, T. J. et al. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 8, 6 (2015).
Zhou, W., Triche, T. J. Jr, Laird, P. W. & Shen, H. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 46, e123–e123 (2018).
Fortin, J.-P., Triche, T. J. Jr & Hansen, K. D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 33, 558–560 (2017).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Touleimat, N. & Tost, J. Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 4, 325–341 (2012).
Morante-Palacios, O. & Ballestar, E. shinyÉPICo: a graphical pipeline to analyze Illumina DNA methylation arrays. Bioinformatics https://doi.org/10.1093/bioinformatics/btaa1095 (2021).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Xu, Z., Niu, L. & Taylor, J. A. ENmix: a comprehensive R package for the analysis of Illumina DNA methylation arrays. https://doi.org/doi:10.18129/B9.bioc.ENmix (2020).
Xu, Z., Niu, L., Li, L. & Taylor, J. A. ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 44, e20–e20 (2016).
Xu, Z., Langie, S. A. S., De Boever, P., Taylor, J. A. & Niu, L. RELIC: a novel dye-bias correction method for Illumina Methylation BeadChip. BMC Genomics 18, 4 (2017).
Niu, L., Xu, Z. & Taylor, J. A. RCP: a novel probe design bias correction method for Illumina Methylation BeadChip. Bioinformatics 32, 2659–2663 (2016).
Pedersen, B. S., Schwartz, D. A., Yang, I. V. & Kechris, K. J. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 28, 2986–2988 (2012).
Xu, Z., Xie, C., Taylor, J. A. & Niu, L. ipDMR: identification of differentially methylated regions with interval P-values. Bioinformatics https://doi.org/10.1093/bioinformatics/btaa732 (2020).
Hu, X., Tang, L., Wang, L., Wu, F.-X. & Li, M. MADA: a web service for analysing DNA methylation array data. BMC Bioinformatics 21, 403 (2020).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA 98, 5116–5121 (2001).
Kolde, R., Märtens, K., Lokk, K., Laur, S. & Vilo, J. seqlm: an MDL based method for identifying differentially methylated regions in high density methylation array data. Bioinformatics 32, 2604–2610 (2016).
FOXO BioScience. methylprep. https://github.com/FOXOBioScience/methylprep (2021).
FOXO BioScience. methylcheck. https://github.com/FOXOBioScience/methylcheck (2021).
FOXO BioScience. methylize. https://github.com/FOXOBioScience/methylize (2021).
Almeida, D. et al. Efficient detection of differentially methylated regions using DiMmeR. Bioinformatics 33, 549–551 (2017).
Almeida, D. et al. Jllumina—A comprehensive Java-based API for statistical Illumina Infinium HumanMethylation450 and Infinium MethylationEPIC BeadChip data processing. J. Integr. Bioinformatics 13, 24–32 (2016).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 208, https://doi.org/10.1186/s13059-016-1066-1 (2016).
Bibikova, M. et al. High density DNA methylation array with single CpG site resolution. Genomics 98, 288–295 (2011).
Moran, S., Arribas, C. & Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 8, 389–399 (2016).
R Core Team. R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, 2020).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Huber, W. et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015).
Grossman, R. L. et al. Toward a shared vision for cancer genomic. Data. N. Engl. J. Med. 375, 1109–1112 (2016).
Gao, G. F. et al. Before and after: comparison of legacy and harmonized TCGA genomic data Commons’ data. Cell Syst. 9, 24–34.e10 (2019).
Mounir, M. et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 15, e1006701 (2019).
Cancer Genome Atlas Research Network et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Hutter, C. & Zenklusen, J. C. The Cancer Genome Atlas: creating lasting value beyond its data. Cell 173, 283–285 (2018).
Zhang, J. et al. International Cancer Genome Consortium Data Portal—a one-stop shop for cancer genomics data. Database J. Biol. Databases Curation, https://doi.org/10.1093/database/bar026 (2011).
International Cancer Genome Consortium et al. International network of cancer genome projects. Nature 464, 993–998 (2010).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Athar, A. et al. ArrayExpress update—from bulk to single-cell expression data. Nucleic Acids Res. 47, D711–D715 (2019).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Iorio, F. et al. A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016).
Xiong, Z. et al. EWAS Data Hub: a resource of DNA methylation array data and metadata. Nucleic Acids Res. 48, D890–D895 (2020).
Lv, J. et al. DiseaseMeth: a human disease methylation database. Nucleic Acids Res. 40, D1030–D1035 (2012).
Xiong, Y. et al. DiseaseMeth version 2.0: a major expansion and update of the human disease methylation database. Nucleic Acids Res. 45, D888–D895 (2017).
Ongenaert, M. et al. PubMeth: a cancer methylation database combining text-mining and expert annotation. Nucleic Acids Res. 36, D842–D846 (2008).
Siegel, E. M. et al. Expanding epigenomics to archived FFPE tissues: an evaluation of DNA repair methodologies. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 23, 2622–2631 (2014).
Eilers, P. H. C. & Goeman, J. J. Enhancing scatterplots with smoothed densities. Bioinformatics Oxf. Engl. 20, 623–628 (2004).
Jolliffe, I. T. in Principal component analysis 129–155 (Springer, 1986).
Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015).
Venet, D., Dumont, J. E. & Detours, V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput. Biol. 7, https://doi.org/10.1371/journal.pcbi.1002240 (2011).
Berglund, A. E., Welsh, E. A. & Eschrich, S. A. Characteristics and validation techniques for PCA-based gene-expression signatures. Int. J. Genomics 2017, 2354564 (2017).
Dhawan, A. et al. Guidelines for using sigQC for systematic evaluation of gene signatures. Nat. Protoc. 14, 1377–1400 (2019).
Berglund, A. E. et al. Accessible pipeline for translational rsearch using TCGA: examples of relating gene mechanism to disease-specific outcomes. Methods Mol. Biol. Clifton NJ 2194, 127–142 (2021).
Ding, W. et al. DNMIVD: DNA methylation interactive visualization database. Nucleic Acids Res. 48, D856–D862 (2020).
Zhang, H. et al. Cooperative genomic alteration network reveals molecular classification across 12 major cancer types. Nucleic Acids Res. 45, 567–582 (2017).
Li, Y., Ge, D. & Lu, C. The SMART App: an interactive web application for comprehensive DNA methylation analysis and visualization. Epigenet. Chromatin 12, https://doi.org/10.1186/s13072-019-0316-3 (2019).
Díez-Villanueva, A., Mallona, I. & Peinado, M. A. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenet. Chromatin 8, 22 (2015).
Koch, A., De Meyer, T., Jeschke, J. & Van Criekinge, W. MEXPRESS: visualizing expression, DNA methylation and clinical TCGA data. BMC Genomics 16, https://doi.org/10.1186/s12864-015-1847-z (2015).
Koch, A., Jeschke, J. Van Criekinge, W., van Engeland, M. & De Meyer, T. MEXPRESS update 2019. Nucleic Acids Res. 47, W561–W565 (2019).
Modhukur, V. et al. MethSurv: a web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 10, 277–288 (2017).
Acknowledgements
This work has been supported in part by the Biostatistics and Bioinformatics Shared Resource at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292). The results shown here are based in part upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Berglund, A., Putney, R.M., Hamaidi, I. et al. Epigenetic dysregulation of immune-related pathways in cancer: bioinformatics tools and visualization. Exp Mol Med 53, 761–771 (2021). https://doi.org/10.1038/s12276-021-00612-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00612-z
This article is cited by
-
Inhibition of USP7 enhances CD8+ T cell activity in liver cancer by suppressing PRDM1-mediated FGL1 upregulation
Acta Pharmacologica Sinica (2024)
-
Novel target and treatment agents for natural killer/T-cell lymphoma
Journal of Hematology & Oncology (2023)
