
Abstract
Nonalcoholic fatty liver disease (NAFLD), the most common cause of chronic liver disease, ranges from simple hepatic steatosis to nonalcoholic steatohepatitis (NASH), which is a more aggressive form characterized by hepatocyte injury, inflammation, and fibrosis. Increasing evidence suggests that NASH is a risk factor for hepatocellular carcinoma (HCC), which is the fifth most common cancer worldwide and the second most common cause of cancer-related death. Recent studies support a strong mechanistic link between the NASH microenvironment and HCC development. The liver has a large capacity to remove circulating pathogens and gut-derived microbial compounds. Thus, the liver is a central player in immunoregulation. Altered immune responses are tightly associated with the development of NASH and HCC. The objective of this study was to differentiate the roles of specific immune cell subsets in NASH and HCC pathogenesis.
Similar content being viewed by others
Introduction
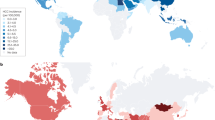
Hepatocellular carcinoma (HCC) is the most common type of liver cancer and accounts for 70–85% of all liver cancer cases1. HCC is the sixth leading cause of cancer-related deaths globally and is expected to become the third leading cause of liver cancer-related deaths by 20302. Such changes in HCC incidence are affected by obesity, type 2 diabetes, and nonalcoholic fatty liver disease (NAFLD), which is the most common liver disease3. Although NAFLD has a spectrum of liver pathologies similar to those of alcohol-induced fatty liver damage, NAFLD can occur in patients even in the absence of alcohol abuse4. NAFLD is characterized by a steatosis or the accumulation of triglycerides in lipid droplets inside hepatocytes (hepatic steatosis)5. Such accumulation of lipids is closely associated with metabolic syndromes such as obesity, type 2 diabetes, hypertension, and dyslipidemia6. NAFLD is highly prevalent on every continent. The global prevalence of NAFLD was ~25%. The Middle East has the highest prevalence rate of 32%, followed by South America (31%). Africa has the lowest prevalence at 14%7. NAFLD can progress to a more severe form called nonalcoholic steatohepatitis (NASH). NASH is marked by abnormal fat accumulation in the liver and immune cell infiltration into the liver due to chronic hepatitis and inflammation. In addition, it seems that most NASH patients develop progressive fibrosis7. NASH can cause liver diseases such as cirrhosis and HCC and is also associated with an increased risk of cardiovascular disease8.
The prevalence of NASH among NAFLD patients in the United States has been estimated to be 21% (95% confidence interval or CI: 19.85–22.95%). The prevalence of NASH in the United States accounts for ~3–4% of the entire population9. NASH is the fastest increasing cause of HCC in the United States10. As such, the incidences of NAFLD and NASH increase each year. Patients with these disorders are highly likely to have more than one metabolic syndrome. These individuals are at high risk of developing HCC11,12. The incidence of NAFLD/NASH-released HCC has continuously increased in many ethnic groups, including in the United States13 Europe14,15,16, South Korea17, and Japan18, over the past decades. A study released in 2010 stated that NAFLD/NASH (59%) was the most common etiological risk factor in the United States, followed by diabetes (36%) and hepatitis C virus (22%)19. Given recent advances in anti-hepatitis C virus (HCV) therapy, NASH is highly likely to become a major cause of progressive liver disease within the next three decades.
Thus, the epidemiology of NASH-associated HCC is continuously changing as the number of patients with metabolic syndrome surges yearly. Compared to patients with other causative factors, patients with NASH-associated HCC are more prone to complications such as diabetes, obesity, dyslipidemia, and hypertension. These factors can exacerbate the clinical complexity of patients and eventually result in a difficult situation for clinical management. Additionally, although patients with lesions caused by HCV or HBV can be partially treated because of the development of treatments, effective treatment is currently unavailable for NASH-associated HCC patients20. To overcome this growing burden of NASH and NAFLD/NASH-HCC, it is crucial to understand the factors associated with NASH and HCC to develop preventive and therapeutic strategies.
Importance of the microenvironment during NASH and HCC pathogenesis
Recent studies have shown that the liver microenvironment may play a crucial role in NAFLD/NASH and HCC progression. The liver provides a unique proinflammatory microenvironment that is composed of a variety of immunologically active cells, including Kupffer cells (KCs), T cells, antigen-presenting cells (APCs), and hepatic stellate cells (HSCs)21,22. In pathological liver injury, these cells are part of a complex proinflammatory and fibrogenic background, and hepatocyte death occurs, promoting disease progression. Various pathobiological factors, including proinflammatory cytokines (such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α), leptin, hyperinsulinemia, the gut microbiota, bile acid, and free fatty acid, can interact with components in the liver microenvironment. These factors may cause inflammation, fibrosis, and lipotoxicity as a result of interactions with the liver microenvironment. In the long term, the interactions of these factors with the liver microenvironment may lead to the progression to NASH and increase the possibility of HCC development21.
A proinflammatory microenvironment created by toxic lipid-induced hepatocyte injury (lipotoxicity), which may occur under NASH conditions, has a significant effect on the deterioration of NASH23. NASH may trigger the death of liver cells by inducing metabolic stress in hepatocytes. The generation of damage-associated molecular patterns (DAMPs) occurs after an influx of activated immune cells and is called sterile chronic inflammation. HCC is an inflammation-related cancer. A chronic inflammatory state can trigger the initiation and development of cancer24. Altered immune responses in NASH with chronic inflammation are also associated with the development of HCC25,26,27.
Various factors and microenvironmental changes have been reported in HCC patients. In particular, cancer-related microenvironmental components such as immune cells, fibroblasts, endothelial cells, and extracellular matrix (ECM) can facilitate tumor cell differentiation, proliferation, and invasion26,28. Thus, it is crucial to understand that the microenvironment of the liver plays a key role in the pathogenesis of NAFLD, NASH, and HCC. The pathogenesis of NASH and HCC is affected by various factors within the microenvironment. However, as inflammation is the most prominent feature of NASH and HCC, the present study reviews the function of immune cells in the respective microenvironments of NASH and HCC.
Immunological differences in the microenvironment associated with NASH and HCC progression
NK cells
Natural killer (NK) cells are a group of innate immune cells that show cytolytic activity against cells under stress, such as tumor cells and virus-infected cells29. NK cells play crucial roles in NAFLD and NASH (Fig. 1). The number of NK cells is increased in hepatic inflammation associated with NASH30. The number of ligands of various NK cells is increased in the liver31. NK cells play an antifibrotic role and prevent the development of liver fibrosis by killing HSCs that cause fibrosis31. The cytolytic activity of NK cells is increased either by upregulating stress ligand activity or by downregulating HSC inhibitory ligands. However, HSCs that fail to undergo senescence can resist NK cell-mediated killing, while senescent HSCs decrease fibrogenesis and increase the inflammatory response. HSCs are chronically activated in NASH due to dysregulated senescence32. HSCs can resist the cytolytic function of NK cells in NASH. Additionally, NK cell activation by inflammatory cytokines such as IL-12, IL-15, and IL-18 contributes to the production of interferon (IFN)-γ and the formation of a proinflammatory environment33,34,35.

The liver has the largest population of resident macrophages (KCs) and high densities of NK cells, NKT cells, various T cells and liver-transiting and/or resident lymphocytes, with a higher ratio of CD8+ T cells to CD4+ T cells than that in the periphery. Liver nonparenchymal cells are associated with innate and adaptive immune systems and control the liver microenvironment. During NASH progression, these cells functionally interact with each other by secreting cytokines and chemokines to induce profibrotic and proinflammatory signals. Immune cells also interact with other cells in the liver microenvironment, especially hepatic stellate cells (HSCs) and hepatocytes, to promote fibrosis and inflammation that can affect disease severity. Th1 cells can affect macrophages by secreting IFN-γ and TNF-α. These macrophages can produce cytokines to activate HSCs. CD8+ cells, Th17 cells, KCs, and neutrophils also contribute to the activation of HSCs. In contrast, NK cells inhibit the activation of HSCs. NKT cells and Th17 cells induce lipid accumulation in hepatocytes. Thus, altered immune regulation in the liver microenvironment can eventually lead to NASH development.
As mentioned above, NK cells usually perform antitumor functions through surveillance. The surveillance of NK cells in tumors is crucial in combating HCC (Fig. 2). However, in HCC conditions, the number of NK cells is reduced. The production of IFN-γ and cytotoxic functions are also impaired. This is due to increased Tregs in HCC patients36. In addition, it seems that NK cell function is decreased by myeloid-derived suppressor cells (MDSCs) during the development of HCC (Fig. 2). Thus, NK cells can exert antitumor effects. However, in the case of HCC, these cells are defective because they exist in an ineffective form, which is unable to remove tumor cells37.

In the HCC liver microenvironment, a large population of immune cells, Th17 cells, Tregs, iNKT cells, TAMs, and neutrophils are activated. However, immune cells with antitumor functions such as NK cells, CD8+ T cells, and KCs are mostly defective. The number of NK cells is decreased, and their function is suppressed by Tregs and MDSCs. KCs have dual roles in the tumor microenvironment. These cells secrete IL-6 to promote tumor development. KCs also have antitumor functions. However, the antitumor function of KCs is suppressed by MDSCs. CD8+ T cells can directly inhibit tumor growth by secreting granular components and cytokines. However, these cells are suppressed by Tregs in the HCC microenvironment. TAMs play various roles during HCC progression, including affecting angiogenesis. Neutrophils can also induce angiogenesis while attracting protumorigenic cells, such as TAMs and Tregs. These immune cells play various roles and can exacerbate HCC development by supporting tumor growth, progression, and angiogenesis.
T lymphocytes
T cells play a regulatory role by controlling cells that are involved in immune responses, including B cells and T cells. There are two subsets of T cells: CD4+ helper T (Th) cells and CD8+ cytotoxic T cells. Th cells can be classified into Th1 and Th2 cells. Th1 cells produce IL-2 and IFN-γ and are involved in cellular immunity by regulating macrophages (Fig. 1). Th2 cells produce IL-4, IL-5, and IL-13 and are involved in humoral immunity by regulating eosinophils38. Another subset of Th cells is Th17 cells that secrete IL-17. Th17 cells are involved in the progression of many autoimmune diseases and inflammatory disorders. Th17 cells also secrete IL-22 and granulocyte-macrophage colony-stimulating factor (GM-CSF) to regulate neutrophils39 (Fig. 1). CD8+ cytotoxic T cells express clonally distributed receptors for foreign antigens, undergo marked proliferation and production of IFN-γ in response to infection, and kill any cell that expresses target antigens as an integral component of immunological memory40,41 (Fig. 1). The importance of these T cells in NAFLD has been suggested. It has been shown that T cell-deficient mice do not develop steatosis, hepatic inflammation or fructose-induced NAFLD42.
The selective loss of CD4+ T cells occurs in NAFLD. NAFLD induced by a methionine/choline-deficient (MCD) diet can trigger the loss of CD4+T lymphocytes and promote the development of HCC in liver-specific MYC oncogenic transgenic mice. In the same context, obesity-induced lipid accumulation can result in the selective loss of CD4+ T lymphocytes and promote disease progression from NAFLD to hepatocellular carcinoma43. These CD4+ T cells are biased to Th1 and Th17 subtypes in NASH conditions44. Th1 cells can promote the differentiation of macrophages into M1 macrophages that play a proinflammatory role by secreting IFN-γ and TNF-α45,46. Th17 cells secrete IL-17, which exacerbates hepatic steatosis and inflammation and induces the transition from simple steatosis to hepatic steatosis47,48. Th17 cells also produce neutrophil-attracting chemokines to recruit neutrophils and lymphocytes in NASH49. Fibrosis progression is further induced through HSCs50. In contrast, the number of hepatic regulatory T cell (Tregs) is decreased in NASH48,51. In mice with HFD-induced steatosis, Treg apoptosis can be induced by increased oxidative stress, whereas liver inflammation is markedly reduced by adoptive transfer of Tregs52. In fact, the ratio of Th17 cells/Tregs is higher in NASH patients than in NAFLD patients and normal controls, indicating that the ratio of Th17/Tregs may play a crucial role in the NASH environment53.
The number of Th17 cells and the level of IL-17 are increased in tumor tissues of patients with HCC (Fig. 2). These increases are associated with low survival and increased postoperative recurrence, indicating that Th17 cells may accelerate HCC progression. Systemic IL-17A causes infiltration of neutrophils into adipose tissue, which generates a feed-forward mechanism that exacerbates NASH by releasing fatty acids and inducing DNA damage in hepatocytes, thus promoting HCC54. The number of Tregs is significantly increased in HCC patients55. An increase in Tregs is mainly observed around tumor sites. Tregs extracted from the tumor site contribute to uncontrolled growth of HCC cells by inhibiting the proliferation of CD8+ cytotoxic T cells (Fig. 2), leading to poor clinical outcome and poor survival of HCC patients56.
The infiltration of CD8+ cells, another T cell subset, occurs in the context of NASH57. The recruitment of hepatic CD8+ T cells is increased NF-κB1-knockout NASH model mice. This increase in hepatic CD8+ T cells exacerbates liver inflammation and fibrosis in NF-κB1-knockout mice, indicating that CD8+ T cells are involved in NASH progression and fibrosis58. The depletion of CD8+ T cells could halt the progression of NASH and decrease fibrosis57. CD8+ T cell activation is involved in the development of not only NASH but is also subsequent liver cancer in mice fed a choline-deficient high-fat diet. In the transition from NASH to HCC, the activation of the NF-κB signaling pathway in hepatocytes is induced by CD8+ T cells via crosstalk with hepatocytes59.
CD8+ T cells are abundant in the early stages of HCC. Their number decreases as the disease progresses. CD8+ cells secrete granule components such as perforin and granzyme B (GrB) and play a cytolytic role (Fig. 2). These cells also possess antitumor functions. However, since these activities are significantly reduced and most of these cells exist in a quiescent state, the expression of cytolytic molecules in CD8+ T cells in HCC patients is markedly low. It appears that impaired CD8+ T cells can produce and release granzyme A (GrA), and GrB production might be interrupted by the increased numbers of Tregs in tumors60 (Fig. 2). Furthermore, the ability of CD8+ T cells to produce IFN-γ is impaired61.
NKT cells
Natural killer T (NKT) cells are unconventional cells that express NK and T cell surface markers and secrete various cytokines, such as IFN-γ and IL-4, to control innate and adaptive immunity. NKT cells account for 30% of hepatic nonparenchymal cells62,63. NKT cells can produce factors that modulate inflammatory and fibrogenic responses in the liver64 (Fig. 1). The numbers of NKT cells and CD8+ T cells are increased in NASH patients59. This accumulation of hepatic NKT cells is also observed in the MCD diet-induced NASH model. Activation of the hedgehog pathway occurs during NASH-related fibrogenesis, leading to the induction of factors involved in the recruitment, retention, and viability of NKT cells. Accumulated NKT cells can stimulate HSCs to become myofibroblasts and promote the progression of NASH-associated fibrosis64 (Fig. 1). Additionally, an increase in CXCR6 expression in NKT cells and subsequent binding to CXCL16, which is expressed in endothelial cells and macrophages, in NASH can lead to the accumulation of NKT cells that produce proinflammatory cytokines such as IL-4, IFN-γ, and TNF-α, thus inducing inflammation via macrophage activation65 (Fig. 1). Moreover, hepatic NKT cells can secrete LIGHT to promote hepatic steatosis and liver damage through inflammatory cytokines and CD8+ T cells, contributing to canonical NF-κB signaling and stimulating the transition from NASH to HCC59.
There are more NKT cells in tumors than in blood in HCC patients66. Based on differences in T cell receptor (TCR) usage, these NKT cells can be classified into two types: Type I and Type II. Type I NKT cells express invariant TCRα chains that are readily detectable by α-galactosylceramide (α-GalCer)-loaded CD1d tetramers, while type II NKT cells express a broader TCR repertoire67,68. However, studies on Type II NKT cells in the liver are insufficient because of the absence of a specific marker. Thus, Type I NKT cells called invariant NKT (iNKT) cells have been mostly studied in patient tumors (Fig. 2). iNKT cells perform dual roles. These cells either promote the antitumor response by activating effector T cells or promote tumor growth by inducing the Th2 response through the recruitment of Tregs (Fig. 2). CD4+ and CD4− iNKT cell subsets both produce Th1-type cytokines such as IFN-γ. However, CD4+ subsets show enhanced secretion of Th2 cytokines and regulatory activity. In the tumor microenvironment, the number of CD4-expressing iNKT cells is increased in HCC tumors, thus producing more Th2 cytokines, preventing the expansion of tumor Ag-specific CD8+ T cells, and promoting tumor growth69,70. On the other hand, iNKT cells exert antitumor effects by inhibiting the inflammatory response through oncogenic β-catenin71.
Macrophages
Macrophages that circulate in the blood or exist in various organs and tissues are the first barriers to any disease. These cells play the most important role in innate and acquired immunity. Macrophages are generally divided into two phenotypes: classically activated (M1) macrophages and alternatively activated (M2) macrophages72. M1 macrophages are characterized by an IL-12hiIL-23hiIL-10lo phenotype. These cells produce toxic effector molecules (ROS and NO) and inflammatory cytokines such as IL-1β, TNF, and IL-6. They participate in Th1 responses and mediate resistance to intracellular parasites and tumors. In contrast, M2 cells are characterized by an IL-12loIL-23loIL-10hiTGF-βhi phenotype in vitro. M2 cells participate in polarized Th2 responses, allergies, parasite clearance, dampening of inflammation, tissue remodeling, angiogenesis, immunoregulation, and tumor promotion73. In the liver, hepatic resident macrophages are called KCs and are involved in inflammation signaling and metabolic changes.
In NASH, various metabolic syndromes and insulin resistance promote the accumulation of free fatty acids (FFAs) in hepatocytes and blood, triggering the innate immune response by stimulating lipotoxin and LPS. In a healthy liver, the KCs primarily function as the body’s frontline defense against phagocytosis and pathogens from the portal vein and arterial circulation74. The level of KCs decreases in NASH, while the infiltration of Ly6C+ monocyte-derived macrophages is increased in the early stage of NASH in an MCD diet model75. In NASH, toll-like receptor (TLR) 4 in KCs is more highly expressed than other TLRs76. When LPS binds to TLR4, KCs are activated, and the production of proinflammatory cytokines such as TNF-α, IL-1β, IL-2, IL-6, IL-10, and IFN-γ is enhanced, triggering the recruitment of lymphocytes and other leukocytes77 and promoting the activation of NF-κB, MAPK, ERK1, p38, JNK, and IRF378. Additionally, the expression of hepatic inflammatory genes is increased by oxidized low-density lipoprotein (LDL) trapped in KC lysosomes in a NASH model79. Recent studies have shown an increase in endolysosomal lipid accumulation in KCs during the progression from NAFLD to NASH80, indicating that KCs play a crucial role in the progression of NASH. Along with KCs, infiltrated Ly6C+ macrophages produce cytokines such as TNF-α and IL-1β, thus promoting inflammation and activating HSCs81.
KCs act as a major mediator in fibrosis (Fig. 1). In vivo KC depletion inhibits HSC activation and fibrosis development in a mouse model of fibrosis82. Indeed, KCs can secrete profibrogenic cytokines such as transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF)78. In other words, KCs contribute to inflammation and fibrosis progression through various processes, exacerbating NASH (Fig. 1). In addition, KCs can recruit CD4 T cells and increase T cell tolerance83. In the late stage of NASH, the release of DAMPs such as intracellular DNA from dying hepatocytes may trigger the activation of KCs. It has been reported that hypoxia in the liver may activate KCs by upregulating transcription factors such as hypoxia inducible factor 1α (HIF-1α)84.
KCs contribute to immune surveillance and play a role in antitumor immunity. However, KCs present in the tumor microenvironment (TME) of HCC have low expression of costimulatory molecules but high expression of coinhibitory molecules. Moreover, due to the presence of MDSCs, the antitumor function of KCs is suppressed85 (Fig. 2). Recently, it has been proven that KCs are involved in the development of HCC22. In a diethylnitrosamine (DEN)-induced HCC model, hepatocellular carcinogenesis was attenuated when the activation of KCs was mitigated by deletion of the proinflammatory myeloid cell surface receptor triggering receptor expressed on myeloid cell-1 (TREM-1), which is expressed by KCs. It has been revealed that the activation of KCs is crucial for tumor development in the early stage of chemical-induced carcinogenesis86. Furthermore, IL-1a secreted either by oncogene-induced senescent hepatocytes or by apoptotic hepatocytes in the DEN model can induce the production of IL-6 in KCs and trigger compensatory proliferation and tumor progression, which are essential for HCC development22. Once a primary tumor is established, liver-infiltrated macrophages play a more prominent role than KCs. Macrophages infiltrate near tumors. Macrophages present in the TME are called tumor-associated macrophages (TAMs)87. A tumor with highly infiltrated TAMs leads to a worse prognosis in HCC88 (Fig. 2). TAMs are attracted by chemokines, such as macrophage colony-stimulating factors (M-CSF), produced by tumor cells. These cells infiltrate the tumor site89. Additionally, glypican-3, which is expressed in HCC, can recruit macrophages90.
In the HCC microenvironment, the transformation of macrophages into the M2 phenotype is further amplified by factors such as CSF1, Wnt, HIFs, IL-8, and HMGB191. Macrophages that are transformed into the M2 phenotype can exacerbate HCC. M2-polarized macrophages enable epithelial–mesenchymal transition (EMT) and the migration of HCC cells via the TLR4/signal transducer and activator of transcription (STAT) 3 signaling pathway92. Moreover, increased stability of HIF-1α in hypoxic conditions in HCC increases the secretion of IL-1β and promotes EMT and metastasis of tumor cells93. TAMs can also secrete IL-6 and promote the expansion of cancer stem cells and tumor formation through IL-6/STAT3 signaling94 (Fig. 2). Additionally, TAMs can secrete a larger amount of TGF-β than other macrophages and facilitate the progression of tumors by obtaining cancer stem cell-like properties through TGF-β1-induced EMT95. Indeed, the level of TGF-β is increased in HCC patients96.
TAMs can interact with various immune cells. Macrophages can trigger the accumulation of Tregs through the secretion of IL-10 and inhibit the activation of other CD4+CD25− cells to induce the progression of HCC97. Macrophages can also promote the expansion of Th17 cells and tumor growth by secreting various proinflammatory cytokines98, causing dysfunction and apoptosis of NK cells through interactions with NK cells via CD48/2B499. In addition to the immune response, TAMs contribute to angiogenesis, which exacerbates cancer. TAMs can produce proangiogenic factors such as VEGF and TNF-α, interact with the ECM, promote angiogenesis, and facilitate tumor cell invasion87 (Fig. 2).
B cells
B cells are immune cells that play a role in antibody secretion, antigen presentation, T cell costimulation, and cytokine secretion. B cells play an immunomodulatory role and contribute to autoimmunity and disease pathogenesis100. Relatively fewer studies have been conducted on B cells than on other kinds of immune cells in the NASH microenvironment. However, the role of B cells in NASH is gradually being elucidated. B cell-activating factor (BAFF), a TNF superfamily member, is secreted by adipocytes. BAFF regulates the maturation and development of B cells101. NASH patients have higher levels of BAFF than control patients with simple steatosis. BAFF is associated with the degree of ballooning hepatocytes and hepatic fibrosis102. In BAFF-knockout mice, the number of mature B cells is decreased. Recent studies have shown that deletion of BAFF in the NAFLD model attenuates hepatic fat accumulation, inhibits inflammation and fibrosis in VAT, improves insulin resistance, and weakens liver steatosis. These results indicate that BAFF plays a role in exacerbating NAFLD and NASH103. BAFF-knockout mice show reduced systemic inflammation104. In response to LPS stimulation, hepatic B cells produce more IFN-γ, IL-6 and tumor necrosis factor (TNF)-α but less IL-10 than those from secondary lymphoid tissue, indicating that hepatic B cells promote inflammatory responses105. Increased TGF-β levels in NASH patients106 trigger the transformation of IgM-expressing B cells into IgA-expressing B cells with regulatory activity. The level of serum IgA is increased in NASH patients. A high level of IgA is associated with the state of fibrosis107,108. Additionally, the accumulation of liver-resident IgA+PDL1+ cells that secrete IL-10 concurrent in NASH can promote tumor growth by inhibiting the function of CD8+ T cells107.
In the development of HCC, B cells can inhibit the growth of established tumors, while T cells prevent the onset of tumor formation109. An increased level of tumor-infiltrating B cells can lead to better clinical outcomes. However, intratumor infiltration of B cells is impaired during the progression of HCC110. B cells secrete immunoglobulin with a direct antitumor effect that is beneficial for HCC patients111 (Fig. 2). When mature B cells are transplanted with hepatoma cells in B cell-depleted mice, tumor growth is not controlled, and the activation of local T cells is decreased, indicating that B cells might play a crucial role in cancer112. In contrast, it has been reported that the infiltration of CD20+ B cells is associated with poor survival in HCC patients. Ablation of CD20+ B cells induces senescence-mediated fibrosis resolution and promotes inhibition of the protumorigenic TNF-α/NF-κB pathway in Mdr2−/− mice, which is an inflammation-associated cancer model113.
Neutrophils
Neutrophils are innate immune cells that account for an overwhelming majority of circulating lymphocytes. Neutrophils serve as the frontline defense of the immune system. They are the first cells to infiltrate in the event of acute liver injury. Neutrophils can capture and destroy invasive microorganisms via phagocytosis, intracellular degradation, granule release, and the formation of neutrophil extracellular traps (NETs). These cells serve as mediators of inflammation114. NASH is characterized by the infiltration of a large number of neutrophils (Fig. 1). In the MCD diet-induced NASH model, when the infiltration of hepatic neutrophils is suppressed, an early stage of NASH is significantly attenuated, resulting in reduced levels of serum alanine transferase (ALT) and proinflammatory mRNA, indicating that neutrophils may exacerbate the early stage of NASH115. Neutrophil-derived myeloperoxidase (MPO) is also associated with hepatic cholesterol accumulation, inflammation, and fibrosis. MPO enhances macrophage cytotoxicity and induces neutrophil activation116. Additionally, neutrophil-derived human neutrophil peptide (HNP)-1 induces hepatic fibrosis through HSC proliferation and exacerbates NASH117 (Fig. 1). Indeed, the neutrophil-to-lymphocyte ratio (NLR) has been found to be high in NASH patients with advanced fibrosis, indicating that neutrophils are a key factor in fibrosis and NASH118.
Neutrophils also play a crucial role in the transition from NASH to HCC (Fig. 2). The levels of markers of NETs (large, extracellular, web-like structures composed of cytosolic and granule proteins that are assembled on a scaffold of decondensed chromatin119) expelled by neutrophils are increased in sera samples from NASH patients. NETs play a key role in the formation of chronic inflammatory liver microenvironment in NASH that promotes the progression to HCC. The formation of NETs has been observed in livers of STAM mice (NASH induced by neonatal streptozotocin and a high-fat diet), followed by an influx of monocyte-derived macrophages, the formation of inflammatory cytokines, and progression to HCC120.
Neutrophils are enriched mainly around the peritumoral stromal region in HCC. The frequency of neutrophil infiltration in the liver is an indicator of poor survival in HCC121,122. Increased levels of IL-17 in HCC promote the secretion of CXC chemokines to recruit neutrophils around the peritumoral stromal regions (Fig. 2). Accumulated neutrophils can promote the secretion of matrix metalloproteinase-9 to induce angiogenesis122. Additionally, CXCR6high tumor cells in HCC can secrete cytokines such as IL-1β, IL-6, and IL-8, induce the infiltration of CD66b+ neutrophils, and promote neutrophil-mediated multiple protumor responses123. Tumor-associated neutrophils in HCC can promote tumor growth and progression by recruiting macrophages and Tregs, which are increased after sorafenib treatment to resist the antitumor effect of treatment124. Additionally, neutrophil infiltration in HCC can produce hepatocyte growth factors (HGFs) and promote the growth and metastasis of malignant cells that are activated by GM-CSF, which is secreted by hepatoma cells in the HCC microenvironment125. Taken together, these findings indicate that neutrophils play a protumorigenic role in HCC progression, growth, and metastasis.
Other cell types
Other myeloid cells also have various roles in NASH and HCC pathogenesis. Particularly, dendritic cells (DCs) are the most powerful APCs and can induce adaptive immune responses and generate tolerance to self-antigens126. Since DCs induce the adaptive immune response, the importance of DCs during hepatic inflammation has emerged. The hepatic DC population becomes expanded and mature in the NASH liver127. When DCs are depleted, intrahepatic fibrosis-associated inflammation is markedly exacerbated. Hence, in NASH, DCs can limit CD8+ T cell expansion and restrict Toll-like receptor expression and cytokine production in innate immune effector cells, including KCs, neutrophils, and inflammatory monocytes127. DCs can capture HCC-related antigens, process antigens, and activate antigen-specific T cells to remove tumors. However, in advanced HCC, the functions of DCs and IL-12 production are impaired. Thus, antigen-specific T cells cannot be activated properly128.
Monocytes play a pivotal role in inflammation and metabolic stresses. The infiltration of monocytes is an important feature of NASH. CD11bintLy6Chi monocytes appear to be the predominant source of TNFα production at a later time point in MCD diet-induced disease, leading to NASH progression129. These infiltrated monocytes have potent proinflammatory activities. In addition, together with KC, infiltrated monocytes can propagate lipid accumulation induced by the MCD diet mainly through the production of TNFα129. In HCC patients, PD-L1+ monocytes are highly enriched in the peritumoral stroma. These monocytes are activated to suppress tumor-specific T cell immunity. In fact, high infiltration of these monocytes is associated with poor survival of HCC patients130.
Therapeutic perspective and conclusion
Acute inflammation is a useful reaction to achieve tissue recovery by promoting regeneration. Conversely, chronic inflammation is maladaptive and provides an environment that is conducive to the development of NASH and HCC. Chronic injury triggers the secretion of significant amounts of proinflammatory molecules, including IL-1, IL-6, TNF-α, and lymphotoxin-β, that can facilitate HCC development24. As mentioned above, NAFLD and NASH are major causes of the increased prevalence of HCC. Additionally, NASH-related HCC mainly occurs in the context of cirrhosis. Although NASH-related HCC cases occur mainly in cirrhotic patients, an increasing number of HCC cases have been reported in NAFLD and NASH patients with little or no cirrhosis131. While many studies on the respective microenvironments of NASH and HCC have been conducted, such studies on changes in the progression from NASH to HCC are insufficient. Since various immune cells have different functions that can affect the development of NASH and HCC, it is critical to develop an experimental model to identify the transition from NASH to HCC.
NASH with advanced cirrhosis is currently the primary etiology for liver transplantation and is estimated to become the leading indication for liver transplantation132. Thus, patients with NASH are at increased risk of adverse liver-related outcomes, with the degree of fibrosis contributing most significantly to this increased risk. However, medications have not yet been approved by the Federal Drug Administration (FDA) or European Medicines Agency (EMA) for the treatment of NASH. Many agents are currently being studied in clinical trials Phase 2 clinical trials have been for the TLR4 antagonist JKB-121 (ClinicalTrials.gov identifier: NCT02442687), which targets cells expressing TLRs, such as KCs, and solithromycin (ClinicalTrials.gov identifier: NCT02510599), which inhibits TNF-α/CXCL8 production and MMP9 activity in monocytic cells. Obeticholic acid (OCA) is a derivative of the primary human bile acid chenodeoxycholic acid (CDCA) and functions as an agonist of farnesoid X receptor (FXR). There was a randomized placebo-controlled, phase IIb trial and a large phase III trial to evaluate the safety and efficacy of OCA for the treatment of NASH and fibrosis133. Elafibranor is an agent currently in a phase III trial for the treatment of NASH (ClinicalTrials.gov Identifier: NCT02704403). Elafibranor, a dual peroxisome proliferator-activated receptor (PPAR)-α/δ agonist133, and cenicriviroc (CVC), a dual CCR2/CCR5 chemokine receptor antagonist that has been shown to play key roles in hepatic inflammation and fibrosis134, are being investigated in current phase III clinical trials in patients with NASH and fibrosis, respectively (ClinicalTrials.gov Identifier: NCT03028740). Additionally, the immune checkpoint inhibitors nivolumab (ClinicalTrials.gov identifier: NCT02576509), pembrolizumab (ClinicalTrials.gov identifier: NCT03062358), and tremelimumab (ClinicalTrials.gov identifier: NCT03298451) are undergoing phase III clinical trials in patients with HCC.
Various mechanisms and microenvironmental changes are involved in the onset and progression of NASH and HCC, raising a fundamental question on whether one targeted treatment would be effective. Indeed, most of the abovementioned drug candidates did not show expected therapeutic efficacies in phase II or phase III clinical trials. Importantly, a combination therapy that uses more than two types of targeted drugs to provide additional and/or synergistic effects could be a more effective strategy for NASH and HCC treatment. For instance, antidiabetic drugs or antifibrotic drugs with an FXR agonist, a well-known drug for NASH, have shown notable effects in clinical trials135. Furthermore, instead of using only checkpoint inhibitors, combined therapies targeting immune cells involved in the progression of HCC or targeting cells participating in angiogenesis with checkpoint inhibitors are now presenting promising outcomes136,137. However, combinational therapy that controls steatosis, chronic inflammation, and fibrosis could be the most efficient therapeutic strategy for NASH treatment. In addition, overcoming the hypocellular hepatic microenvironment is the most important condition for inducing efficient drug effects. Thus, developing effective drug candidates that can reverse the functions of immune cells in the respective microenvironments of NASH and HCC is expected to become a novel therapeutic strategy.
References
Jemal, A. et al. Global cancer statistics. CA Cancer J. Clin. 61, 69–90 (2011).
Rahib, L. et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74, 2913–2921 (2014).
Caldwell, S. H., Crespo, D. M., Kang, H. S. & Al-Osaimi, A. M. Obesity and hepatocellular carcinoma. Gastroenterology 127, S97–S103 (2004).
Michelotti, G. A., Machado, M. V. & Diehl, A. M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 10, 656–665 (2013).
Sunny, N. E., Bril, F. & Cusi, K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 28, 250–260 (2017).
Anstee, Q. M., Targher, G. & Day, C. P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 10, 330–344 (2013).
Younossi, Z. M. et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016).
Arrese, M. & Feldstein, A. E. Nash-related cirrhosis: an occult liver disease burden. Hepatol. Commun. 1, 84–86 (2017).
Younossi, Z. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15, 11–20 (2018).
Younossi, Z. M. The epidemiology of nonalcoholic steatohepatitis. Clin. Liver Dis. (Hoboken) 11, 92–94 (2018).
Welzel, T. M. et al. Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology 54, 463–471 (2011).
Borena, W. et al. Metabolic risk factors and primary liver cancer in a prospective study of 578,700 adults. Int J. Cancer 131, 193–200 (2012).
Wong, R. J., Cheung, R. & Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 59, 2188–2195 (2014).
Adams, L. A. et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129, 113–121 (2005).
Dyson, J. et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 60, 110–117 (2014).
Petrick, J. L. et al. International trends in liver cancer incidence, overall and by histologic subtype, 1978-2007. Int J. Cancer 139, 1534–1545 (2016).
Kim, G. A. et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. S0168-8278, 32294–32298 (2017).
Kawamura, Y. et al. Large-scale long-term follow-up study of Japanese patients with non-alcoholic Fatty liver disease for the onset of hepatocellular carcinoma. Am. J. Gastroenterol. 107, 253–261 (2012).
Sanyal, A., Poklepovic, A., Moyneur, E. & Barghout, V. Population-based risk factors and resource utilization for HCC: US perspective. Curr. Med Res Opin. 26, 2183–2191 (2010).
D’Avola, D., Labgaa, I. & Villanueva, A. Natural history of nonalcoholic steatohepatitis/nonalcoholic fatty liver disease-hepatocellular carcinoma: Magnitude of the problem from a hepatology clinic perspective. Clin. Liver Dis. (Hoboken) 8, 100–104 (2016).
Agosti, P., Sabba, C. & Mazzocca, A. Emerging metabolic risk factors in hepatocellular carcinoma and their influence on the liver microenvironment. Biochim. Biophys. Acta Mol. Basis. Dis. 1864, 607–617 (2018).
Stauffer, J. K., Scarzello, A. J., Jiang, Q. & Wiltrout, R. H. Chronic inflammation, immune escape, and oncogenesis in the liver: a unique neighborhood for novel intersections. Hepatology 56, 1567–1574 (2012).
Ibrahim, S. H., Hirsova, P. & Gores, G. J. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 67, 963–972 (2018).
Capece, D. et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 187204 (2013).
Loomba, R. & Sanyal, A. J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 10, 686–690 (2013).
Hernandez-Gea, V., Toffanin, S., Friedman, S. L. & Llovet, J. M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 144, 512–527 (2013).
Fattovich, G., Stroffolini, T., Zagni, I. & Donato, F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 127, S35–S50 (2004).
Tahmasebi Birgani, M. & Carloni, V. Tumor microenvironment, a paradigm in hepatocellular carcinoma progression and therapy. Int J. Mol. Sci. 18, 405 (2017).
Paul, S. & Lal, G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol. 8, 1124 (2017).
Kahraman, A. et al. Major histocompatibility complex class I-related chains A and B (MIC A/B): a novel role in nonalcoholic steatohepatitis. Hepatology 51, 92–102 (2010).
Stiglund, N. et al. Retained NK cell phenotype and functionality in non-alcoholic fatty liver disease. Front Immunol. 10, 1255 (2019).
Narayanan, S., Surette, F. A. & Hahn, Y. S. The immune landscape in nonalcoholic steatohepatitis. Immune Netw. 16, 147–158 (2016).
Vivier, E., Tomasello, E., Baratin, M., Walzer, T. & Ugolini, S. Functions of natural killer cells. Nat. Immunol. 9, 503–510 (2008).
Cepero-Donates, Y. et al. Interleukin-15-mediated inflammation promotes non-alcoholic fatty liver disease. Cytokine 82, 102–111 (2016).
Gomez-Santos, L. et al. Inhibition of natural killer cells protects the liver against acute injury in the absence of glycine N-methyltransferase. Hepatology 56, 747–759 (2012).
Cai, L. et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin. Immunol. 129, 428–437 (2008).
Hoechst, B. et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 50, 799–807 (2009).
Saito, S., Nakashima, A., Shima, T. & Ito, M. Th1/Th2/Th17 and regulatory T-cell paradigm in pregnancy. Am. J. Reprod. Immunol. 63, 601–610 (2010).
Sandquist, I. & Kolls, J. Update on regulation and effector functions of Th17 cells. F1000Res 7, 205 (2018).
Bevan, M. J. Helping the CD8(+) T-cell response. Nat. Rev. Immunol. 4, 595–602 (2004).
Freeman, B. E., Hammarlund, E., Raue, H. P. & Slifka, M. K. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc. Natl Acad. Sci. USA 109, 9971–9976 (2012).
Bhattacharjee, J. et al. Role of immunodeficient animal models in the development of fructose induced NAFLD. J. Nutr. Biochem. 25, 219–226 (2014).
Ma, C. et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531, 253–257 (2016).
Sutti, S. et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 59, 886–897 (2014).
Ferreyra Solari, N. E. et al. The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J. Clin. Immunol. 32, 611–621 (2012).
Inzaugarat, M. E. et al. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J. Clin. Immunol. 31, 1120–1130 (2011).
Ye, C., Li, W. Y., Zheng, M. H. & Chen, Y. P. T-helper 17 cell: a distinctive cell in liver diseases. Hepatol. Res 41, 22–29 (2011).
Tang, Y. et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 166, 281–290 (2011).
Hubscher, S. G. Histological assessment of non-alcoholic fatty liver disease. Histopathology 49, 450–465 (2006).
Tan, Z. et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J. Immunol. 191, 1835–1844 (2013).
Liu, Y. et al. 3, 3′-Diindolylmethane alleviates steatosis and the progression of NASH partly through shifting the imbalance of Treg/Th17 cells to Treg dominance. Int Immunopharmacol. 23, 489–498 (2014).
Ma, X. et al. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 46, 1519–1529 (2007).
Rau, M. et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory t cell ratio in peripheral blood and in the liver. J. Immunol. 196, 97–105 (2016).
Ringelhan, M., Pfister, D., O’Connor, T., Pikarsky, E. & Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 19, 222–232 (2018).
Lan, Y. T., Fan, X. P., Fan, Y. C., Zhao, J. & Wang, K. Change in the Treg/Th17 cell imbalance in hepatocellular carcinoma patients and its clinical value. Medicine (Baltimore) 96, e7704 (2017).
Yang, X. H. et al. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J. Hepatol. 45, 254–262 (2006).
Bhattacharjee, J. et al. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol. Commun. 1, 299–310 (2017).
Locatelli, I., Sutti, S., Vacchiano, M., Bozzola, C. & Albano, E. NF-kappaB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin. Sci. (Lond.) 124, 279–287 (2013).
Wolf, M. J. et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 26, 549–564 (2014).
Fu, J. et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 132, 2328–2339 (2007).
Flecken, T. et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 59, 1415–1426 (2014).
Gao, B., Radaeva, S. & Park, O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J. Leukoc. Biol. 86, 513–528 (2009).
Bendelac, A., Savage, P. B. & Teyton, L. The biology of NKT cells. Annu Rev. Immunol. 25, 297–336 (2007).
Syn, W. K. et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 51, 1998–2007 (2010).
Wehr, A. et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J. Immunol. 190, 5226–5236 (2013).
Sachdeva, M., Chawla, Y. K. & Arora, S. K. Immunology of hepatocellular carcinoma. World J. Hepatol. 7, 2080–2090 (2015).
Liao, C. M., Zimmer, M. I. & Wang, C. R. The functions of type I and type II natural killer T cells in inflammatory bowel diseases. Inflamm. Bowel Dis. 19, 1330–1338 (2013).
Dasgupta, S. & Kumar, V. Type II NKT cells: a distinct CD1d-restricted immune regulatory NKT cell subset. Immunogenetics 68, 665–676 (2016).
Bricard, G. et al. Enrichment of human CD4+ V(alpha)24/Vbeta11 invariant NKT cells in intrahepatic malignant tumors. J. Immunol. 182, 5140–5151 (2009).
Zhu, S., Zhang, H. & Bai, L. NKT cells in liver diseases. Front Med 12, 249–261 (2018).
Anson, M. et al. Oncogenic beta-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J. Clin. Invest. 122, 586–599 (2012).
Arora, S., Dev, K., Agarwal, B., Das, P. & Syed, M. A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 223, 383–396 (2018).
Italiani, P. & Boraschi, D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 5, 514 (2014).
Cha, J. Y., Kim, D. H. & Chun, K. H. The role of hepatic macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Lab Anim. Res 34, 133–139 (2018).
Reid, D. T. et al. Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PLoS ONE 11, e0159524 (2016).
Ye, D. et al. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 61, 1058–1067 (2012).
Wenfeng, Z. et al. Kupffer cells: increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 13, 489–495 (2014).
Bieghs, V. & Trautwein, C. The innate immune response during liver inflammation and metabolic disease. Trends Immunol. 34, 446–452 (2013).
Bieghs, V. et al. Trapping of oxidized LDL in lysosomes of Kupffer cells is a trigger for hepatic inflammation. Liver Int. 33, 1056–1061 (2013).
Galassi, T. V. et al. An optical nanoreporter of endolysosomal lipid accumulation reveals enduring effects of diet on hepatic macrophages in vivo. Sci. Transl. Med. 10, eaar2680 (2018).
Dong, X., Liu, J., Xu, Y. & Cao, H. Role of macrophages in experimental liver injury and repair in mice. Exp. Ther. Med. 17, 3835–3847 (2019).
Van Hul, N. et al. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am. J. Pathol. 179, 1839–1850 (2011).
You, Q., Cheng, L., Kedl, R. M. & Ju, C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 48, 978–990 (2008).
Koh, M. Y. et al. A new HIF-1alpha/RANTES-driven pathway to hepatocellular carcinoma mediated by germline haploinsufficiency of SART1/HAF in mice. Hepatology 63, 1576–1591 (2016).
Lacotte, S. et al. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology 5, e1234565 (2016).
Wu, J. et al. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 72, 3977–3986 (2012).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Siveen, K. S. & Kuttan, G. Role of macrophages in tumour progression. Immunol. Lett. 123, 97–102 (2009).
Zhu, X. D. et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J. Clin. Oncol. 26, 2707–2716 (2008).
Takai, H. et al. The expression profile of glypican-3 and its relation to macrophage population in human hepatocellular carcinoma. Liver Int. 29, 1056–1064 (2009).
Li, Z., Wu, T., Zheng, B. & Chen, L. Individualized precision treatment: Targeting TAM in HCC. Cancer Lett. 458, 86–91 (2019).
Yao, R. R., Li, J. H., Zhang, R., Chen, R. X. & Wang, Y. H. M2-polarized tumor-associated macrophages facilitated migration and epithelial-mesenchymal transition of HCC cells via the TLR4/STAT3 signaling pathway. World J. Surg. Oncol. 16, 9 (2018).
Zhang, J. et al. Hypoxia-inducible factor-1alpha/interleukin-1beta signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology 67, 1872–1889 (2018).
Wan, S. et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 147, 1393–1404 (2014).
Fan, Q. M. et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 352, 160–168 (2014).
Shen, Y. et al. TGF-beta regulates hepatocellular carcinoma progression by inducing Treg cell polarization. Cell Physiol. Biochem. 35, 1623–1632 (2015).
Zhou, J. et al. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int J. Cancer 125, 1640–1648 (2009).
Kuang, D. M. et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology 51, 154–164 (2010).
Wu, Y. et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 57, 1107–1116 (2013).
Hoffman, W., Lakkis, F. G. & Chalasani, G. B Cells, Antibodies, and More. Clin. J. Am. Soc. Nephrol. 11, 137–154 (2016).
Kim, Y. H., Choi, B. H., Cheon, H. G. & Do, M. S. B cell activation factor (BAFF) is a novel adipokine that links obesity and inflammation. Exp. Mol. Med. 41, 208–216 (2009).
Miyake, T. et al. B cell-activating factor is associated with the histological severity of nonalcoholic fatty liver disease. Hepatol. Int. 7, 539–547 (2013).
Nakamura, Y. et al. Depletion of B cell-activating factor attenuates hepatic fat accumulation in a murine model of nonalcoholic fatty liver disease. Sci. Rep. 9, 977 (2019).
Kim, D. H. & Do, M. S. BAFF knockout improves systemic inflammation via regulating adipose tissue distribution in high-fat diet-induced obesity. Exp. Mol. Med. 47, e129 (2015).
Zhang, H., Stolz, D. B., Chalasani, G. & Thomson, A. W. Hepatic B cells are readily activated by Toll-like receptor-4 ligation and secrete less interleukin-10 than lymphoid tissue B cells. Clin. Exp. Immunol. 173, 473–479 (2013).
Tarantino, G. et al. Enhanced serum concentrations of transforming growth factor-beta1 in simple fatty liver: is it really benign? J. Transl. Med. 6, 72 (2008).
Shalapour, S. et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345 (2017).
McPherson, S., Henderson, E., Burt, A. D., Day, C. P. & Anstee, Q. M. Serum immunoglobulin levels predict fibrosis in patients with non-alcoholic fatty liver disease. J. Hepatol. 60, 1055–1062 (2014).
Schneider, C. et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut 61, 1733–1743 (2012).
Zhang, Z. et al. Landscape of infiltrating B cells and their clinical significance in human hepatocellular carcinoma. Oncoimmunology 8, e1571388 (2019).
Brunner, S. M. et al. Tumor-infiltrating B cells producing antitumor active immunoglobulins in resected HCC prolong patient survival. Oncotarget 8, 71002–71011 (2017).
Garnelo, M. et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 66, 342–351 (2017).
Faggioli, F. et al. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 67, 1970–1985 (2018).
Rosales, C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. 9, 113 (2018).
Zang, S. et al. Neutrophils play a crucial role in the early stage of nonalcoholic steatohepatitis via neutrophil elastase in mice. Cell Biochem Biophys. 73, 479–487 (2015).
Rensen, S. S. et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE 7, e52411 (2012).
Ibusuki, R. et al. Transgenic expression of human neutrophil peptide-1 enhances hepatic fibrosis in mice fed a choline-deficient, L-amino acid-defined diet. Liver Int. 33, 1549–1556 (2013).
Alkhouri, N. et al. Neutrophil to lymphocyte ratio: a new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int. 32, 297–302 (2012).
Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147 (2018).
van der Windt, D. J. et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 68, 1347–1360 (2018).
Li, X. et al. Neutrophil count is associated with myeloid derived suppressor cell level and presents prognostic value of for hepatocellular carcinoma patients. Oncotarget 8, 24380–24388 (2017).
Kuang, D. M. et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J. Hepatol. 54, 948–955 (2011).
Gao, Q. et al. CXCR6 upregulation contributes to a proinflammatory tumor microenvironment that drives metastasis and poor patient outcomes in hepatocellular carcinoma. Cancer Res. 72, 3546–3556 (2012).
Zhou, S. L. et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 150, 1646–1658 e17 (2016).
He, M. et al. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology 5, e1219828 (2016).
Audiger, C., Rahman, M. J., Yun, T. J., Tarbell, K. V. & Lesage, S. The importance of dendritic cells in maintaining immune tolerance. J. Immunol. 198, 2223–2231 (2017).
Henning, J. R. et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 58, 589–602 (2013).
Ormandy, L. A. et al. Direct ex vivo analysis of dendritic cells in patients with hepatocellular carcinoma. World J. Gastroenterol. 12, 3275–3282 (2006).
Tosello-Trampont, A. C., Landes, S. G., Nguyen, V., Novobrantseva, T. I. & Hahn, Y. S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J. Biol. Chem. 287, 40161–40172 (2012).
Kuang, D. M. et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 206, 1327–1337 (2009).
Perumpail, R. B., Wong, R. J., Ahmed, A. & Harrison, S. A. Hepatocellular carcinoma in the setting of non-cirrhotic nonalcoholic fatty liver disease and the metabolic syndrome: US experience. Dig. Dis. Sci. 60, 3142–3148 (2015).
Konerman, M. A., Jones, J. C. & Harrison, S. A. Pharmacotherapy for NASH: current and emerging. J. Hepatol. 68, 362–375 (2018).
Ratziu, V. et al. REGENERATE: design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp. Clin. Trials 84, 105803 (2019).
Fantuzzi, L., Tagliamonte, M., Gauzzi, M. C. & Lopalco, L. Dual CCR5/CCR2 targeting: opportunities for the cure of complex disorders. Cell Mol. Life Sci. 76, 4869–4886 (2019).
Dufour J. F., Caussy C. & Loomba R. Combination therapy for non-alcoholic steatohepatitis: rationale, opportunities and challenges. Gut. (2020). https://doi.org/10.1136/gutjnl-2019-319104.
Cheng, A. L., Hsu, C., Chan, S. L., Choo, S. P. & Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 72, 307–319 (2020).
Zhu, X. D. & Sun, H. C. Emerging agents and regimens for hepatocellular carcinoma. J. Hematol. Oncol. 12, 110 (2019).
Acknowledgements
This study was supported by a grant (2017R1A2B3006776) from the National Research Foundation (NRF) funded by the Ministry of Education, Science, and Technology (MEST) of the Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koo, SY., Park, EJ. & Lee, CW. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp Mol Med 52, 1209–1219 (2020). https://doi.org/10.1038/s12276-020-0480-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0480-3
This article is cited by
-
Differential DNA methylation and changing cell-type proportions as fibrotic stage progresses in NAFLD
Clinical Epigenetics (2021)
-
The impact of endotrophin on the progression of chronic liver disease
Experimental & Molecular Medicine (2020)
