
Abstract
Blinding eye diseases such as corneal neovascularization, proliferative diabetic retinopathy, and age-related macular degeneration are driven by pathological angiogenesis. In cancer, angiogenesis is key for tumor growth and metastasis. Current antiangiogenic treatments applied clinically interfere with the VEGF signaling pathway—the main angiogenic pathway—to inhibit angiogenesis. These treatments are, however, only partially effective in regressing new pathologic vessels, and the disease relapses following cessation of treatment. Moreover, the relapse of pathological angiogenesis can be rapid, aggressive and more difficult to treat than angiogenesis in the initial phase. The manner in which relapse occurs is poorly understood; however, recent studies have begun to shed light on the mechanisms underlying the revascularization process. Hypotheses have been generated to explain the rapid angiogenic relapse and increased resistance of relapsed disease to treatment. In this context, the present review summarizes knowledge of the various mechanisms of disease relapse gained from different experimental models of pathological angiogenesis. In addition, the basement membrane—a remnant of regressed vessels—is examined in detail to discuss its potential role in disease relapse. Finally, approaches for gaining a better understanding of the relapse process are discussed, including prospects for the management of relapse in the context of disease.
Similar content being viewed by others

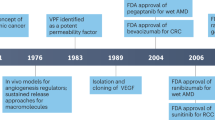
Introduction
Angiogenesis is the growth of new blood vessels from a pre-existing vascular plexus and is an important process characterizing the progression of pathological conditions such as tumors1 and blinding eye diseases such as age-related macular degeneration (AMD), diabetic retinopathy2, and corneal neovascularization3. The angiogenic process involves the breakdown of endothelial cell (EC) junctions, for example, mediated by vascular endothelial growth factor (VEGF)-induced VE–cadherin endocytosis4. This process leads to vessel dilation, degradation of the basement membrane (BM), and EC proliferation in response to this angiogenic growth factor. BM degradation is mediated by the proteolytic activity of matrix metalloproteinases (MMPs), such as MMP-2 and MMP-9, which are largely recognized as key players in angiogenesis5,6. ECs in their quiescent state do not express active MMPs; however, under certain conditions, such as during inflammation, a common denominator in pathological angiogenesis, ECs are activated to express abundant levels of these proteases7. The activity of MMPs is regulated endogenously by tissue inhibitors of metalloproteinases8. The VEGF family of ligands in mammals is composed of secreted related growth factor members, including VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF). The biological function of these ligands is mediated by protein kinase receptors (VEGFRs), i.e., VEGFR-1, −2, and −39. The ligands interact with receptors with varying affinities. In addition, the ligands interact with the nontyrosine kinase receptors neuropilin-1 and neuropilin-2 (Fig. 1). VEGF-A binds principally to VEGFR-2, a cell surface receptor on vascular ECs, leading to increased permeability of vessels, the expression of mediators of BM and ECM degradation, and the proliferation and migration of ECs, leading to sprouting angiogenesis10.

a Simplified schematic illustration of the VEGF family ligands VEGF-A, -B, -C, and -D interacting with their receptors VEGR-1, -2 and -3 to drive lymphangiogenesis and angiogenesis. Neuropilin receptors (NRP1 and NRP2) enhance VEGF signaling by acting as coreceptors for VEGFR-1 and VEGFR-2. b Angiogenesis, the growth of new blood vessels from pre-existing vessels, is regulated mainly by the VEGF signaling pathway. Growth factors in the VEGF family, such as VEGF-A, VEGF-C, VEGF-D, and PlGF, as well as other factors, such as the fibroblast growth factor (FGF) family, angiopoietin-1 (Ang-1), and MMPs, are important for the sprouting of new vessels. The newly formed blood vessels are initially leaky but over time undergo maturation characterized by deposition of BM pericyte and mural cell coverage, regulated by pathways such as PDGF-B signaling via PDGFR-B and ANG-1 signaling via Tie2, and TGF-β133. Mature blood vessels are maintained in a functional state by factors such as VEGF-B. Under pathological conditions, such as in the tumor microenvironment, these processes are dysregulated, leading to poorly developed vessels that leak fluid and form a disorganized vascular network134.
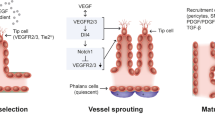
The angiogenic front of the sprouting vessel is characterized by two EC phenotypes: stalk and tip ECs. The acquisition of these cell phenotypes is highly regulated. Upregulation of VEGFR-2 expression promotes tip cell behavior, and within stalk cells, DLL4-Notch signaling is activated to limit VEGFR-2 expression, thus maintaining a proliferative but nonmigratory stalk cell phenotype11,12. The VEGF/Notch signaling pathway regulates the selection of tip and stalk cells to facilitate directionality at the angiogenic front13. The tip EC migrates through the degraded extracellular matrix (ECM) with the aid of filopodia, following the concentration gradient of the secreted growth factor(s), while the stalk cell lags14. The newly formed vessels are initially leaky, immature, and disorganized but become mature over time through the deposition of BM around the growing vessel and by pericyte coverage. The formed vasculature is maintained in a functional state by the expression of genes such as VEGF-B, which has been shown to be important for the maintenance of vessels in the cornea15.
Most studies investigating pathological angiogenesis have focused on how to regress pathological vessels, with little attention given to the concept of disease relapse, even though disease relapse is a major clinical challenge in cancers and in blinding eye diseases16,17. To date, a few studies have investigated this phenomenon, putting forward different theories to explain rapid disease relapse17,18,19. The current knowledge, however, is based on different theories derived from different experimental models of pathological angiogenesis. There is a need to summarize and carefully analyze this knowledge, as understanding it is an important step for advancing the knowledge of disease relapse through further study and developing strategies for more effective therapeutic outcomes when treating disease relapse. This review summarizes the current knowledge of the described mechanisms for rapid disease relapse in different contexts of pathological angiogenesis. In addition, the role of the basement membrane—a remnant of regressed vessels—is examined in detail in relation to its role in promoting or inhibiting rapid disease relapse.
Vessel regression may define revascularization
EC apoptosis is one of the key stages in capillary regression and is thought to be triggered by a lack of hemodynamic forces and by macrophages in response to a diminished angiogenic microenvironment20. Meeson et al. suggested that EC apoptosis during capillary regression is sequential and can be attributed to two causes21. The first cause is described to be macrophage dependent. While studying regression of the pupillary membrane, a transient capillary network located in the anterior chamber of the developing eye, Lang et al. noted that the pupillary membrane consisted of ECs in a network of capillaries and that cell death during capillary regression was caused by apoptosis. They observed that apoptosis occurred either within a single EC in a healthy vessel or synchronously along the entire segment of the capillary segment. They speculated that apoptosis was mediated by macrophages, that elicited cell death of a target EC22. In line with this, liposome-mediated macrophage depletion in the eye resulted in the persistence of ECs and vessels, which would have regressed normally in the presence of macrophages. Rescue experiments involving the introduction of bone marrow-derived macrophages induced apoptosis and capillary regression23. In another study, macrophages were found to induce the apoptosis of ECs and pericytes in a cell cycle-dependent manner, specifically in the G1 phase24.
A second suggested cause of regression by EC death, as proposed by Meeson et al., involves the coordinated apoptosis of vascular endothelial cells mediated by lack of blood flow21. ECs sense shear stress (resulting from flow) through adhesive protein activation, glycocalyx elongation, bending of primary cilia, caveolae-mediated regulation of Ca+, ion channel activation, G protein-coupled receptor activation, and tyrosine kinase receptor activation25,26. These mechanisms are the means by which ECs adopt a specific cell shape and function. In veins, for instance, ECs are polygonal in shape, while arterial ECs are spindle-like and point in the direction of flow27. The magnitude, temporal characteristics and spatial gradient of fluid shear stress are key determinants of the EC transcriptome28,29. Previous studies showed that shear stress-aligned ECs exhibited altered junctional inclination30, reduced the expression of nitric oxide following reversed flow31, and reduced vessel permeability32. A study by Wang et al. showed that the direction of flow was an important stimulus for regulating the EC gene expression profile, as activation of the eNOS pathway was maximal when ECs were parallel to the flow, whereas NFkB signaling was active when the ECs were perpendicular to the direction of the flow33.
In vitro, laminar shear stress was shown to suppress the expression of the proapoptotic Fas receptor and upregulate the antiapoptotic genes FasExo6Del and Bcl-x(L) in HUVECs34. Low shear stress, on the other hand, induces EC apoptosis by activating Akt signaling and producing increased intracellular levels of reactive oxygen species35,36. Shear stress may also regulate apoptosis in ECs through cytochrome C release from mitochondria37 and trophoblast-induced EC apoptosis38 or via PI3K/Akt-dependent signaling39. Interestingly, a recent study showed that blocking EC apoptosis in regressing retinal vessels promotes ischemic tissue revascularization. The preserved ECs were found to reassemble into functional networks to restore the blood supply rapidly following exposure to hypoxia40. Another study showed that the proinflammatory mediators interleukin-1β (IL), tumor necrosis factor α (TNF), and thrombin regulate vessel regression because of increased levels of phospho-p38 and phospho-MLC2 and decreased levels of phospho-Pak2, acetylated tubulin, phospho-cofilin, and pro-caspase341. In general, regressed vessels are devoid of ECs, and they leave behind empty basement sleeves (embs, i.e., the remnants of regressed vessels), which are imprints of a previous vascular network, as shown here by immunohistochemistry to identify the basement membrane marker type IV collagen and the vascular endothelium marker CD31 (Fig. 2).

a–c Regressed blood vessels are devoid of the vascular endothelium (CD31) but retain the basement membrane which is positive for type IV collagen (CoI IV) (arrows) in the magnified region of C as indicated by dashed lines. d Schematic representation of the process of capillary regression as observed in a suture induced model of inflammatory corneal angiogenesis. Functional vessels undergo vasoconstriction, cessation of blood flow, apoptosis of the vascular endothelial cells and loss of supporting pericytes, leaving behind empty basement membrane sleeves18.
Treatment of pathological angiogenesis
Antiangiogenic therapies targeting growth factors such as VEGF are used to induce regression of pathological vessels42,43. Aptamers such as Macugen were developed as antiangiogenic agents that bind human VEGF-A to prevent signaling via VEGF receptors, thus inhibiting activation of a downstream signaling cascade to inhibit pathological angiogenesis44. Bevacizumab is a commonly used antiangiogenic agent. Bevacizumab is a recombinant full-length humanized monoclonal antibody against VEGF-A165 that binds and blocks the biologically active forms of VEGF to inhibit angiogenesis45. Bevacizumab was initially approved for the treatment of colon cancer but is now used as an off-label treatment of various cancers and blinding eye diseases46. Ranibizumab is an anti-VEGF agent with the Fab region of the same monoclonal antibody as bevacizumab and is approved for the treatment of neovascular AMD47. The efficacy of ranibizumab is similar to that of bevacizumab48; however, ranibizumab is more expensive. Aflibercept, also known as VEGF trap, is a decoy receptor for several VEGF family ligands, i.e., VEGF-A, VEGF-B, and PIGF. Aflibercept was created by fusing the immunoglobulin (Ig) domains of VEGFR-1 and VEGFR-2 to the constant region (Fc portion) of human IgG1. Aflibercept binds VEGF-A, VEGF-B, and PlGF with great affinity, thereby inhibiting angiogenesis49. Other antiangiogenic treatments include receptor tyrosine kinase inhibitors (TKIs), which compete with ATP for tyrosine kinase-binding sites to reduce tyrosine kinase phosphorylation and thereby inhibit cancer cell proliferation. VEGFR TKIs, such as sorafenib, inhibit RAF-1, and VEGFR-3 activity50, and sunitinib, a multitarget kinase inhibitor, targets VEGFR, PDGFR-α, and PDGFR-β51. Despite being approved for cancer treatment, rapid emergence of resistance followed by increased tumor invasiveness limits the clinical benefit of sunitinib as an angiogenesis inhibitor52,53, and similarly, innate and acquired resistance to sorafenib is common54. Treatment of pathological angiogenesis remains a major clinical challenge regardless of the drug used. Targeting a specific growth factor (such as VEGF) can lead to toxicity, nonresponse among patients, and resistance of the pathological vessels to the modulation of a specific pathway55,56. Furthermore, relapse of the disease is reported following cessation of treatment in both cancer and eye diseases; however, how relapse occurs is not fully understood.
Relapse of pathological angiogenesis
In many cancers, anti-VEGF treatment must be given continuously, but this leads to toxicity, worsening the quality of life and/or exacerbating drug resistance mediated by treatment-induced epigenetic changes; therefore, a treatment ‘holiday’ may be required to allow reversion to a previous epigenetic profile57. However, relapse of the pathology can occur during these breaks from treatment58,59. In the eye, AMD is a chronic and progressive disease affecting the central retina. AMD causes blindness mainly among the elderly population in the developed world60 and is classified as either nonexudative, also known as dry AMD; exudative, also known as wet AMD; or neovascular (nAMD)61. nAMD is characterized by an ingrowth of pathologic blood vessels from the choroid into the macular region of the retina62 and is the cause of complications such as subretinal hemorrhage, vitreous hemorrhage, fibrosis, and scarring, which lead to vision loss63. nAMD is less frequent than dry AMD; however, nAMD is responsible for the greatest loss of central vision (90%) associated with AMD64. Intravitreal injection of anti-VEGF drugs such as ranibizumab, aflibercept or bevacizumab has become the standard first-line therapy for the management of nAMD16. Anti-VEGF treatments require 3 monthly doses followed by individualized treatment (called Treat and Extend), 7–9 injections/year, fixed dosing over 4–8 weeks, or monthly assessment for lesion activity and treatment pro re nata (PRN—as needed dosing). These repeated treatments are required because as the effect of anti-VEGF molecules subsides, the disease relapses. Repeated therapy places a large economic burden on health care systems65,66, and these treatments maintain but do not improve overall vision. Moreover, anti-VEGF therapies may be associated with adverse side effects such as increased intraocular pressure67, and there is a growing concern of acquired resistance to anti-VEGF56,68. There is also a group with innate resistance, so-called anti-VEGF “nonresponders”, in which vision loss is characterized by subretinal hemorrhage with persistent fluid/blood leakage47. Cessation of regular anti-VEGF treatment, instituted due to side effects or for other reasons, carries a risk of relapse of choroidal neovascularization in nAMD. Numerous studies have reported relapse of pathological angiogenesis following treatment stoppage. For instance, there is a high risk for relapse of retinopathy of prematurity following monotherapy with intravitreal injection of ranibizumab69,70 and after intravitreal bevacizumab monotherapy71. Following a long-term follow-up, myopic choroidal neovascularization was observed to recur in approximately 40% of patients after intravitreal anti-vascular treatment72. Other study estimates show approximately 30% recurrence of choroidal neovascularization in patients treated with anti-VEGF16,73, and despite these reports, little is understood regarding how recurrence occurs.
At the front of the eye, corneal neovascularization is the ingrowth of pathological vessels from the peripheral limbus toward the center of the normally clear and avascular cornea. A vascularized cornea, however, impairs vision by absorbing and scattering light and triggering a fibrotic wound healing response that irreversibly alters the normally transparent collagen structure of the cornea. Estimates indicate that corneal neovascularization affects 1.4 million people annually, with 12% of these people losing their vision74. Currently, there are no approved treatments for corneal neovascularization; however, VEGF is widely targeted to treating corneal neovascularization. Bevacizumab, for example, has been used off-label for treating corneal neovascularization. In clinical and experimental settings, recurrence of corneal neovascularization has been noted75,76. Recurrence of corneal neovascularization as high as 35% was shown in a prospective interventional case series study following the discontinuation of subconjunctival injections of bevacizumab77. Dexamethasone—a corticosteroid—is effective in regressing pathological vessels in the cornea78; however, this treatment was found to be ineffective for regressing recurrent vessels in a cornea model of pathological angiogenesis79.
In the field of cancer therapy, tumor rebound has been documented, for example, in a case report of a patient with recurrent high-grade glioma. Following discontinuation of bevacizumab treatment, tumor regrowth was found to be more aggressive and treatment-resistant80. In another case report, tumor rebound was noted after discontinuation of aflibercept treatment in metastatic colorectal carcinoma81. Similarly, in preclinical models, the discontinuation of anti-VEGF therapy has been shown to promote metastasis via liver revascularization59.
These reports highlight the challenge presented by disease relapse; however, currently there are no treatments specifically designed to address this phenomenon. Furthermore, the process of disease relapse is still poorly understood at the functional level of angiogenic vessels. To better address this situation, it is important to keep in mind how vessels regress (discussed above) and how this regression may contribute to the regrowth of vessels under the appropriate conditions.
The process of pathological angiogenesis relapse and the role of the basement membrane
Little is known about the processes characterizing the rapid recurrence of pathological angiogenesis following cessation of treatment for tumors and ocular angiogenesis. Only a few recent studies have examined the relapse of pathological angiogenesis in great detail. Using the RIP-Tag2 model of tumor angiogenesis in mice, it was shown that pathological vessels regressed following anti-VEGF treatment; however, relapse occurred by EC reoccupation of the ebms once treatment was stopped19. The ebms were postulated to serve as a scaffold for revascularizing ECs, thus facilitating rapid revascularization, as illustrated in Fig. 3.

a Rapid relapse of tumor angiogenesis is hypothesized to be facilitated by the empty basement membrane sleeves (ebms) of previously regressed vessels, which are postulated to serve as scaffolding for ECs during new angiogenic sprouting, following cessation of antiangiogenic treatment19. In contrast, b rapid relapse of corneal neovascularization is facilitated by dilation and reperfusion of the persistent (treatment-resistant vessels) vessels and by de novo sprouting after vessel reperfusion, which do not involve using the ebms as scaffold18.
The study, however, only examined a short-term (7-day) inhibition of pathological angiogenesis, which does not mimic a typical clinical treatment strategy, where treatment typically lasts for several weeks or months. It is of interest to know if a similar rate and pattern of disease relapse are observable after long-term VEGF inhibition. With sustained anti-VEGF treatment over a long period, ebms may not persist long enough to facilitate disease relapse. These aspects are important to address to gain a better understanding of the dynamics of tumor relapse under different treatment time frames. However, despite the lack of detailed knowledge concerning these aspects, the repopulation of initially abandoned ebms is accepted as the de facto mechanism of tumor angiogenic relapse following cessation of treatment.
Tumors treated with combretastatin A4 phosphate, a compound that selectively disrupts blood vessels, were found to be rapidly repopulated with TIE2-expressing macrophages (TEMs), leading to increased expression of the chemokine CXCL12. Depletion of TEMs improved the efficacy of combretastatin A4 phosphate in inhibiting angiogenesis82. This finding illustrates that macrophages promote the rapid relapse of tumor angiogenesis by facilitating a proangiogenic environment by expressing chemokines important for cell migration. Therefore, determining whether TEMs are critical for the relapse of tumor growth following anti-VEGF treatment in various types of cancer is an interesting study objective.
The term tumor microenvironment is often used to describe the tumor in its entirety, including the vascular network, connective tissue, infiltrating immune cells, and ECM. The ECM constitutes up to 60% of the tumor mass, and it differs greatly from that of normal organs. Processes key for tumor survival, such as intertumoral signaling, transport mechanisms, oxygenation, and metabolism, are all potentially regulated by the ECM. Several studies reviewed by Henke et al.83 show that the ECM is important for the outcomes of various forms of cancer therapy, for example, by acting as a barrier protecting tumor cells from the actions of a therapeutic agent.
The extracellular matrix (ECM) is a complex network of polysaccharides, proteoglycans, glycoproteins, and proteins and is a key determinant of the physical and mechanical properties of the tissue, with profound effects on cell behavior84. In terms of angiogenesis, studies have shown that the interaction between ECM components and EC receptors mediates EC growth, survival and differentiation85. The different components of the ECM, such as fibronectin and thrombospondins, are discussed in greater detail in later sections.
We recently investigated in detail the process of relapse of corneal neovascularization using a rat model of suture-induced inflammatory angiogenesis86,87. In this model, a month-long period of vascular regression was induced followed by relapse. The relapse of neovascularization was rapid and more aggressive compared to the initial phase of angiogenesis79, corroborating observations in tumor studies. Initially, the examination of the regressed corneal vascular bed indicated presence of many ebms with a few persisting blood vessels. Examination of the persistent blood vessels by electron microscopy indicated the presence of a degenerating vascular endothelium, basement membrane and supporting pericytes. During a secondary stimulus to induce relapse of corneal neovascularization, the degenerating blood vessels rapidly normalized via EC recovery, vasodilation, and perfusion with tightly packed erythrocytes, facilitating the relapse of corneal neovascularization. In this model, ebms did not serve as conduits for revascularizing ECs18. Rather, during the vascular regression period, the vessels were devoid of ECs, leaving behind ebms. Excess basement membrane probably expressed by the ECs, was deposited at junctional points of the ebms with few remaining nonregressed functional vessels to irreversibly “seal off” the ebms from blood flow, thus preventing sprouting into these structures during revascularization18. In the RIP-Tag2 tumor model from which revascularization hypothesis II was generated, a similar basement membrane deposition pattern was noticeable; however, this excess basement membrane deposition was interpreted to represent basement membrane heterogeneity19. Although observations in the cornea model appear to contradict observations previously reported for the RIP-Tag2 tumor model, it is notably important to note that the two studies were conducted in different models, and importantly, the longer period for vascular regression in the cornea model appeared to allow irreversible changes in the ebms. When recapitulated in the cornea model, a shorter 7-day period of regression led to only partial abandonment of the basement membrane sleeves, which retained some ECs and were not sealed off from the blood flow18. These ECs would presumably have the potential, as in the tumor model, to rapidly repopulate the still functional basement membrane sleeves. Therefore, the duration of antiangiogenic treatment, and not necessarily the model or tissue, may play a key role in determining the functionality of basement membrane sleeves in facilitating relapse. In support of the idea of EC recovery for revascularization as observed in corneal revascularization, a recent study showed that preventing EC apoptosis was key for the revascularization of an ischemic retina. It was observed that the preserved ECs reassembled and sprouted to revascularize the tissue following exposure to hypoxia40.
During the relapse of corneal neovascularization, several proinflammatory and proangiogenic pathways are more strongly activated than are activated during the initial phase of neovascularization. Some of these pathways include leukocyte extravasation, LPS/IL-1–mediated inhibition of RXR function, ILK signaling, production of nitric oxide and ROS signaling, and innate and adaptive immune responses. Genes such as Il-1β and Cxcl2 were increasingly expressed during relapse than during initial angiogenesis79. These findings hint at a potential regulatory basis for the relapse of pathological angiogenesis and a potential memory effect, and some of these pathways can be investigated as targets to inhibit or delay angiogenesis relapse. Investigating whether these pathways and inflammation generally have a similar expression pattern in relapsing tumor angiogenesis is an interesting study direction.
In retinopathy of prematurity, failure of the retina to revascularize was associated with senescence, preceded by oxidative stress in the choroid and in the retinal pigment epithelium. These aspects of the disease were thought to be regulated by the suppressed expression of insulin-like growth factor 1 receptor and by the upregulation of the tumor suppressor p5388. Therefore, determining whether, for example, promoting p53 signaling can be a means of suppressing revascularization in other tissues and in other contexts of pathological angiogenesis is of interest.
It is unclear whether ebms play any direct role in the relapse of nAMD. A recent study investigating revascularization in a mouse model of ischemic retinopathy showed that preventing EC apoptosis delays vessel regression; however, when the vessels eventually regress, isolated clusters of ECs are preserved within the avascular zone of the tissue. Following a second wave of hypoxia stimulation, these EC clusters rapidly formed vascular networks to revascularize the tissue independent of VEGF-A40. In that study, notably, ebms were not used as conduits for revascularization, and sprouting originated from these EC clusters. This mechanism is likely similar to that observed in the cornea model of pathological angiogenesis, and the nonregressed (resistant or persistent) pathological vessels in nAMD may be critical for the relapse of the pathology by dilating and breaking down endothelial barriers leading to the sprouting of new vessels. Recent advancements in retinal imaging provide a more detailed investigation into the mechanisms driving recurrent nAMD. Numerous studies have, for example, used optical coherence tomography angiography (OCT-A) imaging to investigate the vascular response to antiangiogenic treatment by monitoring blood flow patterns. In one study investigating the early effects of treatment discontinuation in choroidal neovascularization, it was noted that anti-VEGF treatment enhanced closure of terminal vessels and that treatment caused increased flow within the trunk during choroidal neovascularization reactivation89. In another study, Alexandra et al., using OCT-A, found that after long-term VEGF inhibition, relapse was characterized by sub-RPE neovascularization90. Studies such as these are shedding light on the processes of pathological angiogenesis relapse in the retina, and this knowledge may influence future treatment regimens for pathological angiogenesis in this tissue.
Why empty basement membrane sleeves may or may not facilitate rapid disease relapse
Ebms have long been thought to be merely dormant structures that remain following regression of vessels; however, these structures are now hypothesized to potentially play roles in the rapid relapse of tumor angiogenesis following cessation of short-duration treatment19. After a longer duration of antiangiogenic treatment, however, such as in the corneal neovascularization model, ebms were found not to be important for the rapid relapse of the pathology cessation of treatment18. It is therefore of interest to examine the potential mechanisms by which the BM—a component of embs—can either support or hinder rapid relapse of pathologic angiogenesis.
The BM is a thin, dense and cross-linked component of the extracellular matrix that borders all cellular monolayers, including the epithelia and endothelia91. The BM is synthesized within and secreted by the epithelia or endothelia. BMs were initially identified in muscle and later in nearly all tissues92. The BM consists of members of the laminin family, nidogens, heparan sulfate proteoglycans, perlecan, and collagens91. Figure 4 shows a schematic of the four major structural components of the BM93.

Schematic illustration of the four major components of the basement membrane (collagen IV, perlecan, nidogen, and laminin).
The BM is highly heterogeneous as a result of variations in the relative amounts of the different BM components and the subtypes participating in BM assembly. Variations in type IV collagen and laminin are the main factors influencing tissue-specific BM composition and are thus critical for the functional diversity of the BM. Mechanisms such as splice variants, tissue-specific gene regulation and posttranscriptional modifications define the composition of a specific BM. Proteolytic posttranslational processing of laminins, for instance, modulates their function and binding affinities for other components of the BM, thus influencing overall BM composition94.
Under normal physiological conditions, the vascular basement membrane supports the vascular endothelium and acts as a barrier to diapedesis of leukocytes; however, transendothelial migration occurs during embryogenesis or in pathologies such as cancer, during cancer cell metastases95, and/or in much more common situations such as inflammation. Migration of cells across the BM is thought to involve several processes, including ICAM-1-mediated leukocyte rolling96, local degradation of the BM via expression of matrix metalloproteinases, or cellular invadopodia breaching small imperfections in the BM barrier97. Among the many functions of BMs, the ability to sequester and act as a growth factor reservoir is relevant for revascularization. Growth factors such as fibroblast growth factor bind heparan sulfate proteoglycan (HSPG) and are enzymatically cleaved and released as soluble ligands98. Cleavage, however, is not believed to be a prerequisite for the functionality of HSPG-bound growth factors, as HS may act as a cofactor presenting ligands during signaling. A previous study analyzing the growth factors in reconstituted basement membrane found that bFGF, EGF, and PDGF were all present in this basement membrane and that, when released, these growth factors promoted cell proliferation99. In the bovine cornea, bFGF was found to bind to heparan sulfate, and this binding was speculated to be an inherent mechanism for regulating angiogenesis100. This ability of BM to bind and store growth factors may support the theory of its role in facilitating rapid revascularization by acting as scaffold for revascularization by endothelial cells following cessation of antiangiogenic treatment. The rapid rate of the observed revascularization, however, indicate that ECs must divide and migrate very rapidly to repopulate ebms.
Collagen—a major component of the BM
Collagens constitute a group of proteins characterized by Gly-X-Y motifs, with types I, II, and III being classical examples of collagens containing long Gly-X-Y motifs. The vascular BM consists of varying amounts of type IV, XV, and XVIII collagen. One study found that type XVIII collagen is the predominant collagen type in the BM of capillaries in the lung alveolus. In addition, BM with type XV collagen is predominant in capillaries of the heart, muscle layer of small intestines, and skeletal muscle101. Of the collagens, type IV collagen makes up 50% of the entire BM and is essential for BM assembly and stability102. Type IV collagen is expressed and secreted as protomers, which are heterodimers composed of three α chains.
Type XV collagen
Type XV collagen is expressed in various tissues, muscle cells, fibroblasts and ECs and is in the basement membrane of vessels103,104. Type XV collagen-deficient mice were shown to develop normal vasculature and vascular BMs105. Abnormal capillaries were, however, detected in the heart and skeletal muscle in the bacterium C. elegans, which possesses a gene homologous to that coding for Type XV in vertebrates106. Cleavage of the C-terminal NCI domain of type XV collagen results in the production of restin, a molecule shown to possess antiangiogenic properties in vitro by inhibiting bFGF-induced EC migration and in a xenograft carcinoma model in vivo107,108. Therefore, the degradation of type XV collagen would likely result in the generation of molecules such as restin and endostatin, which would exert antiangiogenic effects during disease relapse.
Type XVIII collagen
Type XVIII collagen is a precursor of endostatin, a molecule known to possess antiangiogenic properties109. Endostatin is a carboxyl-terminal fragment of collagen XVIII generated from protease activity. Similar to angiostatin, endostatin inhibits EC proliferation and was shown to mediate antiangiogenic properties by inhibiting EC proliferation and tumor growth109,110. Endostatin is proposed to function by preventing TNF-induced activation of JNK signaling111 and by causing cell cycle arrest of ECs112. In vitro, endostatin was shown to specifically induce apoptosis of ECs by increasing the activity of caspase 3, leading to DNA degradation. In addition, the expression of the antiapoptotic proteins Bcl2 and Bcl-XL was reduced with endostatin treatment110. In another study, endostatin in the presence of bFGF was shown to induce tyrosine kinase phosphorylation of shb adapter protein to enhance the apoptosis rate of ECs113. These reports demonstrate the antiangiogenic role of endostatin. If ebms, as remnants of regressed vessels, are digested by proteases during a vascular regression phase, antiangiogenic molecules would be expected to be present in the local ECM during disease relapse, possibly favoring ebms abandonment over their reuse as scaffolding for revascularizing ECs.
Type IV collagen
Type IV collagen is a major structural component of the BM consisting of six separate α-chains, α1 (IV)–α6 (IV), which assemble into three distinct promoters114. During angiogenesis, type IV collagen forms gradually around the developing vascular endothelium, appearing initially in patchy deposits, which later develop into a continuous stabilization structure;115 however, the exact regulatory mechanisms underlying this process are not well understood. In one study, type IV collagen was shown in vitro to promote both the proliferation and stabilization of endothelial sprouts in a rat aortic ring assay116. The function of type IV collagen in the elongation and stabilization of microcapillaries was dose-dependent; however, with low concentrations of type IV collagen promoting elongation, and high concentrations of type IV collagen stabilizing microcapillaries116. In this study, the authors speculated that type IV collagen induced elongation by enhancing morphogenetic recruitment of ECs from the aortic explant, given that previous reports had shown that ECM molecules induced capillary morphogenesis in a planar culture system117,118. The role of stabilizing vessels at high concentrations is not limited to type IV collagen, as studies have shown that laminin-entactin also enhances the stability of vessels119.
Canstatin is a fragment of the α2 chain of type IV collagen and has been shown to possess antiangiogenic properties. For instance, canstatin reduced the expression of angiopoietin‐1 to inhibit lymphangiogenesis in vitro and in vivo120. Similar antiangiogenic and antilymphatic activities have been reported in tumors (reviewed by Mundel et al.)121,122. Additionally, Magnon et al. showed that the antiangiogenic properties of canstatin are mediated via mitochondrial damage as a result of integrin-induced caspase-8-promoted apoptosis123. A similar mode of action was proposed through the use of a gastric cancer xenograft model124. Mechanistically, canstatin inhibits the phosphorylation of key elements involved in the VEGF signaling pathway, such as Akt and focal adhesion kinase, to inhibit cell proliferation and migration125. Other described modes of action of canstatin involve the induction of Fas-dependent apoptosis of ECs126. Degradation of the BM by proteases results in the generation of canstatin, which mediates angiogenic properties, making the BM less favorable for reuse by revascularizing ECs during disease relapse.
Tumstatin is a 28 kD noncollagenous domain fragment (NC1) cleaved from the α-3 chain of collagen IV and is largely known as an endogenous inhibitor of angiogenesis127. A tumstatin peptide was shown to prevent hypertrophy in diabetic nephropathy by inhibiting alterations in glomerular hypertrophy, hyperfiltration and albuminuria127. In a streptozotocin-induced diabetes mouse model, tumstatin inhibited inflammatory cell infiltration and the expression of angiogenesis-associated molecules such as VEGF and ANG-2127. In another study, tumstatin was shown to bind the collagen-binding domain of MMP2, inhibiting its activation and thus mediating antiangiogenic properties in a laser-induced choroidal neovascularization model128. In ECs, tumstatin mediates antiangiogenic properties and promotes apoptosis via the inhibition of Cap-dependent translation (protein synthesis) mediated by the focal adhesion kinase/phosphatidylinositol 3-kinase/Akt/mTOR/4E-BP1 pathway129. Ascertaining the exact role of tumstatin during relapse of pathological angiogenesis is an interesting goal, given that this antiangiogenic peptide is produced by MMP-9 proteolysis of the α-3 chain of collagen IV130.
Fibronectin, a glycoprotein component of the ECM, is a provisional matrix highly expressed around developing vessels during embryogenesis but is almost undetectable in adult vasculature. The expression of this component of the ECM is, however, found to be reactivated under pathological angiogenesis. A study showed that fibronectin, acting via its receptor integrin α5β1, regulates angiogenic pathways that are distinct from VEGF-mediated angiogenic pathways83. In the OIR model where the VEGF receptor tyrosine kinase inhibitor KRN633 was used to block VEGF signaling to establish capillary-free zones in the central and peripheral retinas of neonatal mice, revascularization was found to occur following the cessation of treatment, with the re-formed fibronectin (provisional matrix) serving as a scaffold for revascularization131.
Another glycoprotein component of the ECM, thrombospondins (TSP 1 and 2), are known to exhibit antiangiogenic properties. A study showed that TSP1 and TSP2 induce the apoptosis of microvascular ECs by binding to the transmembrane glycoprotein CD36 receptor, impairing EC function and tube formation132.
Following the regression of blood vessels, it is not yet clear whether the ebms remain as hollow tubes or whether they collapse soon after EC abandonment. Ascertaining the behavior of these structures over time is of interest because the results will lead to better understanding of the rapid disease relapse process in tumors and corneal neovascularization following the cessation of treatment. It is likely that the reoccupation of collapsed ebms may require activation of a subset of genes different from those required for the reoccupation of hollow ebms. Since embs composed of collagen IV dominate the regressed vascular bed, the facilitation, not necessarily the inhibition of disease relapse, deserves closer investigation given the abundant antiangiogenic properties described for collagen IV and its various protein derivatives, including canstatin, tumstatin, endostatin, and restin.
Conclusions and future prospects
Pathological angiogenesis is a major clinical challenge in cancer and in eye disease. As the search for more effective therapies for pathological angiogenesis continues, future approaches should aim to inhibit initial angiogenesis more completely to prevent relapse of the pathology. Persistent, incompletely regressed vessels likely serve as the main sources of vessel regrowth and reactivation and ultimately disease relapse. Therefore, a sustained period within an antiangiogenic environment is required to regress active angiogenic vessels and deactivate the remaining ebms. Disease relapse is observed clinically in ocular and tumor angiogenesis and is often managed by a similar regimen as that used to treat the initial angiogenesis. Disease relapse, however, can be more rapid and aggressive than disease in the initial phase. For example, in the cornea, dexamethasone, a corticosteroid shown to efficiently inhibit initial angiogenesis, was found to be ineffective in regressing pathological vessels during disease relapse79. This observation may indicate that potentially different regulatory mechanisms drive the process of initial angiogenesis and relapse79. Although higher therapeutic doses can be given, the higher levels may increase toxicity and side effects associated with the treatment. Here, two hypotheses were discussed to explain the rapid relapse of pathology after terminating antiangiogenetic treatment. One hypothesis described in the context of tumor angiogenesis implicated ebms as key structures driving rapid revascularization by acting as conduits for EC migration during relapse after a short-duration treatment19. Several questions, however, remain unanswered regarding this hypothesis; for example, if ebms act as conduits after several weeks of sustained antiangiogenic treatment, what are the roles of persistent vessels and their vasodilation response in relapse? How fast must ECs migrate and proliferate to rapidly repopulate ebms? Why have attempts to degrade ebms using inhibitors been ineffective in preventing rapid relapse19? In addition, numerous components of the basement membrane possess antiangiogenic properties, which may interfere with inhibiting relapse.
An alternative hypothesis is that partially regressed, persistent vessels, and not ebms, are the main drivers of the relapse of pathologic angiogenesis18. Persistent vessels have been shown to facilitate rapid relapse of corneal neovascularization upon return to a proangiogenic environment after four weeks of sustained vessel regression18. In the corneal model, ebms were not found to be reoccupied by ECs during relapse; instead, new capillary sprouts emerged alongside dormant ebms18. Rapid and aggressive relapse was instead shown to be linked to hyperdilation, hyperperfusion of the persisting vessels, and elevated inflammatory and angiogenic signaling during the relapse phase. The results differ with respect to ebms used as scaffolds during rapid relapse of pathology; however, these differences need to be interpreted with caution because MMPs, which are important for angiogenesis, are proteolytic enzymes that degrade BM into its derivatives, which possess antiangiogenic properties. MMPs also digest the ECM, releasing ECM-bound growth factors, which in turn attract inflammatory cells that express additional growth factors to drive angiogenesis. Degradation of the BM, as expected during the initial phase of angiogenesis, however, may result in the generation of BM-derived antiangiogenic fragments such as canstatin, tumstatin, and arrestin, whose antiangiogenic functions were discussed in detail above. These fragments may slow down or even inhibit angiogenesis to prevent relapse of the pathology. MMPs in relation to the BM, ECM and angiogenesis can therefore be considered “double-edged swords”; on the one hand they initiate angiogenesis through the proteolytic release of ECM-bound growth factors, and on the other hand, they inhibit angiogenesis via the generation of BM-derived anti-angiogenic fragments. It is likely that a fine balance is maintained to tightly regulate proangiogenic and antiangiogenic effects. Furthermore, observations that persistent corneal vessels (treatment-resistant vessels) may facilitate rapid relapse18 warrant further investigation to ascertain why these vessels do not fully regress in response to antiangiogenic treatment, such as anti-VEGF or dexamethasone treatment. Addressing this issue is important, as immunologic rejection of grafts following corneal transplantation and regrowth of choroidal neovessels in nAMD can occur via the reactivation of pre-existing pathologic vessels.
Regardless of whether ebms are reused, relapse after cessation of antiangiogenic treatment is an established fact. Where the reuse of ebms during relapse was not apparent, angiogenesis proceeded in the usual manner, with rapid vasodilation of persistent vessels followed by sprouting angiogenesis, albeit this process progressed away from the abandoned ebms. Angiogenesis relapses, regardless of the fate of ebms, are critical and warrant new strategies to inhibit relapse. In this context, observations that ebms are not reused as scaffolding by revascularizing EC is an important finding. In the study by Mukwaya et al., dense collagen IV deposits were found at the junction points between the embs and the persisting perfused vessels, which presumably completely seal off the ebms18 from the flow, thereby leading to irreversible EC apoptosis and the abandonment of vessel segments. Understanding this sealing process may lead to new treatment approaches by, for example, inducing excess collagen IV deposition in new sprouts to block new sprout growth and the connection of sprouts to parent perfused vessels. Rather than promoting rapid revascularization, collagen IV may be useful in the quest to inhibit pathologic angiogenic sprouting and relapse. This possibility leads to an interesting line of inquiry for future studies.
In summary, knowledge of the mechanisms of relapse of pathological angiogenesis is still in its infancy. Relapse should therefore be examined in greater detail at the phenotypic and regulatory levels, ideally in a disease- and tissue-specific manner. Models should be developed to recapitulate the conditions and timing of treatment in the clinical context, and the mechanism for relapse should eventually guide possible treatment strategies.
References
Mousa, S. A. Mechanisms of angiogenesis in vascular disorders: potential therapeutic targets. Drugs Future 23, 51–60 (1998).
Miller, J. W., Le Couter, J., Strauss, E. C. & Ferrara, N. Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology 120, 106–114 (2013).
Epstein, R. J., Stulting, R. D., Hendricks, R. & Harris, D. M. Corneal neovascularization. Pathogenesis and inhibition. Cornea 6, 250–257 (1987).
Gavard, J. & Gutkind, J. S. VEGF controls endothelial-cell permeability by promoting the β-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 8, 1223–1234 (2006).
Van Hinsbergh, V. W., Engelse, M. A. & Quax, P. H. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler. Thromb. Vasc. Biol. 26, 716–728 (2006).
Egeblad, M. & Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 (2002).
Hanemaaijer, R., Koolwijk, P., Le Clercq, L., De Vree, W. & Van Hinsbergh, V. Regulation of matrix metalloproteinase expression in human vein and microvascular endothelial cells. Effects of tumour necrosis factor α, interleukin 1 and phorbol ester. Biochem. J. 296, 803–809 (1993).
Handsley, M. M. & Edwards, D. R. Metalloproteinases and their inhibitors in tumor angiogenesis. Int. J. Cancer 115, 849–860 (2005).
Holmes, D. I. & Zachary, I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol. 6, 1–10 (2005).
Senger, D. R., Perruzzi, C. A., Feder, J. & Dvorak, H. F. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res. 46, 5629–5632 (1986).
Liu, Z.-J. et al. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol. Cell. Biol. 23, 14–25 (2003).
Blanco, R. & Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 3, a006569 (2013).
Hellström, M. et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776–780 (2007).
Benedito, R. et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137, 1124–1135 (2009).
Potente, M., Gerhardt, H. & Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887 (2011).
Adrean, S. D., Chaili, S., Grant, S. & Pirouz, A. Recurrence rate of choroidal neovascularization in neovascular age-related macular degeneration managed with a treat–extend–stop protocol. Ophthalmol. Retin. 2, 225–230 (2018).
Yang, Y. et al. Discontinuation of anti-VEGF cancer therapy promotes metastasis through a liver revascularization mechanism. Nat. Commun. 7, 1–13 (2016).
Mukwaya, A. et al. Revascularization after angiogenesis inhibition favors new sprouting over abandoned vessel reuse. Angiogenesis 22, 553–567 (2019).
Mancuso, M. R. et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J. Clin. Investig. 116, 2610–2621 (2006).
Lang, R. A. & Bishop, J. M. Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell 74, 453–462 (1993).
Meeson, A., Palmer, M., Calfon, M. & Lang, R. A relationship between apoptosis and flow during programmed capillary regression is revealed by vital analysis. Development 122, 3929–3938 (1996).
Lang, R., Lustig, M., Francois, F., Sellinger, M. & Plesken, H. Apoptosis during macrophage-dependent ocular tissue remodelling. Development 120, 3395–3403 (1994).
Diez-Roux, G. & Lang, R. A. Macrophages induce apoptosis in normal cells in vivo. Development 124, 3633–3638 (1997).
Diez-Roux, G., Argilla, M., Makarenkova, H., Ko, K. & Lang, R. A. Macrophages kill capillary cells in G1 phase of the cell cycle during programmed vascular regression. Development 126, 2141–2147 (1999).
Ando, J. & Yamamoto, K. Vascular mechanobiology. Circulation J. 73, 1983–1992 (2009).
Barakat, A. I., Lieu, D. K. & Gojova, A. Secrets of the code: do vascular endothelial cells use ion channels to decipher complex flow signals? Biomaterials 27, 671–678 (2006).
Levesque, M. J., Liepsch, D., Moravec, S. & Nerem, R. M. Correlation of endothelial cell shape and wall shear stress in a stenosed dog aorta. Arteriosclerosis 6, 220–229 (1986).
Zhang, J. & Friedman, M. H. The adaptive response of vascular endothelial cells to an acute increase in shear stress magnitude. Am. J. Physiol.—Heart Circulatory Physiol. 302, H983–H991 (2011).
Blackman, B. R., Garcıa-Cardena, G. & Gimbrone, M. A. A new in vitro model to evaluate differential responses of endothelial cells to simulated arterial shear stress waveforms. J. Biomech. Eng. 124, 397–407 (2002).
Melchior, B. & Frangos, J. A. Shear-induced endothelial cell-cell junction inclination. Am. J. Physiol.—Cell Physiol. 299, C621–C629 (2010).
Lu, X. & Kassab, G. Nitric oxide is significantly reduced in ex vivo porcine arteries during reverse flow because of increased superoxide production. J. Physiol. 561, 575–582 (2004).
Adamson, R. H. et al. Microvascular permeability to water is independent of shear stress, but dependent on flow direction. Am. J. Physiol.-Heart Circ. Physiol. 304, H1077–H1084 (2013).
Wang, C., Baker, B. M., Chen, C. S. & Schwartz, M. A. Endothelial cell sensing of flow direction. Arterioscler. Thromb. Vasc. Biol. 33, 2130–2136 (2013).
Bartling, B. et al. Shear stress-dependent expression of apoptosis-regulating genes in endothelial cells. Biochem. Biophys. Res. Commun. 278, 740–746 (2000).
Dimmeler, S., Assmus, B., Hermann, C., Haendeler, J. & Zeiher, A. M. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ. Res. 83, 334–341 (1998).
Zhang, J. et al. Low shear stress induces human vascular endothelial cell apoptosis by activating Akt signal and increasing reactive oxygen species. Nan Fang. Yi Ke Da Xue Xue Bao 33, 313–317 (2013).
Hu, Y.-L., Hur, S. S., Lei, L., Wang, Y. & Chien, S. Shear stress induces apoptosis via cytochrome c release from dynamic mitochondria in endothelial cells. FASEB J. 31, 689.614–689.614 (2017).
James, J. L., Whitley, G. S. & Cartwright, J. E. Shear stress and spiral artery remodelling: the effects of low shear stress on trophoblast-induced endothelial cell apoptosis. Cardiovasc. Res. 90, 130–139 (2010).
Kim, S. & Woo, C.-H. Laminar flow inhibits ER stress-induced endothelial apoptosis through PI3K/Akt-dependent signaling pathway. Mol. Cells 41, 964 (2018).
Grant, Z. L. et al. Blocking endothelial apoptosis revascularises the retina in a model of ischemic retinopathy. J Clin. Invest. 130, 4235–4251 (2020).
Koller, G. M. et al. Proinflammatory mediators, IL (Interleukin)-1β, TNF (Tumor Necrosis Factor) α, and thrombin directly induce capillary tube regression. Arterioscler. Thromb. Vasc. Biol. 40, 365–377 (2020).
Moshfeghi, A. A. et al. Systemic bevacizumab (Avastin) therapy for neovascular age-related macular degeneration: twenty-four–week results of an uncontrolled open-label clinical study. Ophthalmology 113, 2002–2011. e2002 (2006).
Ferrara, N., Hillan, K. J., Gerber, H.-P. & Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 3, 391 (2004).
Gragoudas, E. S., Adamis, A. P., Cunningham, Jr,E. T., Feinsod, M. & Guyer, D. R. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 351, 2805–2816 (2004).
Presta, L. G. et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 57, 4593–4599 (1997).
Rosenfeld, P. J., Moshfeghi, A. A. & Puliafito, C. A. Optical coherence tomography findings after an intravitreal injection of bevacizumab (Avastin®) for neovascular age-related macular degeneration. Ophthalmic Surg., Lasers Imaging Retin. 36, 331–335 (2005).
Brown, D. M. et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N. Engl. J. Med. 355, 1432–1444 (2006).
Group, C. R. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 364, 1897–1908 (2011).
Holash, J. et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc. Natl Acad. Sci. 99, 11393–11398 (2002).
Jiao, Q. et al. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 17, 36 (2018).
Imbulgoda, A., Heng, D. Y. & Kollmannsberger, C. Sunitinib in the treatment of advanced solid tumors. in Small Molecules in Oncology 165-184 (Springer, 2014).
Ebos, J. M. et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239 (2009).
Pàez-Ribes, M. et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231 (2009).
Zhu, Y.-j, Zheng, B., Wang, H.-y & Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharm. Sin. 38, 614–622 (2017).
Kane, R. C. et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 12, 7271–7278 (2006).
Yang, S., Zhao, J. & Sun, X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: a comprehensive review. Drug Des. Dev. Ther. 10, 1857 (2016).
Tonini, G., Imperatori, M., Vincenzi, B., Frezza, A. M. & Santini, D. Rechallenge therapy and treatment holiday: different strategies in management of metastatic colorectal cancer. J. Exp. Clin. Cancer Res. 32, 92 (2013).
Kuroda, Y. et al. Factors associated with recurrence of age-related macular degeneration after anti-vascular endothelial growth factor treatment: a retrospective cohort study. Ophthalmology 122, 2303–2310 (2015).
Yang, Y. et al. Discontinuation of anti-VEGF cancer therapy promotes metastasis through a liver revascularization mechanism. Nat. Commun. 7, 12680 (2016).
Lim, L. S., Mitchell, P., Seddon, J. M., Holz, F. G. & Wong, T. Y. Age-related macular degeneration. Lancet 379, 1728–1738 (2012).
Bhutto, I. & Lutty, G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 33, 295–317 (2012).
Gass, J. D. M., Agarwal, A., Lavina, A. M. & Tawansy, K. A. Focal inner retinal hemorrhages in patients with drusen: an early sign of occult choroidal neovascularization and chorioretinal anastomosis. Retina 23, 741–751 (2003).
Rasmussen, A. & Sander, B. Long-term longitudinal study of patients treated with ranibizumab for neovascular age-related macular degeneration. Curr. Opin. Ophthalmol. 25, 158–163 (2014).
Feder, R. S. et al. Comprehensive adult medical eye evaluation Preferred Practice Pattern® guidelines. Ophthalmology 123, 209–236 (2016).
Hollingworth, W., Jones, T., Reeves, B. C. & Peto, T. A longitudinal study to assess the frequency and cost of antivascular endothelial therapy, and inequalities in access, in England between 2005 and 2015. BMJ Open 7, e018289 (2017).
Patel, S. Medicare spending on anti–vascular endothelial growth factor medications. Ophthalmol. Retin. 2, 785–791 (2018).
Silva, R. et al. The SECURE study: long-term safety of ranibizumab 0.5 mg in neovascular age-related macular degeneration. Ophthalmology 120, 130–139 (2013).
Bergers, G. & Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592 (2008).
Chan, J. J. et al. Risk of recurrence of retinopathy of prematurity after initial intravitreal ranibizumab therapy. Sci. Rep. 6, 1–7 (2016).
Wong, R. K., Hubschman, S. & Tsui, I. Reactivation of retinopathy of prematurity after ranibizumab treatment. Retina 35, 675–680 (2015).
Hu, J. et al. Reactivation of retinopathy of prematurity after bevacizumab injection. Arch. Ophthalmol. 130, 1000–1006 (2012).
Kang, H. M. & Koh, H. J. Ocular risk factors for recurrence of myopic choroidal neovascularization: long-term follow-up study. Retina 33, 1613–1622 (2013).
Potter, M. J. & Szabo, S. M. Recurrence of choroidal neovascularization after photodynamic therapy in patients with age-related macular degeneration. Br. J. Ophthalmol. 91, 753–756 (2007).
Lee, P., Wang, C. C. & Adamis, A. P. Ocular neovascularization: an epidemiologic review. Surv. Ophthalmol. 43, 245–269 (1998).
Chen, W.-L. et al. Subconjunctival injection of bevacizumab (avastin) on corneal neovascularization in different rabbit models of corneal angiogenesis. Investig. Ophthalmol. Vis. Sci. 50, 1659–1665 (2009).
Lin, C.-T. et al. The different effects of early and late bevacizumab (Avastin) injection on inhibiting corneal neovascularization and conjunctivalization in rabbit limbal insufficiency. Investig. Ophthalmol. Vis. Sci. 51, 6277–6285 (2010).
Chu, H.-S., Chen, T.-C., Hu, F.-R. & Chen, W.-L. Recurrence of corneal neovascularization associated with lipid deposition after subconjunctival injection of bevacizumab. Cornea 32, 1446–1453 (2013).
Mirabelli, P., Peebo, B. B., Xeroudaki, M., Koulikovska, M. & Lagali, N. Early effects of dexamethasone and anti-VEGF therapy in an inflammatory corneal neovascularization model. Exp. Eye Res. 125, 118–127 (2014).
Mukwaya, A. et al. Repeat corneal neovascularization is characterized by more aggressive inflammation and vessel invasion than in the initial phase. Investig. Ophthalmol. Vis. Sci. 60, 2990–3001 (2019).
Zuniga, R. M. et al. Rebound tumour progression after the cessation of bevacizumab therapy in patients with recurrent high-grade glioma. J. Neuro-Oncol. 99, 237–242 (2010).
Oumrani, S. et al. Tumour growth increased following antiangiogenic interruption: the challenge of tumour evaluation. Anti-Cancer Drugs 28, 1062–1065 (2017).
Welford, A. F. et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J. Clin. Investig. 121, 1969–1973 (2011).
Kim, S., Bell, K., Mousa, S. A. & Varner, J. A. Regulation of angiogenesis in vivo by ligation of integrin α5β1 with the central cell-binding domain of fibronectin. Am. J. Pathol. 156, 1345–1362 (2000).
Hynes, R. O. The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219 (2009).
Varner, J. The role of vascular cell integrins αvß3 and αvß5 in angiogenesis. in Regulation of Angiogenesis 361-390 (Springer, 1997).
Peebo, B. B., Fagerholm, P., Traneus-Röckert, C. & Lagali, N. Cellular level characterization of capillary regression in inflammatory angiogenesis using an in vivo corneal model. Angiogenesis 14, 393 (2011).
Mukwaya, A. et al. Factors regulating capillary remodeling in a reversible model of inflammatory corneal angiogenesis. Sci. Rep. 6, 32137 (2016).
Zhou, T. E. et al. The inability of the choroid to revascularize in oxygen-induced retinopathy results from increased p53/miR-Let-7b activity. Am. J. Pathol. 189, 2340–2356 (2019).
Lumbroso, B., et al. Optical coherence tomography angiography study of choroidal neovascularization early response after treatment. In OCT Angiography in Retinal and Macular Diseases, Vol. 56 77-85 (Karger Publishers, 2016).
Miere, A. et al. Optical coherence tomography angiography changes in early type 3 neovascularization after anti-vascular endothelial growth factor treatment. Retina 37, 1873–1879 (2017).
Yurchenco, P. D. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 3, a004911 (2011).
Bowman, W. XXI. On the minute structure and movements voluntary muscle. Philos. Trans. R Soc. Lond. 130, 457–501 (1840).
Paulsson, M. Basement membrane proteins: structure, assembly, and cellular interactions. Crit. Rev. Biochem. Mol. Biol. 27, 93–127 (1992).
Tunggal, P., Smyth, N., Paulsson, M. & Ott, M. C. Laminins: structure and genetic regulation. Microsc. Res. Technol. 51, 214–227 (2000).
Sherwood, D. R., Butler, J. A., Kramer, J. M. & Sternberg, P. W. FOS-1 promotes basement-membrane removal during anchor-cell invasion in C. elegans. Cell 121, 951–962 (2005).
Springer, T. A. Adhesion receptors of the immune system. Nature 346, 425–434 (1990).
Huber, A. R. & Weiss, S. J. Disruption of the subendothelial basement membrane during neutrophil diapedesis in an in vitro construct of a blood vessel wall. J. Clin. Investig. 83, 1122–1136 (1989).
Vlodavsky, I., Miao, H.-Q., Medalion, B., Danagher, P. & Ron, D. Involvement of heparan sulfate and related molecules in sequestration and growth promoting activity of fibroblast growth factor. Cancer Metastasis Rev. 15, 177–186 (1996).
Vukicevic, S. et al. Identification of multiple active growth factors in basement membrane Matrigel suggests caution in interpretation of cellular activity related to extracellular matrix components. Exp. Cell Res. 202, 1–8 (1992).
Folkman, J. et al. A heparin-binding angiogenic protein-basic fibroblast growth factor-is stored within basement membrane. Am. J. Pathol. 130, 393 (1988).
Tomono, Y. et al. Epitope-defined monoclonal antibodies against multiplexin collagens demonstrate that type XV and XVIII collagens are expressed in specialized basement membranes. Cell Struct. Funct. 27, 9–20 (2002).
Kühn, K. et al. Macromolecular structure of basement membrane collagens: Identification of 7 S collagen as a crosslinking domain of type IV collagen. FEBS Lett. 125, 123–128 (1981).
Kivirikko, S., Saarela, J., Myers, J. C., Autio-Harmainen, H. & Pihlajaniemi, T. Distribution of type XV collagen transcripts in human tissue and their production by muscle cells and fibroblasts. Am. J. Pathol. 147, 1500 (1995).
Myers, J. C., Dion, A. S., Abraham, V., Amenta, P. S. & Type, X. V. collagen exhibits a widespread distribution in human tissues but a distinct localization in basement membrane zones. Cell Tissue Res. 286, 493–505 (1996).
Muona, A., Eklund, L., Väisänen, T. & Pihlajaniemi, T. Developmentally regulated expression of type XV collagen correlates with abnormalities in Col15a1−/− mice. Matrix Biol. 21, 89–102 (2002).
Eklund, L. et al. Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc. Natl Acad. Sci. 98, 1194–1199 (2001).
Ramchandran, R. et al. Antiangiogenic activity of restin, NC10 domain of human collagen XV: comparison to endostatin. Biochem. Biophys. Res. Commun. 255, 735–739 (1999).
John, H., Radtke, K., Ständker, L. & Forssmann, W.-G. Identification and characterization of novel endogenous proteolytic forms of the human angiogenesis inhibitors restin and endostatin. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 1747, 161–170 (2005).
O’Reilly, M. S. et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88, 277–285 (1997).
Dhanabal, M. et al. Endostatin induces endothelial cell apoptosis. J. Biol. Chem. 274, 11721–11726 (1999).
Yin, G. et al. Endostatin gene transfer inhibits joint angiogenesis and pannus formation in inflammatory arthritis. Mol. Ther. 5, 547–554 (2002).
Scappaticci, F. A. et al. Combination angiostatin and endostatin gene transfer induces synergistic antiangiogenic activity in vitro and antitumor efficacy in leukemia and solid tumors in mice. Mol. Ther. 3, 186–196 (2001).
Dixelius, J. et al. Endostatin-induced tyrosine kinase signaling through the Shb adaptor protein regulates endothelial cell apoptosis. Blood 95, 3403–3411 (2000).
Boutaud, A. et al. Type IV collagen of the glomerular basement membrane evidence that the chain specificity of network assembly is encoded by the noncollagenous nc1 domains. J. Biol. Chem. 275, 30716–30724 (2000).
Nicosia, R. & Madri, J. The microvascular extracellular matrix. Developmental changes during angiogenesis in the aortic ring-plasma clot model. Am. J. Pathol. 128, 78 (1987).
Bonanno, E., Iurlaro, M., Madri, J. A. & Nicosia, R. F. Type IV collagen modulates angiogenesis and neovessel survival in the rat aorta model. Vitr. Cell. Dev. Biol.—Anim. 36, 336–340 (2000).
Kubota, Y., Kleinman, H. K., Martin, G. R. & Lawley, T. J. Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J. Cell Biol. 107, 1589–1598 (1988).
Madri, J. A. & Williams, S. K. Capillary endothelial cell cultures: phenotypic modulation by matrix components. J. Cell Biol. 97, 153–165 (1983).
Nicosia, R. F., Bonanno, E., Smith, M. & Yurchenco, P. Modulation of angiogenesis in vitro by laminin-entactin complex. Dev. Biol. 164, 197–206 (1994).
Hwang‐Bo, J., Yoo, K. H., Park, J. H., Jeong, H. S. & Chung, I. S. Recombinant canstatin inhibits angiopoietin‐1‐induced angiogenesis and lymphangiogenesis. Int. J. Cancer 131, 298–309 (2012).
Monboisse, J. C., Oudart, J. B., Ramont, L., Brassart-Pasco, S. & Maquart, F. X. Matrikines from basement membrane collagens: a new anti-cancer strategy. Biochim. Biophys. Acta (BBA)—Gen. Subj. 1840, 2589–2598 (2014).
Mundel, T. M. & Kalluri, R. Type IV collagen-derived angiogenesis inhibitors. Microvasc. Res. 74, 85–89 (2007).
Magnon, C. et al. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with αvβ3 and αvβ5 integrins. Cancer Res. 65, 4353–4361 (2005).
Xing, Y.-N., Deng, P. & Xu, H.-M. Canstatin induces apoptosis in gastric cancer xenograft growth in mice through the mitochondrial apoptotic pathway. Biosci. Rep. 34, e00106 (2014).
Cai, H. et al. Roundabout4 suppresses glioma-induced endothelial cell proliferation, migration and tube formation in vitro by inhibiting VEGR2-mediated PI3K/AKT and FAK signaling pathways. Cell. Physiol. Biochem. 35, 1689–1705 (2015).
Panka, D. J. & Mier, J. W. Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. J. Biol. Chem. 278, 37632–37636 (2003).
Yamamoto, Y. et al. Tumstatin peptide, an inhibitor of angiogenesis, prevents glomerular hypertrophy in the early stage of diabetic nephropathy. Diabetes 53, 1831–1840 (2004).
Akulapalli, S., Gunda, V., Laknaur, A. & Shekhar, B. C. Mechanism of regulation of laser induced choroidal neovascularisation by tumstatin. Investig. Ophthalmol. Vis. Sci. 52, 1794–1794 (2011).
Sudhakar, A. et al. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by αvβ3 and α5β1 integrins. Proc. Natl Acad. Sci. 100, 4766–4771 (2003).
Hamano, Y. et al. Physiological levels of tumstatin, a fragment of collagen IV α3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via αVβ3 integrin. Cancer Cell 3, 589–601 (2003).
Morita, A. et al. The process of revascularization in the neonatal mouse retina following short-term blockade of vascular endothelial growth factor receptors. Cell Tissue Res. 382, 529–549 (2020).
Dawson, D. W. et al. CD36 mediates the in vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol. 138, 707–717 (1997).
Jain, R. K. Molecular regulation of vessel maturation. Nat. Med. 9, 685–693 (2003).
Noguera-Troise, I. et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444, 1032–1037 (2006).
Funding
This work was supported by scholarships from Ögonfonden, from the charity Synskadades Väl, and from the Swedish society of ophthalmology. Open Access funding provided by Linköping University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mukwaya, A., Jensen, L. & Lagali, N. Relapse of pathological angiogenesis: functional role of the basement membrane and potential treatment strategies. Exp Mol Med 53, 189–201 (2021). https://doi.org/10.1038/s12276-021-00566-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00566-2
This article is cited by
-
Modeling early pathophysiological phenotypes of diabetic retinopathy in a human inner blood-retinal barrier-on-a-chip
Nature Communications (2024)
-
Aberrant lncRNA expression in patients with proliferative diabetic retinopathy: preliminary results from a single-center observational study
BMC Ophthalmology (2023)
-
The Significant Role of microRNAs in Gliomas Angiogenesis: A Particular Focus on Molecular Mechanisms and Opportunities for Clinical Application
Cellular and Molecular Neurobiology (2023)
-
Decoding dysregulated angiogenesis in HTLV-1 asymptomatic carriers compared to healthy individuals
Medical Oncology (2023)
