
Abstract
Posttranslational modifications of proteins, such as acetylation, are essential for the regulation of diverse physiological processes, including metabolism, development and aging. Autophagy is an evolutionarily conserved catabolic process that involves the highly regulated sequestration of intracytoplasmic contents in double-membrane vesicles called autophagosomes, which are subsequently degraded after fusing with lysosomes. The roles and mechanisms of acetylation in autophagy control have emerged only in the last few years. In this review, we describe key molecular mechanisms by which previously identified acetyltransferases and deacetylases regulate autophagy. We highlight how p300 acetyltransferase controls mTORC1 activity to regulate autophagy under starvation and refeeding conditions in many cell types. Finally, we discuss how altered acetylation may impact various neurodegenerative diseases in which many of the causative proteins are autophagy substrates. These studies highlight some of the complexities that may need to be considered by anyone aiming to perturb acetylation under these conditions.
Similar content being viewed by others


Introduction
Macroautophagy (hereafter autophagy) is a process mediating the delivery of cytoplasmic components to the lysosome for degradation via double-membrane vesicles called autophagosomes1. In mammalian cells, autophagosomes are formed from cup-shaped precursor structures called phagophores, which include a complex of autophagy proteins, including ATG5, ATG12 and ATG16L12. The membranes of phagophores expand and form enclosed autophagosomes, and completed autophagosomes subsequently fuse with lysosomes2,3. Lysosomal digestion of autophagic cargoes protects cells against starvation and related stresses by releasing recycled building blocks from autophagic substrates.
Acetylation is a major posttranslational modification (PTM) and affects diverse aspects of protein function by altering properties such as stability, hydrophobicity, enzymatic activity, subcellular localization and interactions with other substrates and cofactors in the cell4. In acetylation, the acetyl group of an acetyl-coenzyme (Ac-CoA) can be co- or posttranslationally transferred to either the α-amino group of the N-terminus of a protein (Nt-acetylation) or to the ε-amino group of a lysine residue (K-acetylation). Nt-acetylation is catalyzed by highly conserved Nt-acetyltransferases (NATs) and is considered irreversible. On the other hand, K-acetylation is a reversible modification mediated by lysine acetyltransferases (KATs) at the ε-amino group of lysine residues. The tight regulation of acetylation by these enzymes plays fundamental regulatory roles in development and diverse human diseases, including diabetes and neurodegenerative conditions4.
In this review, we describe how autophagy is regulated by acetylation, particularly K-acetylation, by previously identified KATs and deacetylases (KDACs). We also summarize the therapeutic targeting of acetylation, which may potentially lead to effective strategies to treat neurodegenerative diseases.
Introduction to autophagy
Under normal conditions, cells sustain basal levels of autophagy to maintain homeostasis. However, a variety of stimuli, including nutrient deprivation, metabolic imbalance or cellular stress, can activate autophagy2,3. Autophagosome biogenesis includes three early stages: initiation, nucleation, and expansion of the isolation membrane (Fig. 1), and the process is mediated by autophagy-related proteins (ATGs)2, which were initially discovered in yeast5. Many ATG proteins can be regulated by PTMs, such as phosphorylation, ubiquitination and acetylation6. The Unc-51-like autophagy-activating kinase (ULK) 1/2 complex (consisting of ULK1, ATG13, RB1-inducible coiled-coil protein 1 (FIP200) and ATG101) plays a major role in autophagy as a signaling node for several pathways and by phosphorylating downstream effectors. During the initiation of autophagosome formation, this complex acts as a serine/threonine kinase that phosphorylates Beclin 1 in the vacuolar protein-sorting 34/PI3-kinase (VPS34)/PI3K complex7. Additionally, the ULK1 complex recruits ATG9, which is thought to be involved in delivering membranes to autophagosomal structures and may act as a lipid scramblase8,9,10. The VPS34/PI3K complex generates phosphatidylinositol 3-phosphate (PI3P), which facilitates the recruitment of WD-repeat protein-interacting with phosphoinositides (WIPI2) that recruits the ATG5-ATG12-ATG16L1 complex to the sites of phagophore formation11. This complex enables the conjugation of LC3 and its family members to phosphatidylethanolamine in phagophore membranes12. To degrade the autophagosomal content, autophagosomes must fuse with a functional lysosome, and SNARE proteins mediate this fusion13.

mTORC1 inhibition and AMPK activation positively regulate the ULK1 complex through a series of phosphorylation events. The ULK1 complex subsequently activates VPS34/PI3K complexes, which leads to PI3P synthesis and the nucleation of preautophagosomal structures. PI3P then recruits PI3P effector proteins, namely, WIPIs and the ATG12-ATG5-ATG16L1 complex, which is essential for autophagosome membrane elongation. Subsequent fusion to lysosomes results in the degradation of a variety of substrates, such as protein aggregates, infectious agents, and damaged mitochondria.
Autophagy is tightly regulated by intracellular and extracellular signals14. Mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) integrates signals related to growth and metabolism in response to nutrient and energy levels and negatively regulates autophagy15 through the hosphorylation of ULK1, ATG13, transcription factor EB (TFEB) and other autophagy-related proteins under nutrient-rich conditions.
Regulation of acetylation in cells
Nt-acetylation
Six different NATs have been identified in mammals (NatA to NatF)16. NATs regulate the transfer of an acetyl group from Ac-CoA to the free α-amino group of a polypeptide chain that is being synthesized. NATs can differ in their subunit composition and substrate specificity17. Nt-acetylation can regulate the subcellular localization of proteins, protein stability, and protein-protein interactions4.
K-acetylation
Lysine (K) acetylation depends on the use of Ac-CoA, and also nicotine adenine dinucleotide (NAD+) in the case of sirtuins, a class of lysine deacetylases (KDACs; HDACs), which use it as a co-substrate. This type of acetylation links metabolism with cell signaling, as Ac-CoA and NAD+ are key metabolites18, and modifies pathways that can be reversibly altered by deacetylases (Fig. 2). The K-acetylation and deacetylation of proteins were first studied in histones because of their roles in gene regulation. However, KATs and KDACs also acetylate nonhistone proteins in the nucleus or cytoplasm to regulate major biological processes19. Acetylation also occurs through nonenzymatic mechanisms and is affected by the availability of Ac-CoA20,21.

a Lysine acetylation is a reversible posttranslational modification of proteins, including histones. Proteins can be acetylated at lysine residues (Ac-K) by specific enzymes, i.e., KATs, or deacetylated by KDACs. b Most canonical mammalian KATs are classified into three major families: The p300/CBP, GNAT and MYST family. KDACs are divided into two categories: classical Zn2+ -dependent HDACs and NAD+ -dependent sirtuin deacetylases. KDACs can be further grouped into class I, class IIa, class IIb, class III and class IV.
KATs and KDACs and autophagy regulation
To date, approximately 40 mammalian proteins have been proposed to possess endogenous KAT activity. Thirteen are well characterized (canonical) and can be classified into three major families: the GCN5 (also known as KAT2A) and PCAF (also known as KAT2B) family (together members of the overarching GNAT family); the E1A binding protein p300 (encoded by EP300, also known as KAT3B) and CREB-binding protein (CBP, also known as KAT3A) family; and the MYST family, named for its founding members MOZ (also known as KAT6A), yeast Ybf2, Sas2, and Tip6 (also known as KAT5)22 (Fig. 2). All canonical KATs are predominantly localized in the nucleus and acetylate histones and nonhistone proteins. However, some KATs, such as p300, are nuclear but can be exported to the cytoplasm depending on intracellular signaling23. The substrate specificities of KATs are thought to be defined by their specific subcellular localization, their interacting proteins and the accessibility of lysine residues in substrate proteins19. KATs are found in unique complexes that influence their target specificities and their abilities to interact with other proteins22. More than 2,000 acetylation targets in the nucleus, cytoplasm, mitochondria and endoplasmic reticulum have been identified in human cells24.
The human genome encodes 18 KDACs, and they are divided into two major categories: zinc-dependent KDACs and NAD + -dependent sirtuin deacetylases (Table 1). On the basis of phylogenetic conservation and sequence similarities, zinc-dependent KDACs are further divided into four classes: class I, class IIa, class IIb and class IV25. Class I and class IV KDACs are localized in the nucleus, class IIb KDACs are cytoplasmic, and class IIa KDACs are primarily localized in the nucleus but are also found in the cytoplasm. Sirtuin (SIRT) deacetylases localize to different cellular compartments26, including the nucleus (SIRT1, SIRT6 and SIRT7), cytoplasm (SIRT2) and mitochondria (SIRT3, SIRT4 and SIRT5).
In mammalian cells, KATs and KDACs play pivotal roles in autophagy regulation at multiple steps27 (Table 1). As protein acetylation is a major regulator of gene transcription, the epigenetic regulation of autophagy genes by KATs or KDACs may be important for autophagy regulation. Depending on the target proteins of KATs and KDACs, acetylation has the potential to induce or inhibit autophagy (Table 1).
Regulation of autophagy by p300-dependent acetylation
Among KATs, p300 appears to acetylate many ATG proteins that regulate autophagy at multiple steps27,28,29 (Table 1). p300 depletion or specific p300 inhibitors can induce autophagy, whereas the overexpression of p300 inhibits autophagy28,29.
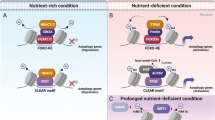
Recently, we reported that under nutrient-depleted conditions, such as amino acid (AA) or leucine starvation, p300-dependent acetylation regulates autophagy through the acetylation of the mTORC1 component Raptor at K109730. This acetylation of Raptor enables the interaction of mTORC1 with the Rag complex on the lysosomal membrane, where mTORC1 is activated. In this way, Raptor acetylation is mediated by leucine, and p300 activation results in mTORC1 activation and autophagy repression. Cells expressing an acetylation-dead mutant of Raptor (Raptor K1097R; KR) manifested autophagy activation without altered acetylation of autophagy-related proteins. Furthermore, p300 activation had no discernible effects on autophagy levels in Raptor KR-expressing cells or in cells where mTORC1 was inhibited. Thus, our data suggest that p300 activity (and leucine) inhibits autophagy primarily by activating mTORC1 rather than by altering the acetylation of other proteins30. This Ac-CoA-p300-Raptor regulation of autophagy via mTORC1 occurs in most cell types, including neurons (Fig. 3).

Ac-CoA activates p300, which acetylates Raptor, leading to mTORC1 activation, which inhibits autophagy. Under nutrient (e.g., leucine) deprivation conditions, autophagy activation is mainly mediated by decreased Raptor acetylation to inhibit mTORC1.
The regulation of autophagy by other KATs
-
1.
GNAT family
In mammalian cells and Drosophila, GCN5 inhibits the biogenesis of autophagosomes and lysosomes by regulating the acetylation of TFEB at lysine 274 and lysine 279. The acetylation of TFEB disturbs its dimerization and its subsequent binding to target gene promoters, many of which regulate autophagy or lysosomal biogenesis31. PCAF, another member of the GNAT family, is reported to regulate autophagy through the inhibition of the mTORC1 pathway in some cancers, such as hepatocellular carcinoma32.
-
2.
MYST family
The MYST acetyltransferase family also appears to regulate autophagy. Serum deprivation leads to the association of the protein kinase GSK3β with TIP60/KAT5 and subsequent phosphorylation at serine 86 of TIP60. Phosphorylated TIP60 acetylates and activates ULK133, which is essential for serum deprivation-induced autophagy. Additionally, under nutrient starvation, the induction of autophagy is coupled to a reduction in histone H4 lysine 16 (H4K16) acetylation through the downregulation of MOF/KAT834.
The regulation of autophagy by KDACs
-
1.
KDACs family
KDAC family members are important for the regulation of autophagy at several levels. HDAC1 has been reported to be overexpressed in hepatocellular carcinoma, and inhibition of HDAC1 induces autophagy to repress tumor cell growth35. Chemical or genetic HDAC1 inhibition also induces autophagy and lysosomal activity in HeLa cells36. However, knocking down HDAC2, but not HDAC1, inhibited autophagy in cardiomyocytes37. In contrast, the deletion of both HDAC1 and HDAC2 in mice blocked autophagic flux in skeletal muscle38. Knocking down class IIa HDAC4 leads to autophagy induction by increasing the expression levels of ATG proteins, including Beclin 1 and ATG739.
MicroRNA-dependent protein acetylation can also regulate autophagy. The regulation of HDAC4 and HDAC5 by miRNA-9 increased total LC3B and Rab7 levels40. Another study showed that the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) decreased HDAC7 protein levels in endometrial stromal sarcoma cells, producing an accumulation of autophagic vacuoles41. Recently, the HDAC8-selective inhibitor HMC was shown to induce autophagy in MCF-7 cells42. In response to hypoxia in a myoblast cell line, class IIa HDAC9 was significantly increased, thereby inhibiting intracellular autophagy through direct binding to the promoter regions of Beclin 1, ATG7 and LC343.
Class IIb HDAC6 is the only mammalian deacetylase that contains a ubiquitin-binding domain; therefore, when the ubiquitin-proteasome system is impaired, this HDAC has an important role in autophagy-dependent protein degradation44. Furthermore, HDAC6 overexpression increases autophagosome formation in various liver cancer cells by activating c‐Jun NH2‐terminal kinase (JNK)45. Additionally, HDAC6 depletion impairs serum starvation-induced autophagy. In serum-starved cervical carcinoma cells, increased LC3 acetylation resulting from HDAC6 inhibition correlated with decreased autophagic flux46. HDAC6 is also important for autophagosome-lysosome fusion. HDAC6 knockout impaired the fusion of autophagosomes and lysosomes by perturbing the formation of F-actin networks mediated by acetylation of cortactin47. Another class IIb deacetylase, HDAC10, promotes autophagy in neuroblastoma cells, and its knockdown disrupts autophagic flux48. In most studies, depletion of class I and IIa HDACs is associated with the enhanced expression of autophagy regulators involved in the induction steps, which results in the upregulation of autophagy. By contrast, inhibition of class IIb HDACs, such as HDAC6 and HDAC10, is more associated with the blockade of autophagic flux.
-
2.
Sirtuins
The sirtuin family of class III HDACs are NAD + -dependent deacetylases that modulate a variety of cellular processes, including energy metabolism, stress responses, cell survival and proliferation. The deacetylation reactions catalyzed by sirtuins are coupled to the cleavage of NAD + into nicotinamide and 1-O-acetyl-ADP ribose. Therefore, sirtuin activities are dependent on the availability of cellular NAD + and are influenced by cellular metabolic status. Seven sirtuins (SIRT1 to SIRT7) have been identified in the human genome, and recent studies have proposed important roles for all sirtuins in the regulation of autophagy49,50,51,52,53,54,55,56. In particular, SIRT1 deacetylates ATG5, ATG7 and LC3 and appears to positively regulate autophagy at several steps49.
KATs and KDACs in neurodegenerative disease
Most of the neurodegenerative diseases in humans are caused by toxic intracytoplasmic, aggregate-prone proteins. Alzheimer’s disease (AD) pathology is characterized by amyloid-beta, an extracellular product derived from amyloid precursor protein (APP), and intracellular aggregated tau57. Parkinson’s disease (PD) is associated with the accumulation of alpha-synuclein (α-syn), and excess levels of this protein are sufficient to cause disease58. Huntington’s disease is a monogenic autosomal dominant disease caused by polyglutamine tract expansions in the huntingtin protein, while amyotrophic lateral sclerosis (ALS) can be either monogenic or complex. The monogenic causes of ALS include mutations in SOD1, FUS and TDP-4359. Importantly, all of these disease-causing intracytoplasmic proteins are autophagy substrates, and autophagy-upregulating drugs and genes enhance the clearance of these proteins and attenuate their toxicities in a range of animal models (flies, zebrafish and mice)60,61,62,63,64,65,66,67,68,69. Autophagy may also protect against neurodegeneration by dampening inflammatory-type processes and apoptosis70,71.
The importance of acetylation regulated by KATs and KDACs in neurodegenerative diseases has been highlighted by observations that imbalanced acetylation causes progressive neuron-specific loss, impaired neuronal function, and eventual neuronal death72. Many studies have reported that abnormal acetylation and deacetylation are linked to the pathogenesis of a variety of neurodegenerative diseases73 (Table 2). We briefly review the relationships between acetylation and different neurodegenerative diseases to reveal some of the complexities that may emerge when perturbing relevant modifying enzymes, as these may impact not only autophagy but also numerous other cellular processes pertinent to neurodegeneration.
-
1.
Alzheimer’s disease (AD)
p300-mediated histone H3 acetylation at the presenilin 1 (PS1) and beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) promoters is upregulated, consequently enhancing the expression levels of these genes in an AD model cell line74. Interestingly, p300 levels are significantly increased in an AD model cell line, suggesting that p300 regulates the expression of AD-related genes by controlling acetylation or their promoters. The overexpression of CBP leads to recovered loss of learning and memory in AD triple transgenic mice75. On the other hand, acetylation of tau can be modulated by p300 and SIRT1, and excess acetylated tau may contribute to tau-mediated neurodegeneration76. Interestingly, hyperactivation of p300/CBP activity has been reported to disrupt autophagic flux and cause excessive tau secretion77. PCAF knockout mice are resistant to Aβ-induced toxicity and memory deficits, an effect that has been attributed to the upregulation of the activity of the Aβ-degrading enzyme Neprilysin78. Impaired function of TIP60 has been described in the human AD hippocampus, and imbalanced TIP60/HDAC2 activity is observed in the brain of an APP Drosophila AD model, suppressing the activities of neuroplasticity genes, which can be rescued by overexpression of TIP6079.
Inactivation of HDAC1 activity by the p25/Cdk5 complex, which is involved in neurodegenerative diseases, including AD, causes double-strand DNA breaks and neurotoxicity, which can be restored by HDAC1 overexpression80. Moreover, HDAC3 promotes tauopathy, whereas suppression of HDAC3 may affect not only nonamyloidogenic APP processing but also neuroprotective gene expression in vitro and in an AD mouse model81. Nuclear translocation of class II HDACs such as HDAC4 and HDAC6 is regulated by Aβ oligomers and the apolipoprotein E ε4 allele (apoE4), which is a critical AD risk factor, resulting in the downregulation of BDNF expression, which is important for controlling synaptic repair and synaptic plasticity82. HDAC6 binds to tau in the perinuclear aggresomal compartment, and HDAC6 levels are upregulated in the hippocampus of AD patients and AD mice83. By contrast, loss of HDAC6 improves learning and memory, α-tubulin acetylation and cognitive function in an AD mouse model84. In the brains of an AD mouse model, overexpression of SIRT1 inhibits Aβ oligomers and plaque burden and ameliorates behavioral deficits, suggesting a neuroprotective role for SIRT1 in AD85. Loss of SIRT2 induces microtubule stabilization and initiation of the subsequent autophagic-lysosomal pathway to degrade toxic Aβ oligomers in an AD-derived cell model85.
-
2.
Parkinson’s disease (PD)
While α-syn is believed to be an important effector of PD because of its activities in the cytoplasm, α-syn may also mediate neurotoxicity by interacting with histone H3, thereby inhibiting histone acetylation by inactivating several KATs, including CBP, p300 and PCAF86. The suppression of SIRT2 by either siRNA or a potent inhibitor prevents α-syn-dependent neurotoxicity, as well as the formation of α-syn inclusions in vitro and in a Drosophila model of PD87. However, the relationship between SIRT2 inhibition and α-syn aggregation is still unclear. In cortical Lewy bodies, α-syn colocalizes with the microtubule-binding proteins MABP1 and tau. Increased acetylation of α-tubulin by the inhibition of SIRT2 may promote the formation of α-syn aggregates by binding to microtubules, suggesting that stabilized microtubules can play an important role in neuroprotection87. Furthermore, the upregulation of SIRT2 prevents microtubule hyperacetylation and axonal degeneration88. The overexpression of SIRT1 increases the lifespan of an α-syn A53T PD mouse model and prevents the formation of α-syn aggregates. SIRT1 deacetylates HSF1 (heat shock factor 1) and thereby increases Hsp70 levels, suggesting that Hsp70 activation can inhibit the α-syn-mediated neurotoxicity of Hsp7089.
-
3.
Huntington’s disease (HD)
CBP is observed in the aggregates formed by mutant huntingtin (mHtt)90. PolyQ expansions, the mutations in the huntingtin protein, directly interact with and sequester CBP and PCAF in animal models, leading to transcriptional dysregulation91. In addition, loss of CBP from the nucleus impairs HAT activity and CBP-mediated gene expression, resulting in neuronal dysfunction and neuronal death92,93. Furthermore, soluble mHtt may enhance ubiquitination to accelerate CBP degradation via the ubiquitin-proteasome system94. mHtt acetylation at lysine 444 (K444) by CBP activation or HDAC1 inhibition promotes its trafficking to autophagosomes and subsequent clearance in primary neurons and a C. elegans HD model, suggesting a role in neuroprotection95. Similarly, HDAC6-mediated retrograde transport on microtubules may facilitate mHtt degradation through autophagy44. Several studies have reported that SIRT1 activity ameliorates mHtt-mediated toxicity in both cellular and animal models. In addition, mHtt suppresses SIRT1 deacetylase activity through a direct interaction causing SIRT1 to remain hyperacetylated, leading to the attenuation of SIRT1-regulated neuroprotective effects96. However, SIRT2 controls HD-related metabolism, such as cholesterol biosynthesis, leading to increased production of cholesterol, further increasing mHtt aggregation97.
-
4.
Amyotrophic lateral sclerosis (ALS)
Transgenic mice expressing the disease-causing mutant protein SOD1 G86D have low levels of histone H3 acetylation and CBP in motor neurons98. Similar to SOD ALS mouse models, low levels of CBP are found in the motor neurons of sporadic ALS patients99. Furthermore, SOD1 mutants may cause disrupted axonal transport and contribute to the loss of mitochondria from axons because of defective microtubule-dependent trafficking100. Interestingly, decreased acetylation of α-tubulin is observed in HAT Elp3-deficient cortical neurons101. Knocking out HDAC6 in SOD1 G93A-expressing mice reduces motor neuron degeneration and increases acetylated α-tubulin without affecting disease onset102. However, conflicting functions of HDAC6 in mice harboring mutant SOD1 have also been reported. Inhibition of HDAC6 promotes the formation of large mutant SOD1 aggregates, which is accompanied by the increased acetylation of α-tubulin and enhanced microtubule retrograde transport. Interestingly, HDAC6 specifically binds to mutant SOD1 through SOD1 mutant interaction region (SMIR) motifs103. Other ALS-causing proteins, namely, TDP-43 and FUS/TLS, appear to interact with HDAC6 to control mRNA expression levels. Moreover, the downregulation of TDP-43 lowers the levels of HDAC6, leading to disrupted aggregate formation104. G93A SOD1 induces DNA damage and subsequently facilitates apoptosis by activating p53105. p53 K320 acetylation is modulated by p300/CBP and PCAF and produces neuroprotective effects, including neurite outgrowth and axon regeneration106. Furthermore, p53 K382 acetylation is controlled by p300/CBP and SIRT1, thereby facilitating neuronal apoptosis106.
Concluding remarks
Although the mechanisms remain incompletely understood, accumulating evidence indicates that different KATs and KDACs play pivotal roles in autophagy regulation at multiple steps of the pathway. New links between protein acetylation and autophagy control are likely to emerge. Acetylation also plays a crucial regulatory role in pathological conditions, particularly in neurodegenerative diseases and cancer. Thus, identifying how acetylation impacts various processes involved in neurodegenerative diseases, including autophagy, will help to inform suitable therapeutic strategies.
References
Mizushima, N. & Komatsu, M. Autophagy: renovation of cells and tissues. Cell 147, 728–741 (2011).
Bento, C. F. et al. Mammalian autophagy: how does it work? Annu Rev. Biochem. 85, 685–713 (2016).
Stamatakou, E. et al. Mendelian neurodegenerative disease genes involved in autophagy. Cell Discov. 6, 24 (2020).
Drazic, A., Myklebust, L. M., Ree, R. & Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta 1864, 1372–1401 (2016).
Tsukada, M. & Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169–174 (1993).
Klionsky, D. J. et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 7, 1273–1294 (2011).
Russell, R. C. et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 15, 741–750 (2013).
Maeda, S. et al. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat. Struct. Mol. Biol., https://doi.org/10.1038/s41594-020-00520-2 (2020).
Matoba, K. et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol., https://doi.org/10.1038/s41594-020-00518-w (2020).
Karanasios, E. et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 126, 5224–5238 (2013).
Axe, E. L. et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701 (2008).
Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 63, 1–10 (2019).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 (2012).
Ravikumar, B. et al. Mammalian macroautophagy at a glance. J. Cell Sci. 122, 1707–1711 (2009).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017).
Aksnes, H., Hole, K. & Arnesen, T. in International Review of Cell and Molecular Biology, Vol 316 Vol. 316 International Review of Cell and Molecular Biology (ed. K. W. Jeon) 267–305 (2015).
Starheim, K. K., Gevaert, K. & Arnesen, T. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 (2012).
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E. & Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 (2014).
Narita, T., Weinert, B. T. & Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 20, 156–174 (2019).
Pougovkina, O. et al. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet 23, 3513–3522 (2014).
Davies, M. N. et al. The acetyl group buffering action of carnitine acetyltransferase offsets macronutrient-induced lysine acetylation of mitochondrial proteins. Cell Rep. 14, 243–254 (2016).
Sheikh, B. N. & Akhtar, A. The many lives of KATs - detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet 20, 7–23 (2019).
Son, S. M. et al. Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab. 29, 192–201 e197 (2019).
Kori, Y. et al. Proteome-wide acetylation dynamics in human cells. Sci. Rep. 7, 10296 (2017).
Haberland, M., Montgomery, R. L. & Olson, E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 (2009).
Houtkooper, R. H., Pirinen, E. & Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238 (2012).
Banreti, A., Sass, M. & Graba, Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 9, 819–829 (2013).
Lee, I. H. & Finkel, T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 284, 6322–6328 (2009).
Huang, R. et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell 57, 456–466 (2015).
Son, S. M. et al. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 11, 3148 (2020).
Wang, Y. et al. Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep., e48335, https://doi.org/10.15252/embr.201948335 (2019).
Jia, Y. L. et al. P300/CBP-associated factor (PCAF) inhibits the growth of hepatocellular carcinoma by promoting cell autophagy. Cell Death Dis. 7, e2400 (2016).
Lin, S. Y. et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336, 477–481 (2012).
Fullgrabe, J. et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 500, 468–471 (2013).
Koeneke, E., Witt, O. & Oehme, I. HDAC family members intertwined in the regulation of autophagy: a druggable vulnerability in aggressive tumor entities. Cells 4, 135–168 (2015).
Oh, M., Choi, I. K. & Kwon, H. J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 369, 1179–1183 (2008).
Cao, D. J. et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl Acad. Sci. USA 108, 4123–4128 (2011).
Moresi, V. et al. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl Acad. Sci. USA 109, 1649–1654 (2012).
Kang, Z. H. et al. Histone deacetylase HDAC4 promotes gastric cancer SGC-7901 cells progression via p21 repression. PLoS ONE 9, e98894 (2014).
Roccaro, A. M. et al. microRNA-dependent modulation of histone acetylation in Waldenstrom macroglobulinemia. Blood 116, 1506–1514 (2010).
Hrzenjak, A. et al. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 216, 495–504 (2008).
Chiu, C. F. et al. Induction of apoptosis and autophagy in breast cancer cells by a novel HDAC8 inhibitor. Biomolecules 9, https://doi.org/10.3390/biom9120824 (2019).
Zhang, Z. et al. Increase in HDAC9 suppresses myoblast differentiation via epigenetic regulation of autophagy in hypoxia. Cell Death Dis. 10, 552 (2019).
Pandey, U. B. et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 (2007).
Jung, K. H. et al. Histone deacetylase 6 functions as a tumor suppressor by activating c-Jun NH2-terminal kinase-mediated beclin 1-dependent autophagic cell death in liver cancer. Hepatology 56, 644–657 (2012).
Liu, K. P. et al. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 441, 970–975 (2013).
Lee, J. Y. et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980 (2010).
Oehme, I. et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl Acad. Sci. USA 110, E2592–E2601 (2013).
Lee, I. H. et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl Acad. Sci. USA 105, 3374–3379 (2008).
Hariharan, N. et al. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 107, 1470–1482 (2010).
Zhao, Y. et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675 (2010).
Mukherjee, S. et al. Effects of Longevinex (modified resveratrol) on cardioprotection and its mechanisms of action. Can. J. Physiol. Pharm. 88, 1017–1025 (2010).
Shi, L. et al. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 13, 358–375 (2019).
Iachettini, S. et al. Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis. 9, 996 (2018).
Lang, A. et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging 9, 2163–2189 (2017).
Ding, M. et al. SIRT7 depletion inhibits cell proliferation and androgen-induced autophagy by suppressing the AR signaling in prostate cancer. J. Exp. Clin. Cancer Res 39, 28 (2020).
Gallardo, G. & Holtzman, D. M. Amyloid-beta and Tau at the crossroads of Alzheimer’s disease. Adv. Exp. Med Biol. 1184, 187–203 (2019).
Ross, O. A. et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 63, 743–750 (2008).
Shatunov, A. & Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis., 105156, https://doi.org/10.1016/j.nbd.2020.105156 (2020).
Ravikumar, B., Duden, R. & Rubinsztein, D. C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117 (2002).
Ravikumar, B. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 (2004).
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N. & Rubinsztein, D. C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013 (2003).
Lopez, A. et al. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 140, 1128–1146 (2017).
Sarkar, S. et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 3, 331–338 (2007).
Williams, A. et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 4, 295–305 (2008).
Menzies, F. M. et al. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 133, 93–104 (2010).
Rose, C. et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet 19, 2144–2153 (2010).
Siddiqi, F. H. et al. Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat. Commun. 10, 1817 (2019).
Menzies, F. M. et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034 (2017).
Ravikumar, B., Berger, Z., Vacher, C., O’Kane, C. J. & Rubinsztein, D. C. Rapamycin pre-treatment protects against apoptosis. Hum. Mol. Genet 15, 1209–1216 (2006).
Rubinsztein, D. C., Bento, C. F. & Deretic, V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med. 212, 979–990 (2015).
Peserico, A. & Simone, C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 371832 (2011).
Saha, R. N. & Pahan, K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 13, 539–550 (2006).
Lu, X. et al. Histone acetyltransferase p300 mediates histone acetylation of PS1 and BACE1 in a cellular model of Alzheimer’s disease. PLoS ONE 9, e103067 (2014).
Caccamo, A., Maldonado, M. A., Bokov, A. F., Majumder, S. & Oddo, S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107, 22687–22692 (2010).
Irwin, D. J. et al. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135, 807–818 (2012).
Chen, X. et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol. Neurodegener. 15, 2 (2020).
Duclot, F., Meffre, J., Jacquet, C., Gongora, C. & Maurice, T. Mice knock out for the histone acetyltransferase p300/CREB binding protein-associated factor develop a resistance to amyloid toxicity. Neuroscience 167, 850–863 (2010).
Panikker, P. et al. Restoring Tip60 HAT/HDAC2 balance in the neurodegenerative brain relieves epigenetic transcriptional repression and reinstates cognition. J. Neurosci. 38, 4569–4583 (2018).
Kim, D. et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60, 803–817 (2008).
Janczura, K. J. et al. Inhibition of HDAC3 reverses Alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc. Natl Acad. Sci. USA 115, E11148–E11157 (2018).
Sen, A., Nelson, T. J. & Alkon, D. L. ApoE4 and Abeta oligomers reduce BDNF expression via HDAC nuclear translocation. J. Neurosci. 35, 7538–7551 (2015).
Ding, H., Dolan, P. J. & Johnson, G. V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 106, 2119–2130 (2008).
Govindarajan, N. et al. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 5, 52–63 (2013).
Jesko, H., Wencel, P., Strosznajder, R. P. & Strosznajder, J. B. Sirtuins and their roles in brain aging and neurodegenerative disorders. Neurochem. Res. 42, 876–890 (2017).
Kontopoulos, E., Parvin, J. D. & Feany, M. B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023 (2006).
Outeiro, T. F. et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519 (2007).
Suzuki, K. & Koike, T. Mammalian Sir2-related protein (SIRT) 2-mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: a crucial role of tubulin deacetylation. Neuroscience 147, 599–612 (2007).
Donmez, G. et al. SIRT1 protects against alpha-synuclein aggregation by activating molecular chaperones. J. Neurosci. 32, 124–132 (2012).
Bonnaud, E. M., Suberbielle, E. & Malnou, C. E. Histone acetylation in neuronal (dys)function. Biomol. Concepts 7, 103–116 (2016).
McCampbell, A. et al. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 9, 2197–2202 (2000).
Giampa, C. et al. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington’s disease. Eur. J. Neurosci. 29, 902–910 (2009).
Jiang, H., Nucifora, F. C. Jr., Ross, C. A. & DeFranco, D. B. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet. 12, 1–12 (2003).
Choi, Y. J. et al. Suppression of aggregate formation of mutant huntingtin potentiates CREB-binding protein sequestration and apoptotic cell death. Mol. Cell Neurosci. 49, 127–137 (2012).
Jeong, H. et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137, 60–72 (2009).
Jiang, M. et al. Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat. Med. 18, 153–158 (2011).
Naia, L. & Rego, A. C. Sirtuins: double players in Huntington’s disease. Biochim. Biophys. Acta 1852, 2183–2194 (2015).
Rouaux, C. et al. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 22, 6537–6549 (2003).
Jiang, Y. M. et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251 (2005).
De Vos, K. J. et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728 (2007).
Creppe, C. et al. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell 136, 551–564 (2009).
Taes, I. et al. Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum. Mol. Genet. 22, 1783–1790 (2013).
Gal, J. et al. HDAC6 regulates mutant SOD1 aggregation through two SMIR motifs and tubulin acetylation. J. Biol. Chem. 288, 15035–15045 (2013).
Kim, S. H., Shanware, N. P., Bowler, M. J. & Tibbetts, R. S. Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 285, 34097–34105 (2010).
Barbosa, L. F. et al. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim. Biophys. Acta 1802, 462–471 (2010).
Pearson, M. et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406, 207–210 (2000).
Aubry, S. et al. Assembly and interrogation of Alzheimer’s disease genetic networks reveal novel regulators of progression. PLoS ONE 10, e0120352 (2015).
Schueller, E. et al. Dysregulation of histone acetylation pathways in hippocampus and frontal cortex of Alzheimer’s disease patients. Eur. Neuropsychopharmacol. 33, 101–116 (2020).
Pirooznia, S. K. et al. Tip60 HAT activity mediates APP induced lethality and apoptotic cell death in the CNS of a Drosophila Alzheimer’s disease model. PLoS ONE 7, e41776 (2012).
Guan, J. S. et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60 (2009).
Mahady, L. et al. Frontal cortex epigenetic dysregulation during the progression of Alzheimer’s disease. J. Alzheimers Dis. 62, 115–131 (2018).
Julien, C. et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 48–58 (2009).
Giralt, A. et al. Long-term memory deficits in Huntington’s disease are associated with reduced CBP histone acetylase activity. Hum. Mol. Genet. 21, 1203–1216 (2012).
Bodai, L., Pallos, J., Thompson, L. M. & Marsh, J. L. Pcaf modulates polyglutamine pathology in a Drosophila model of Huntington’s disease. Neurodegener. Dis. 9, 104–106 (2012).
Iwata, A., Riley, B. E., Johnston, J. A. & Kopito, R. R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280, 40282–40292 (2005).
Jeong, H. et al. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 18, 159–165 (2011).
Luthi-Carter, R. et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl Acad. Sci. USA 107, 7927–7932 (2010).
Janssen, C. et al. Differential histone deacetylase mRNA expression patterns in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 573–581 (2010).
Fiesel, F. C. et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 29, 209–221 (2010).
Acknowledgements
We are grateful for funding from the UK Dementia Research Institute (funded by the MRC, Alzheimer’s Research UK and the Alzheimer’s Society), Roger de Spoelberch Foundation, Alzheimer’s Research UK, Cambridge Centre for Parkinson-Plus, National Institute for Health Research Cambridge Biomedical Research Centre (D.C.R.). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
D.C.R. is a consultant for Aladdin Healthcare Technologies, Nido Biosciences and Drishti Discoveries. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Son, S.M., Park, S.J., Fernandez-Estevez, M. et al. Autophagy regulation by acetylation—implications for neurodegenerative diseases. Exp Mol Med 53, 30–41 (2021). https://doi.org/10.1038/s12276-021-00556-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00556-4
This article is cited by
-
p300 nucleocytoplasmic shuttling underlies mTORC1 hyperactivation in Hutchinson–Gilford progeria syndrome
Nature Cell Biology (2024)
-
Neuroprotective Effect of a Multistrain Probiotic Mixture in SOD1G93A Mice by Reducing SOD1 Aggregation and Targeting the Microbiota-Gut-Brain Axis
Molecular Neurobiology (2024)
-
Cyanidin-3-O-glucoside protects the brain and improves cognitive function in APPswe/PS1ΔE9 transgenic mice model
Journal of Neuroinflammation (2023)
-
PFKM inhibits doxorubicin-induced cardiotoxicity by enhancing oxidative phosphorylation and glycolysis
Scientific Reports (2022)
-
Modulation of cellular processes by histone and non-histone protein acetylation
Nature Reviews Molecular Cell Biology (2022)
