
Abstract
Over 90 years ago, Otto Warburg’s seminal discovery of aerobic glycolysis established metabolic reprogramming as one of the first distinguishing characteristics of cancer1. The field of cancer metabolism subsequently revealed additional metabolic alterations in cancer by focusing on central carbon metabolism, including the citric acid cycle and pentose phosphate pathway. Recent reports have, however, uncovered substantial non-carbon metabolism contributions to cancer cell viability and growth. Amino acids, nutrients vital to the survival of all cell types, experience reprogrammed metabolism in cancer. This review outlines the diverse roles of amino acids within the tumor and in the tumor microenvironment. Beyond their role in biosynthesis, they serve as energy sources and help maintain redox balance. In addition, amino acid derivatives contribute to epigenetic regulation and immune responses linked to tumorigenesis and metastasis. Furthermore, in discussing the transporters and transaminases that mediate amino acid uptake and synthesis, we identify potential metabolic liabilities as targets for therapeutic intervention.
Similar content being viewed by others
Introduction
The near century of progress1 in cancer metabolism continues to impact our understanding of oncology. Each decade has yielded new discoveries and engendered additional avenues of research and approaches to therapy. Classically, cancer metabolism has focused on central carbon metabolism, including glycolysis and the tricarboxylic acid cycle (the citric acid cycle, TCA cycle). However, new studies have shed light on the important role of amino acids in cancer metabolism. Amino acids also serve the essential purposes of redox balance, energetic regulation, biosynthetic support, and homeostatic maintenance. This wide breadth of metabolic activities has made amino acid metabolism increasingly popular in cancer research.
While glucose is a renowned energy source for cancer growth, amino acids are also important fuels supporting cancer development. Glutamine, for example, is largely anaplerotic and relinquishes both amine groups to support the TCA cycle2. In addition to glutamine, other amino acids can function as opportunistic fuel sources for cells. Branched-chain amino acids (BCAAs; valine, leucine, and isoleucine) are alternative sources of organic molecules that can also fuel the TCA cycle3.
Biosynthetic pathways, like bioenergetic pathways, also rely on various amino acid contributors. Through acetyl-CoA synthesis, lipogenesis can be mediated by catabolism of BCAAs3. Nucleotide synthesis, which can be delineated into purine and pyrimidine biosynthesis, is another amino acid-dependent process4. Glycine, glutamine, and aspartate serve as carbon and nitrogen donors for purine biosynthesis5. Additional sources of carbon for nucleobases as a form of formate include glycine, serine, and methionine, which provide one-carbon units through the methionine–folate cycle6,7,8.
Amino acids produce derivatives that also support cancer growth and metastatic potential. Arginine-derived polyamines alter gene expression by modulating global chromatin structure and cancer cell proliferation9. Kynurenine produced from tryptophan induces immunosuppression10,11 by binding to and activating the transcription factor aryl hydrocarbon receptor (AhR)12,13,14. This impairs the ability of immune-tolerant dendritic cells (DCs) and regulatory T cells to target and eliminate cancer cells11,15.
Cancer cell proliferation results in the accumulation of reactive oxygen species, which can damage macromolecules and ultimately lead to cell death. To counter this, cancer cells are reliant upon the synthesis of glutathione from glutamate, glycine, and cysteine to mediate redox balance16,17,18. While NADPH sourced from glycolysis and the pentose phosphate pathway are commonly identified as the main contributors to redox balance, a significant amount of NADPH is also produced by the folate cycle, which is primarily driven by serine-derived one-carbon units19.
In addition to the direct integration of amino acids and their derivatives in metabolic reprogramming processes, amino acids are also fundamental in mediating epigenetic regulation and posttranscriptional modification. For example, DNA and histone methylation are regulated by balanced metabolite levels in the methionine cycle, which are influenced by methionine, serine, and glycine20,21. Similarly, histone acetylation events promoting gene expression and cancer progression require acetyl-CoA, which can be derived from BCAAs and lysine22. Amino acid-derived acetyl-CoA is also relevant for protein acetylation that can support tumor growth23,24.
Transaminases are an important class of enzymes for cancer. By facilitating the interconversion of amino acids, transaminases allow for the exploitation of the diverse functions of amino acids from irregular sources. Aspartate transaminase (AST, or glutamic oxaloacetic transaminase (GOT1 and 2)) is essential for redox balance and growth in pancreatic cancer cells25. In colorectal cancer, phosphoserine aminotransferase 1 (PSAT1) levels have been implicated in poor prognosis26. The high demand for essential amino acids (EAAs) to support cancer cell growth leads to the upregulation of EAA transporters to meet these requirements, a common feature of many cancers27,28,29.
The wide range of amino acid functions, including amino acid synthesis, breakdown, and transport, has provided numerous targets for the development of drugs. Amino acid-degrading enzymes such as arginase and asparaginase have been shown to exert antitumor effects in preclinical and clinical settings;30,31,32,33,34 however, additional pathways involving amino acid transport and synthesis suggest effective therapies. Transporters are attractive therapeutic targets given the availability of various transport inhibitors; this method could be applied to ASCT2 (neutral amino acid transporter B(0), also known as alanine, serine, cysteine transporter 2), which regulates glutamine transport35, or xCT (cystine/glutamate antiporter), which provides cysteine for glutathione synthesis36. Other avenues of intervention include amino acid synthesis pathways such as targeting the rate-limiting enzyme phosphoglycerate dehydrogenase (PHGDH) of the serine synthesis pathway37,38 or indoleamine 2,3-dioxygenase 1 (IDO1), which generates kynurenine from tryptophan39,40. Overall, multiple factors involved in the regulation and synthesis of amino acids could be subject to potential therapeutic intervention.
The principal focus of this review is the specific relationship between amino acids and cancer metabolism. Relevant work detailing nonessential amino acid (NEAA) metabolism, transporters, and transaminase in cancer is covered in several reviews and in the cited articles2,6,41,42,43,44. These in combination with recently published studies supplement the discussion of the roles of amino acids, which include bioenergetic formation, biosynthetic activity, and resistance to metabolic stress. This review also discusses the regulation of amino acid metabolism in cancer, therapies that are under investigation, and aspects of amino acid metabolism relevant to inhibition with a specific focus on transaminases and amino acid transporters.
Amino acids as alternative fuels
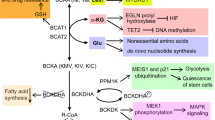
In cancer cells, glutamine is the major amino acid that serves as an anaplerosis metabolite and drives the tricarboxylic acid (TCA) cycle to sustain mitochondrial ATP production. Anaplerotic metabolism of glutamine generates α-ketoglutarate (α-KG) and subsequently oxaloacetate (OAA) and fuels the TCA cycle through a series of biochemical reactions termed glutaminolysis2 (Figs. 1, 4b). Under glucose-deprived conditions, glutamine-derived fumarate, malate, and citrate are significantly increased45. Similarly, under hypoxia or in cancer cells with mitochondrial dysfunctions, the direction of metabolic flow and utilization of glutamine is drastically changed. In such conditions, α-KG from glutamine can undergo reductive carboxylation to generate isocitrate, which is then converted into citrate46,47. Upon glutamine deprivation, asparagine plays a critical role in suppressing apoptotic cell death48,49. The positive regulator that transduces a signal from glutamine depletion to apoptosis is citrate synthase (CS)48. Under normal circumstances, CS condenses OAA from glutamine with acetyl-CoA to maintain the TCA cycle function. Silencing CS leads to enhanced OAA diversion from the TCA cycle to aspartate and asparagine biosynthesis, protecting cells from glutamine depletion-mediated apoptosis48. Exogenous asparagine completely restored cell survival under glutamine-depleted conditions, whereas silencing asparagine synthetase (ASNS) led to apoptosis even in the presence of glutamine. These observations highlight the potential importance of ASNS during tumor cell accumulation and progression (when glutamine availability is limited). Indeed, ASNS expression is associated with poor prognosis in brain tumors, such as glioma and neuroblastoma48.

Metabolic reprogramming is a staple of cancer cell growth and proliferation. Both essential and nonessential amino acids (EAAs and NEAAs) support altered metabolism by serving as energy sources, biosynthetic molecules, and mediators of redox balance. Amino acids produce metabolic intermediates, such as acetyl-CoA, that sustain energy synthesis through the citric acid cycle. Amino acids also provide building blocks for nucleotide synthesis and lipogenesis that are critical to a cell’s ability to grow and develop. To circumvent the effects of oxidative stress, amino acids can regulate redox balance through their production of glutathione. Furthermore, EAA catabolism contributes to the generation of NEAAs through chemical reactions, including those mediated by transaminases. Amino acids are in green, and other metabolites are in red. Orange represents transporters. Yellow boxes signify enzymes. SHMT1 serine hydroxymethyltransferase, cytosolic, BCAT branched-chain amino acid transaminase, mitochondrial, BCAA branched-chain amino acid (valine, leucine, isoleucine), BCKA branched-chain ketoacid, GOT1 aspartate transaminase, cytosolic (AST), GLS glutaminase, GS glutamine synthetase (cytosolic and mitochondrial), ASNS asparagine synthetase, PRODH pyrroline-5-carboxylate dehydrogenase, PYCR pyrroline-5-carboxylate reductase, P5C pyrroline-5-carboxylate, GSH glutathione, Gly glycine, Ser serine, Met methionine, Met cycle methionine cycle, Gln glutamine, Cys cysteine, Glu glutamate, Asp aspartate, Pro proline, Asn asparagine, Arg arginine, PRPP phosphoribosyl pyrophosphate, acetyl-coA acetyl-coenzyme A, α-KG alpha-ketoglutaric acid, OAA oxaloacetic acid, LAT1 large-neutral amino acid transporter 1, SLC25A44 solute carrier family 25 member 44, GLUT glucose transporter, TCA cycle the tricarboxylic acid (also known as the citric acid cycle).
In addition to glutamine, cancer cells also utilize other amino acids as “alternative fuels” to compete for energy with various cells in tumor stroma and to optimize nutrient utilization during the evolution of the tumor41. This contrasts with prevailing views that transforming mutations impose a rigid dependence on specific nutrients, such as glucose50. In human pancreatic ductal adenocarcinoma (PDAC), elevation of circulating BCAAs (leucine, isoleucine, and valine) is an early event of tumor development51. Given that 95% of PDAC is KRAS-driven and that KRAS plays an important role in scavenging nutrients52,53, elevated plasma levels of BCAAs may indicate a prominent role of protein breakdown in KRAS-mutant PDAC. BCAAs can provide an anaplerotic substrate in some tissues (Fig. 1)3. Elevated BCAAs in the plasma of patients with pancreatic cancers51 can potentially be converted into acetyl-CoA and other organic molecules that also enter the TCA cycle. Among BCAAs, leucine is important for melanoma cell survival54. Hyperactivation of the RAS–MEK pathway in melanoma renders cancer cells dependent on leucine, and leucine deprivation in melanoma cells fails to appropriately activate autophagy, subsequently leading to apoptotic death54. Although not in a cancer setting, threonine can yield acetyl-CoA to feed the TCA cycle. Threonine catabolism mediated by threonine dehydrogenase (TDH) produces glycine and acetyl-CoA, which is then utilized to sustain mitochondrial ATP production in mouse embryonic stem cells (eSCs)55.
Amino acids as biosynthetic materials
Enhanced biosynthetic activities are an essential feature of metabolic reprogramming in cancer: they support cells to produce the macromolecules required for DNA replication, cell division, and subsequent tumor growth. Biosynthetic pathways or anabolic pathways convert simple metabolites (e.g., sugars and amino acids) to complex molecules through ATP-dependent processes. Amino acids are involved in synthesizing three primary macromolecules: proteins, lipids, and nucleic acids (Fig. 1). All 20 canonical amino acids are proteinogenic, but only a subset of amino acids is involved in nonessential amino acid (NEAA) synthesis (e.g., glutamine, glutamate, methionine, and phenylalanine). Among the amino acids involved in NEAA synthesis, glutamine supplies nitrogen for synthesizing the amide group of asparagine. It also contributes to the synthesis of several amino acids through its catabolism to glutamate. Glutamine is converted to glutamate, mainly by glutaminase (GLS) activity (Figs. 1, 4b)42. Glutamate can then be further converted to α-KG and other amino acids, such as alanine, aspartate, and phosphoserine, by aminotransferase reactions (Figs. 1, 4b)43.
Unlike the aforementioned glutamine-derived amino acids, which need glutamate as a nitrogen donor, asparagine requires glutamine for de novo synthesis (Figs. 1, 2e, 4b). Glutamine is a substrate for asparagine synthetase (ASNS), providing amide nitrogen to aspartate to produce asparagine. Arginine is another amino acid used to synthesize nonessential amino acids. It can serve as the precursor to proline or as an additional source of glutamate, both via the intermediacy of 1-pyrroline-5-carboxylate (P5C) (Fig. 1)56. 1-Pyrroline-5-carboxylate reductase (PYCR) converts P5C into proline, and 1-pyrroline-5-carboxylate dehydrogenase (P5CDH encoded by ALDH4A1) catalyzes the conversion of P5C into glutamate57. Serine and glycine are closely related and can be interconverted by serine hydroxymethyltransferase (SHMT1 and 2) (Fig. 1)58. SHMT is one of the key enzymes in folate-mediated one-carbon metabolism. One-carbon metabolism encompasses both the folate and methionine cycle and provides methyl groups for the one-carbon pools that are required for de novo nucleotide biosynthesis and DNA methylation59. Tetrahydrofolate (THF) serves as a universal one-carbon acceptor and can accept one-carbon from the conversion of serine to glycine (folate cycle), the oxidation of glycine to CO2 and NH3 by the glycine cleavage system (GCS)6 or methionine to homocysteine conversion (methionine cycle)59. One-carbon bound THF exists in different oxidation states (5,10-methylene tetrahydrofolate (5,10-meTHF), 5-methyl-THF, formate (10-formyl THF)), and supports distinct biosynthetic functions, for example, 5,10-meTHF for pyrimidine biosynthesis and 5-methyl-THF for purine biosynthesis59.

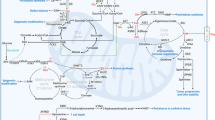
a Reverse-transsulfuration pathway: Cysteine can be produced from methionine through the reverse-transsulfuration pathway. This pathway is a combination of the methionine cycle and transsulfuration pathway. Homocysteine, the intermediate of the first step in the transsulfuration pathway, is generated from the methionine cycle. Serine condenses with homocysteine, producing cystathionine. Cystathionine is then converted to cysteine and alpha-ketobutyrate by CGL. Key enzymes are in red circle. THF tetrahydrofolate, CBS cystathionine β-synthase, SAM S-adenosylmethionine, CGL cystathionine γ-lyase. b Polyamine synthesis: Polyamines (putrescine, spermine, and spermidine) are synthesized from the amino acid arginine, and are converted from one to another (in the order of putrescine to spermidine to spermine). SAM, as the precursor of dcSAM, is the major donor for constructing polyamine structures. Key enzymes are in red circle. ODC ornithine decarboxylase, AMD S-adenosylmethionine decarboxylase, SAM S-adenosylmethionine, dcSAM decarboxylated S-adenosylmethionine. c Nitrogen and carbon source for nucleic acids: Aspartate, glycine, and glutamine provide nitrogen, and glycine and one-carbon units from the folate cycle (as a form of formate) provide carbon for purines. Glycine is formate’s indirect precursor through one-carbon metabolism, providing formate for biochemical reactions in purine biosynthesis. Aspartate and glutamine are the main amino acids involved in pyrimidine synthesis. Carbon (C) is in yellow, and nitrogen (N) is in green. d GSH and NADPH as antioxidants: Reactive oxygen species (ROS) bind and damage cellular macromolecules. The oxidation of NADPH and GSH allows ROS to be reduced to an inactive state. GSH reduces hydrogen peroxide to water and becomes oxidized to GSSG by GPX. Oxidized glutathione (GSSG) is then reduced back to GSH by GR in the presence of NADPH. Enzymes are shown in red circles. GPX glutathione peroxidase, GR glutathione reductase, GSH reduced glutathione, GSSG oxidized glutathione, NADPH reduced nicotinamide adenine dinucleotide phosphate, NADP+ oxidized nicotinamide adenine dinucleotide phosphate. e Amidation reaction for asparagine synthesis: Asparagine is synthesized by an amidotransferase reaction, catalyzed by asparagine synthetase (ASNS). The conserved amide group nitrogen is in a red box, while the enzyme is in a red circle.
The sulfur containing amino acid methionine can provide cysteine by the reverse-transsulfuration pathway (Fig. 2a). Homocysteine generated from the methionine cycle is condensed with serine to become cystathionine by cystathionine β-synthase (CBS), and is then transformed to cysteine by cystathionine γ-lyase (CGL) (Fig. 2a). Methionine to cysteine conversion is connected to serine–glycine interconversion because the methionine cycle is part of one-carbon metabolism coupled with the folate cycle, where SHMT catalyzes serine–glycine synthesis (Fig. 1)6. While asparagine is not directly involved in NEAA biosynthesis, it acts as an amino acid exchange factor and promotes amino acid uptake, preferentially of serine/threonine and nonpolar amino acids60, and can potentially support further building block synthesis.
Amino acids provide both carbon and nitrogen for nucleic acid synthesis (Fig. 2c). Purine biosynthesis requires formate, bicarbonate, and three amino acids: aspartate, glycine, and glutamine. While glutamine and aspartate act as the nitrogen source for both nucleobases (N1 from aspartate and N3 and N9 from glutamine) and the amino group of purines (glutamine for adenine and aspartate for guanine), glycine can contribute to purine biosynthesis in two ways: by direct incorporation into the purine backbone (C4, C5, and N7) or by producing one-carbon units for biochemical reactions involved in purine biosynthesis (C2 and C8) (Fig. 2c)5. The critical carrier of one-carbon units in the latter process is 5,10-meTHF. 5,10-meTHF is further converted to formate (10-formyl THF), contributing C2 and C8 carbons to the nucleobase4,7,61. Pyrimidine biosynthesis is simpler than that of purine. In contrast to purines that are synthesized as ribonucleotides rather than as nucleobases, pyrimidines are synthesized first as nucleobases and then conjugated to phosphoribosyl pyrophosphate (PRPP) to yield the corresponding ribonucleotide. The pyrimidine ring is derived from glutamine, aspartate, and bicarbonate. For pyrimidine synthesis, aspartate acts as both carbon and nitrogen donors (N1, C4, C5, and C6), whereas glutamine contributes to N3 of the nucleobase and amino group of cytosine (Fig. 2c)4. The one-carbon unit derived from serine to glycine conversion is required for thymidylate synthesis. 5,10-meTHF serves as a one-carbon donor to transfer a methyl group to deoxyuridine monophosphate (dUMP) and produce deoxythymidine monophosphate (dTMP), a reaction catalyzed by thymidylate synthase (TS)7,8.
In addition to their primary role in the biosynthesis of nitrogenous metabolites, amino acids can supply carbon atoms for lipid biosynthesis. Under hypoxia, glutamine contributes to the acetyl-CoA pools needed for lipogenesis by being converted into pyruvate that reenters the TCA cycle46,62. BCAAs can also contribute to lipogenesis. In differentiated adipocytes, BCAA catabolic flux increases, and BCAA-derived acetyl-CoA accounts for approximately 30% of the lipogenic acetyl-CoA pools3,63. Essential amino acids (EAAs) act not only as carbon donors, but their ratio can also regulate lipogenesis by impacting lipogenic gene expression64. In bovine mammary epithelial cells, the “optimal” amino acid (AA) ratio (OPAA = Lys:Met 2.9:1; Thr:Phe 1.05:1; Lys:Thr 1.8:1; Lys:His 2.38:1; Lys:Val 1.23:1) upregulates lipogenic gene expression and alters the expression of key miRNAs involved in the control of lipogenic balance, implying a potentially important role of EAA ratios in lipid synthesis64. It would be interesting to see if this is conserved in other species as well as in cancer.
Tumor-associated amino acid derivatives
Amino acid catabolism produces metabolic intermediates affecting tumor cell growth and survival. Polyamines (putrescine, spermine, and spermidine) might be the best-known metabolites to promote tumor proliferation and aggressiveness65. Polyamine synthesis starts from arginine conversion to ornithine through the action of arginase, which is then decarboxylated by the rate-limiting step enzyme, ornithine decarboxylase (ODC), to produce putrescine (Fig. 2b). Decarboxylated S-adenosylmethionine, catalyzed by S-adenosylmethionine decarboxylase (AMD)66, then donates its propyl amine moiety to putrescine and spermidine for the formation of spermidine and spermine, respectively (Fig. 2b)67. Elevated polyamine levels have been observed in patients with cancer. Polyamines and their metabolites in urine and plasma can be useful in both cancer diagnosis and as markers of tumor progression in lung and liver cancers68,69. Polyamines affect numerous processes in tumorigenesis, in part by regulating specific gene expression transcriptionally. As charged cations at physiological pH, polyamines can associate with nucleic acids70, which in turn can affect global chromatin structure71 as well as specific DNA–protein interactions72, leading to impacts on gene transcription. Posttranscriptional aspects of polyamine-mediated gene regulation are associated with the eukaryotic translation initiation factor 5A (eIF5A), whose expression/function is strongly correlated with unfavorable prognostic implications for several cancers73,74,75. The spermidine-derived amino acid hypusine, a unique eIF5A posttranslational modification of lysine residue 50, is essential for eIF5A functions76. Polyamines also exist as a polyamine-RNA complex77. Polyamine binding to RNA leads to structural changes, which stimulate and increase the efficiency of protein synthesis.
Nitric oxide (NO) is another metabolic consequence of arginine catabolism. Depending on its timing, location, and concentration, it has both tumor-suppressive and tumor-promoting effects78. It promotes tumor growth through multiple mechanisms, including increasing angiogenesis and limiting the host immune response against tumors. It can, however, also act as a tumor-suppressive molecule by activating caspases and upregulating tumor suppressor p5378. Thus, a better understanding of NO biology and further validation with molecular and clinical studies are necessary to develop NO-based strategies for cancer prevention and treatment.
Kynurenine is a tumor-associated metabolite that is catabolized from tryptophan by tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO)79. An increased kynurenine to tryptophan ratio has been observed in various tumors, including Hodgkin lymphoma, lung cancer, and ovarian cancer80,81,82. The important role of kynurenine is linked to its ability to suppress antitumor immune responses. Kynurenine secreted from tumors induces cytotoxic CD8 T-cell death, enhancing immune evasion during metastasis10. Importantly, kynurenine-mediated immunosuppression is not limited to cross talk between cancer cells and immune cells (Fig. 3c). Communication within the different immune cells also regulates their kynurenine synthesis11,15. Regulatory T (TR) cells activate IDO in DCs, priming DCs for tolerance induction through CTLA-4 (resting TR cells) or IFN-γ (CD3-activated TR cells) (Fig. 3c)11,15. As immunometabolic adjuvants to widen therapeutic windows, IDO inhibitors may leverage not only immuno-oncology modalities but also conventional chemotherapy and/or radiotherapy.

a Amino acids provide metabolic intermediates for epigenetic regulation. One-carbon units from the methionine (shown here) and folate cycle serve as a methyl donor for DNA and histone methyltransferases, while acetyl-CoA from BCAAs and leucine can be utilized for histone acetylation. b Amino acid-derived acetyl-CoA is also involved in protein acetylation modification; a thrombopoietin (TPO)-responsive homodimeric receptor, CD110, activates lysine catabolism, which generates acetyl-CoA for LRP6 (a Wnt signaling protein) acetylation and promotes the self-renewal of tumor-initiating cells of colorectal cancer24. c Elevated kynurenine (Kyn) levels originating from tryptophan via the enzymes tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO) have been shown in several cancers, including Hodgkin lymphoma, lung cancer, and ovarian cancer. Kynurenine promotes tumor cell survival by both inducing T-cell death and inducing immune tolerance in dendritic cells (DCs). Methylation and acetylation are represented by red Me and blue Ac circles, respectively. Histone methylation and acetylation are represented by curved lines. DNA methylation is represented by a straight line. Amino acids are in green, and other metabolites are in red. Orange represents receptors. Yellow boxes signify proteins. SAM S-adenosylmethionine, SAH S-adenosyl homocysteine, Met methionine, Thr threonine, BCAAs branched-chain amino acids, Leu leucine, Lys lysine, Acetyl-CoA acetyl-coenzyme A, Trp tryptophan, Kyn kynurenine, IFN-γ interferon gamma, mTORC1 mammalian target of rapamycin complex 1, TDH threonine dehydrogenase, EP300 histone acetyltransferase p300, HAT histone acetyltransferase, CD110 myeloproliferative leukemia protein (thrombopoietin receptor), TPO thrombopoietin, IDO indoleamine 2,3-dioxygenase, TDO tryptophan 2,3-dioxygenase, CTLA-4 cytotoxic T-lymphocyte-associated protein 4, TR cell, regulatory T cell.
The proline downstream metabolite, 1-pyrroline-5-carboxylic acid (P5C, also known as hydroxyproline), has implications for tumor aggressiveness. Hydroxyproline provides a direct bridge between the TCA (glutamate and α-KG) and urea cycle (ornithine) (Fig. 1)83. It is correlated with hepatocellular carcinoma (HCC) clinical pathogenesis84. Mechanistically, hypoxic microenvironments activate proline metabolism, resulting in the accumulation of hydroxyproline that promotes HCC tumor progression and induces sorafenib resistance by modulating hypoxia-inducible factor 1-alpha (HIF1α)84.
Amino acids for redox balance
Cancer cells inevitably produce high levels of reactive oxygen species (ROS) due to their highly proliferative nature. ROS are intracellular chemical species containing oxygen and include the superoxide anion (O2−), hydrogen peroxide (H2O2), and the hydroxyl radical (OH·). Oxygen radicals produced from ROS can covalently bind to and oxidize macromolecules (lipids, proteins, and DNA), leading to cellular damage (Fig. 2d). Consequently, increased ROS production requires coupling with increased antioxidant defense production to protect cancer cells from ROS-mediated demise; thus, cancer cells allocate significant energy to maintain their intracellular redox balance. Key metabolic players that control the redox state are reduced nicotinamide adenine dinucleotide phosphate (NADPH) and reduced glutathione (GSH) (Fig. 2d). NADPH is a cofactor that not only provides reducing power for macromolecule biosynthesis, but also functions as an antioxidant by acting as a hydride (hydrogen anion) donor in various enzymatic processes, including reduction of glutathione disulfide (GSSG) back to GSH (Fig. 2d). GSH is an essential thiol antioxidant and plays a key role in controlling the redox state of all subcellular compartments85. GSH reduces hydrogen peroxide to water and becomes oxidized to GSSG in the presence of GSH peroxidase (GPX) (Fig. 2d). Oxidized glutathione (GSSG) is then reduced back to GSH by glutathione reductase (GR) and NADPH. Amino acids are the main elements for GSH synthesis and NADPH generation. Glutathione synthesis requires three amino acids: glutamate, glycine, and cysteine. Among them, cysteine is the key element because of its thiol group (R-SH), which has redox properties. Inhibiting cysteine uptake reduces viability due to cell death caused by uncontrolled oxidative stresses17,18,86. Cysteine can be imported into cells either directly or in its oxidized form, cystine, through the cystine/glutamate antiporter system xc− (xCT). Once in the cell, cystine is immediately reduced to cysteine either by intracellular GSH via the formation of a mixed disulfide intermediate or by thioredoxin reductase 1 (TRR1)87. The rate-limiting step of glutathione synthesis is the ATP-dependent condensation of cysteine and glutamate to form the dipeptide γ-glutamylcysteine by glutamate cysteine ligase (GCL)88. Glycine is then added to the C-terminal of γ-glutamylcysteine to produce glutathione.
The reducing equivalent NADPH is required to maintain multiple antioxidant defense systems. It has been generally accepted that the main route to produce cellular NADPH is glucose via the pentose phosphate pathway (PPP). However, a growing body of research has uncovered that serine-driven one-carbon metabolism via the folate cycle contributes nearly as much to NADPH production as the PPP and malic enzymes in proliferating cells19. In the folate cycle, serine to glycine conversion produces 5,10-meTHF, which is oxidized to 10-formyl-tetrahydrofolate (formate/10-formyl THF)6. The latter reaction is coupled to the reduction of NADP+ to NADPH. SHMT-mediated serine catabolism, especially mitochondrial SHMT2 reaction, is critical for redox regulation under hypoxia89. SHMT2 is induced by hypoxic stress through HIF1α and is involved in maintaining the cellular NADPH/NADP+ ratio89. Serine is also involved in GSH synthesis via the folate cycle90. In addition to NADPH production, the folate cycle contributes to the production of GSH by intersecting with the methionine cycle6. Thus, it is not surprising that serine depletion results in glutathione reduction91. Indeed, activation of serine synthesis is now well identified as a bypass of glycolytic flux contributing to GSH synthesis92,93. Considering the importance of amino acids in redox homeostasis, the transport and internal synthesis pathways for cysteine, serine, glutamine, and to some extent glycine would be legitimate targets for the development of novel redox-based therapeutics.
Amino acids as epigenetic and posttranscriptional regulators
Epigenetic alterations are heritable features that affect cellular phenotypes by modifying gene expression independent of the DNA sequence94,95. Epigenetic control of gene expression is fine-tuned by a balance between enzymes that “write” regulatory marks onto DNA and histone proteins (e.g., DNA- and histone methyltransferases and histone acetyltransferases) and other enzymes that “erase” these same marks (e.g., histone deacetylases, histone demethylases, and DNA demethylases)96. DNA methylation is mediated by DNA methyltransferases (DNMTs), which catalyze the covalent addition of a methyl group to cytosine to form 5-methylcytosine in the context of CpG dinucleotides97. In general, methylation of CpG islands in promoter regions inhibits transcription. Global DNA hypomethylation and hypermethylation of promoters of tumor suppressor genes and homeobox genes is a ubiquitous feature of the cancer genome of DNA98,99,100. Histone methyltransferases (HMTs) catalyze the transfer of mono- to tri-methyl groups to lysine and arginine residues of histone proteins101. Histone methylation can be involved in either activation or repression of gene expression, depending on which residue is modified and how many methyl groups are incorporated. Methionine, by serving as the methyl group donor for methylation, is the major amino acid that contributes to epigenetic regulation (Fig. 3a). Both DNMTs and HMTs utilize S-adenosylmethionine (SAM) as a methyl donor. SAM is generated in the methionine cycle by methionine adenosyltransferase (MAT) using methionine and ATP as substrates102. After donating a methyl group, SAM becomes S-adenosyl-homocysteine (SAH), which inhibits both DNMTs and HMTs (Fig. 3a). Consequently, alterations in the SAM/SAH ratio regulate these methyltransferases’ activity103. The importance of SAM in tumor survival has been observed in various studies where cancer cells’ unique requirement for methionine is based on SAM dependence, not methionine dependence104,105. The enhanced methionine cycle leads to an excess supply of SAM. This in turn causes DNA hypermethylation and inappropriate gene silencing as well as aberrant histone methylation20,106 and enhanced tumor growth. Threonine catabolism mediated by threonine dehydrogenase (TDH) can also provide a precursor for SAM (Fig. 3a)107. Glycine from the TDH reaction facilitates one-carbon metabolism via the glycine cleavage system. Threonine depletion or TDH deletion reduces SAM levels and decreases trimethylation of histone H3 on lysine residue 4 (H3Kme3)107. Although TDH enzyme function is lost in humans, it certainly suggests that human tumorigenesis can be related to a dysregulation in this metabolic pathway (e.g., upregulation of threonine uptake).
Histone acetylation increases DNA accessibility by reducing the attractive electronic interactions between histones and DNA, thereby supporting gene expression. It is regulated by opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs) that catalyze the addition and removal of the acetyl group on lysine residues, respectively108. Acetyl-CoA is an essential metabolic intermediate that regulates acetylation status. HATs utilize the acetyl group of acetyl-CoA to form ε-N-acetyl lysine, and evidence indicates that both acetyl-CoA abundance and the ratio of acetyl-CoA to coenzyme A regulate histone acetylation in cancer109.
BCAAs (leucine, isoleucine, and valine) and lysine are catabolized to acetyl-CoA that can potentially be utilized by HATs22. Importantly, the acetyl-CoA pools from amino acids also modulate protein acetylation and can promote tumor growth. Leucine provides acetyl-CoA to the EP300 acetyltransferase, which mediates inhibitory acetylation of the mTORC1 regulator Raptor at K1097 and leads to mTORC1 activation (Fig. 3a)23. Lysine-derived acetyl-CoA plays an important role in the self-renewal of tumor-initiating cells (TICs) of colorectal cancer (Fig. 3b)24. CD110, a thrombopoietin (TPO)-responsive homodimeric receptor, is a key molecule expressed in colon TICs that drives liver metastasis24. Upon TPO binding, CD110 activates lysine degradation, which generates acetyl-CoA for acetylation of LRP6 (Fig. 3b), a coreceptor of Wnt signaling crucial for self-renewal of colon TICs110, in an EP300-dependent manner. Acetylation of LRP6 stimulates LRP6 activity and self-renewal of CD110+ colon TICs. Collectively, amino acid catabolism involves both epigenetic regulation and posttranslational modification of key proteins in the survival and proliferation signaling pathways, impacting tumor aggressiveness.
Regulation of amino acid metabolic enzymes and transporters
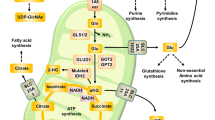
Cancer cells reprogram metabolism in ways that can meet their increased demand for amino acids37,111,112,113. Amino acids can be taken up from the extracellular environment or, in the case of NEAAs, be synthesized by various reactions, including transaminase reactions. Cancer cells display enhanced amino acid uptake114,115. Because of their hydrophilic nature, amino acids require a transporter system to cross the plasma membrane. Most amino acid transporters recognize more than one amino acid as a substrate. Among the transporters, SLC6A14 shows the most broad substrate selectivity encompassing all essential amino acids as well as glutamine (Fig. 4a) and is upregulated in several cancers of epithelial origin, such as colon cancer27, cervical cancer28, and certain subtypes of breast cancer29.

a, b Cells can either import amino acids from the extracellular environment or synthesize them inside the cell. Amino acid transporters are required to transfer them across the plasma membrane (shown in a), and transaminases are required to generate amino acids from sugar precursors and nitrogen from other amino acids (shown in b). Amino acid transporters and transaminases contribute to metabolic reprogramming in cancer through manipulation of the amino acid pool inside and outside cancer cells. Their impact on the tumor microenvironment is associated with tumor aggressiveness; some induce tumor cell growth and expansion through activation of signaling pathways (e.g., mTORC1 activation) or inactivation of tumor suppressors, while others result in apoptotic cell death. Amino acids are in green, and other metabolites are in red. Bright red represents transporters. Yellow boxes signify enzymes. The relative sizes of Lys and Arg represent the tendency to be transferred across CAT1 in a cancer setting. SLC6A14 solute carrier family 6 member 14, LAT1 large-neutral amino acid transporter 1, ASCT2 alanine, serine, cysteine-preferring transporter 2, xCT/CD98hc (4FC2) cystine/glutamate heterodimeric antiporter, CAT2B cationic amino acid transporter 2B, CAT1 cationic amino acid transporter 1, GLS glutaminase, GS glutamine synthetase, PSAT1 phosphoserine aminotransferase 1, AST aspartate aminotransferase, also known as glutamic oxaloacetic transaminase (GOT). GOT1 is in the cytosol and GOT2 in mitochondria, ASNS asparagine synthetase, ALT alanine aminotransferase, EAA essential amino acid, Na+, sodium ion, Cl− chloride ion, Gln glutamine, Leu leucine, AA amino acid, Gly glycine, Glu glutamic acid, Cys cysteine, Cys cystine, Lys, lysine, NH4+ ammonium ion, Asn asparagine, Ala alanine, Asp aspartic acid, Ser serine, PHP 3-phosphohydroxypyruvate, α-KG alpha-ketoglutarate, e− electron, NADPH reduced nicotinamide adenine dinucleotide phosphate, NAD+ oxidized NADPH, OAA oxaloacetate.
The system L-type (leucine-preferring) amino acid transporter LAT1 is an amino acid antiporter, mediating the influx of BCAAs and bulky amino acids (phenylalanine, methionine, histidine, tryptophan, and tyrosine) into cells116,117 in exchange for efflux of intracellular substrates (EAA or glutamine) (Fig. 4a)118,119. The expression of SLC7A5, which encodes LAT1, is elevated in various cancers, including bladder, breast, cervical, and skin cancer, through the action of oncogenes and miRNAs. Hypoxia-induced factor 2α (HIF2α) and oncogenic c-MYC transactivate SLC7A5, whereas miR-126 inhibits SLC7A5 expression120,121. The functional role of LAT1 in cancer, at least in part, is its ability to take in leucine with high affinity. Leucine is a well-known activator of mTOR signaling122,123 (Fig. 4a), and pharmacologic inhibition of LAT1 suppresses mTOR signaling and tumor growth (e.g., non-small cell lung cancer, oral cancer)124,125. LAT1-mediated leucine influx is coupled with another amino acid antiporter, ASCT2, which is encoded by SLC1A5 (Fig. 4a)126. ASCT2 mediates the Na+-coupled influx of neutral amino acids (alanine, serine, cysteine, and glutamine) in mandatory exchange for the Na+-coupled efflux of other amino acids126. In the functional coupling between LAT1 and ASCT2, glutamine enters the cancer cell through ASCT2, which then effluxes out of the cell via LAT1 coupled to the entry of leucine115 (Fig. 4a). Consequently, genetic or pharmacological inhibition of ASCT2 impedes LAT1-mediated leucine uptake, leading to inactivation of mTOR signaling in cancer cells127,128. Notably, SLC1A5 is also a target for c-MYC111,112, implying that cancer cells induce the two transporters (LAT1 and ASCT2) in a coordinated manner to enhance the functional coupling necessary for proliferation. SLC1A5 expression is decreased by the tumor suppressor RB, supporting its role in cancer growth129. The amino acid transport system x−c (xCT), which is encoded by SLC7A11, imports cystine and exports glutamate (Fig. 4a). The primary role of xCT is to provide cells with cysteine for glutathione synthesis. Similar to LAT1′s functional coupling with ASCT2, xCT appears to be functionally coupled to glutamine transporters36. In order to maintain glutathione levels, optimal xCT function requires intracellular glutamate pools to exchange with extracellular cysteine. Glutamine import from the extracellular environment, likely via ASCT2, can provide those glutamate pools through the GLS reaction. In addition to cysteine influx, xCT-mediated glutamate efflux also contributes to tumor aggressiveness by impacting the tumor microenvironment. Glutamate secreted to the extracellular environment via xCT causes peritumoral excitotoxic neuronal cell loss, creating room for tumor cell expansion130. Glutamate released through xCT can also act on tumor cells and promote their growth. In glioma, the secreted glutamate can activate AMPA receptors in both an autocrine and paracrine manner and cause intracellular Ca2+ oscillations, leading to tumor invasion131. In triple-negative breast cancer (TNBC), xCT-mediated glutamate release inhibits xCT and leads to intracellular cysteine depletion. In the absence of cysteine, EglN1, the main prolyl hydroxylase for the HIFα subunit, undergoes oxidative self-inactivation, resulting in HIF1α accumulation and tumor growth132. The TNBC study suggests that blocking xCT in TNBC can aggravate the disease, and thus, caution should be taken when targeting xCT. This notion is further supported by the findings that Slc7a11-null mice exhibit increased sensitivity to chemically induced carcinogenesis133. Basic amino acids, arginine and lysine, are transported via CAT1 and CAT2B, which are encoded by SLC7A1 and SLC7A2, respectively (Fig. 4a). In a cancer setting, CAT1′s ability to transport arginine, rather than lysine, appears to be more relevant to tumor growth and survival. CAT1 suppression reduces arginine uptake and NO production, resulting in apoptotic cell death in breast cancer cells134.
Unlike essential amino acids whose source is solely the extracellular environment, nonessential amino acids can be produced, in most cases, through transamination reactions, which transfer the amino group from glutamate to a sugar precursor and generate α-KG. Aspartate aminotransferase (AST, also known as glutamic oxaloacetic transaminase (GOT), GOT1 in the cytosol and GOT2 in the mitochondria) generates aspartate from oxaloacetate and glutamate (Fig. 4b). Interestingly, recent studies have discovered an important role for aspartate synthesis in maintaining reducing potential. GOT1 consumes aspartate in the cytosol and transfers electrons into the mitochondria, which are accepted by the electron transport chain (ETC) and consume nicotinamide adenine dinucleotide (NADH) to regenerate NAD+135,136 (Fig. 4b). NAD+ can then be utilized for OAA generation and aspartate biosynthesis135,136. In PDAC, GOT1-derived oxaloacetate (OAA) fuels the TCA cycle, which is further converted to malate and pyruvate to produce NADPH from NADP+ to maintain the cellular redox state25. Pathophysiologically, GOT is closely related to alanine aminotransferase (ALT). ALT generates alanine from pyruvate and the nitrogen of glutamate. Under normal physiology, the AST (GOT)/ALT ratio is <1, but upon liver damage, including hepatocellular carcinoma (HCC), AST levels become higher than ALT (AST/ALT ratio>1). In addition to serving as a liver damage marker, ALT has implications for tumor growth. Inhibition of ALT induces oxidative phosphorylation and a subsequent increase in mitochondrial ROS, suggesting ALT as a putative target to promote mitochondrial metabolism and inhibit tumor growth137. A transaminase for serine synthesis is phosphoserine aminotransferase 1 (PSAT1) (Fig. 4b). It transfers nitrogen from glutamate to 3-phosphohydroxypyruvate to make phosphoserine. Similar to other transaminases, PSAT1 is associated with tumor aggressiveness, especially in breast cancer. Both the mRNA and protein levels of PSAT1 in ER-positive primary tumors are associated with poor clinical outcomes following tamoxifen treatment, suggesting that combination with a regimen targeting PSAT1 might enable therapeutic efficacy in this subset of breast cancer26.
Some amino acids are produced by non-transaminase reactions. The best-known non-transaminase NEAA-synthesizing enzyme would be glutaminase (GLS). This amidohydrolase generates glutamate and ammonia from glutamine. GLS activity has been shown to be critical for most cancer cell growth, at least in monolayer culture138,139. Because glutamate serves as a nitrogen donor for transaminase reactions, inhibition of GLS can potentially impact NEAA synthesis. Glutamine and asparagine are synthesized by amidation reactions (Fig. 2e). Glutamine synthetase (GS) ligates ammonia to glutamate and produces glutamine, whereas asparagine synthetase (ASNS) generates asparagine from aspartate and the amide nitrogen of glutamine (Fig. 4b). Asparagine synthesis is important for acquiring a tolerance to nutrient deficiency. It is enhanced upon glucose deprivation via induction of ASNS expression in PDAC140, and inhibition of ASNS leads to glutamine-withdrawal-induced cell death48. Glycine is synthesized from serine by the SHMT reaction; SHMT transfers a one-carbon unit from serine to THF and generates 5,10-meTHF.
In addition to amino acid transporter-mediated influx or biosynthetic pathways, macropinocytosis and proteolytic degradation of extracellular proteins can serve as a source of amino acids52. Inhibition of these processes impairs tumor growth, especially in KRAS-mediated PDAC, which uses macropinocytosed protein as a nutrient source. A recent study uncovered SLC38A9 as the mediator required to release essential amino acids from lysosomes, including leucine (Fig. 4a). PDAC utilizes SLC38A9, an arginine-sensing lysosomal protein with homology to amino acid transporters, to enable leucine generated via lysosomal proteolysis to exit lysosomes and activate mTORC1 and drive cell growth141. Collectively, these cancer cell dependencies on amino acids point to potential vulnerabilities that could be exploited in the design of anticancer therapies.
Therapeutic applications from amino acid metabolism
A large body of work has demonstrated the diverse and important roles of amino acids in cancer metabolism30,142. Since the initial studies demonstrating the critical role of glutamine in the pathology of cancer, the field has continued to uncover the significance of other amino acids in metabolic alterations in cancer, and many efforts have been devoted to developing therapeutic agents targeting amino acid transport and catabolic/biosynthetic pathways. Although some amino acid-degrading enzymes, such as asparaginase and PEGylated arginine deiminase, have already been used in the clinic to treat tumors (asparaginase) and show antitumor efficacy in human patients (arginase), additional therapeutic targeting of cancer metabolism has led to surprisingly few new drugs31,32,33,34. In this section, we highlight a few promising targets in amino acid metabolism (Table 1).
Among the 20 canonical amino acids, glutamine metabolism has been a focal point in cancer therapy given cancer cells’ overreliance on glutamine. Both glutamine uptake and glutaminase activity have been actively investigated as oncological targets. Glutaminase inhibitors have been shown to reduce tumor burden45,143,144, and among the inhibitors, CB-839, the most advanced, is in clinical trials for multiple cancer types as a combination therapy. The glutamine (neutral amino acid) transporter ASCT2 is also an attractive target. The recently discovered V-9302, a selective inhibitor of ASCT2, showed in vivo antitumor efficacy, demonstrating the utility of a pharmacological inhibitor of glutamine transport in oncology35. Approaches targeting glutamate metabolism have been explored in several tumor models145,146. Glutamate conversion into α-KG through glutamate dehydrogenase (GDH) supports cancer cell growth in two ways: providing nitrogen for NEAA biosynthesis as well as an anaplerotic intermediate for the TCA cycle. Several inhibitors for GDH are available for experimental use and have been shown to inhibit tumor growth145,146,147.
Cysteine uptake plays an important role in cancer by maintaining redox balance, and numerous studies have shown the efficacy of xCT inhibition on tumor growth36,148,149. Importantly, a recent study revealed that xCT suppression acts synergistically with the immunotherapeutic agent anti-CTLA-4150. This study laid the groundwork for the clinical utilization of specific xCT inhibitors to expand the efficacy of existing anticancer immunotherapeutics150. Efforts have been made to target serine metabolism, with a specific focus on phosphoglycerate dehydrogenase (PHGDH), a rate-limiting enzyme in the serine de novo synthesis pathway38,151,152,153. Inhibition of PHGDH as a single agent, however, appears to only be effective under conditions of low serine availability154. In addition to the inhibition of serine biosynthesis, dietary restriction of serine and glycine has been explored in mice and was shown to be effective at limiting tumor growth in certain conditions where de novo serine synthesis activity is low21,154. Given the complex interplay between serine biosynthesis and environmental serine availability, selecting the proper tumor types would be critical for successful targeting of serine metabolism as a method of therapeutic intervention.
Manipulation of aspartate availability by dietary restriction or chemical inhibition is an emerging form of therapy. Initial high-throughput compound screening and further medicinal chemistry-based optimization developed two novel series of phenylurea-based compounds inhibiting GOT1 enzymatic activity with potencies in the low micromolar IC50 range155. This work lays the foundation for the development of potential therapeutics for the treatment of tumors that are highly dependent upon metabolic pathways involving GOT1 for the maintenance of redox homeostasis and sustained proliferation (e.g., PDAC).
In essential amino acid metabolism, tryptophan catabolism has been in the spotlight for cancer immunotherapy. Kynurenine produced from tryptophan by IDO1 and TDO2 binds to and activates the transcription factor aryl hydrocarbon receptor (AhR)12,13,14. AhR activation then leads to the generation of immune-tolerant DCs and regulatory T cells15, which foster a tumor immunological microenvironment that is defective in recognizing and eradicating cancer cells. Multiple IDO1 inhibitors have been actively evaluated in clinical trials39, and importantly, combination treatment of IDO inhibitor with the PD-1 immune checkpoint inhibitor pembrolizumab shows durable responses40. Although it is still in the early stages of development, targeting the IDO1/TDO2–KYN–AhR signaling pathway would potentially open new avenues for developing cancer immunotherapies.
BCAA metabolism has also received notable attention as a drug target. The BCAA catabolic enzyme BCAT1 has emerged as a useful prognostic cancer marker156,157,158. In glioblastoma, BCAA catabolism is upregulated, and BCATs are required for their growth156. The fact that Bcat1-knockout mice are viable159,160, suggests that there may be a good therapeutic window for targeting BCAT1-dependent tumors161. Inhibiting BCAA uptake also seems to be beneficial in certain tumors with high LAT1 expression (e.g., glioma)162. Indeed, in vitro screening has found a highly selective compound for LAT1, JPH203, which shows in vivo efficacy in reducing tumor growth163. When selecting patients for therapies targeting BCAA metabolism, however, tumor heterogeneity should be considered. PDAC shows decreased utilization of circulating BCAAs, and BCAA catabolism is dispensable for PDAC growth161. While BCAA catabolism is upregulated in glioblastoma and NSCLC, leukemia displays increased reverse reaction and generates BCAAs from branched-chain ketoacids (BCKAs)161, which supports the disease progression164. In liver cancer, BCAA supplementation has been shown to improve patient prognosis165,166,167. Understanding how the tissue-of-origin and surrounding environment interact and influence metabolic requirements in cancer will be critical to selecting the appropriate therapeutic regimens for patients.
Concluding remarks
Studies from the past decades have proven the important role of amino acids in cancer metabolism in both a tumorigenic and tumor-suppressive way. Amino acids are involved in pathways that feed cancer cells and provide building blocks for cancer cell growth. The TCA cycle highlights an important mechanistic example of amino acid involvement supporting cancer. Amino acids such as glutamate, BCAAs, and threonine fuel the TCA cycle intermediates, resulting in the release of ATP and providing the required energy for oncogenic activities. Nucleotides, a critical building material for growth in normal and cancer cells, require amino acids as nitrogen and/or one-carbon donors (as a form of formate) for their biosynthesis. Some amino acids can regulate lipid biosynthesis by filling the acetyl-CoA pool or altering lipogenic gene expression. Amino acids also influence ROS homeostasis and epigenetic regulation through methylation and acetylation, all of which can enhance tumor aggressiveness. On the other hand, certain metabolic intermediates from amino acids can contribute to both tumorigenic and anti-tumorigenic activities. Nitric oxide (NO), a product of arginine metabolism (citrulline–NO pathway), can support tumor growth by promoting angiogenesis, but it can also act as a tumor suppressor, at least in part, by upregulating p5378.
Inhibition of amino acid metabolism is an active area of study in cancer metabolism; the field has yielded much success for cancer medications in vitro, but still faces many challenges to realize them in vivo. Some drugs targeting amino acid metabolism have been applied in a clinical setting and highlight the therapeutic potential of this mode of inhibition. However, a few things must be considered when targeting proteins involved in amino acid metabolism. Inhibition of the enzymes and/or transporters, especially related to EAA metabolism, is likely to be systemically toxic168 because of their physiological functions in normal tissues. Inhibition of NEAA metabolism, on the other hand, may not show full therapeutic efficacy in vivo. Although GLS inhibition is effective in hematologic and some solid cancers, PDAC displays adaptive metabolic networks that sustain proliferation during GLS inhibition and rarely shows any beneficial therapeutic effects. Thus, in the case of PDAC, targeting glutamine metabolism while taking into account these adaptive responses may yield clinical benefits for patients. In the same vein, PHGDH inhibition is not always sufficient to inhibit tumor growth169,170. Tumors arising in serine-limited environments can acquire a fitness advantage by upregulating serine synthesis pathway enzymes, implying that inhibition of serine metabolism can only be achieved by both PHGDH inhibition and dietary restriction of serine. Inhibiting amino acid transporters reduces amino acid availability to cells and thus depresses their function. However, targeting transporters might be challenging because of their broad specificity. It will be interesting to see if the newly developed LAT1 selective inhibitor shows in vivo efficacy171.
Because most metabolic inhibitors are unlikely to be effective cancer therapies as single agents, combination therapy is likely the best approach. Indeed, several studies have shown promising results using combination treatment of immunotherapeutic agents with metabolic inhibitors, for example, anti-CTLA-4 inhibitor with xCT inhibition150 and the PD-1 immune checkpoint inhibitor pembrolizumab with IDO1 inhibition40,172. All combinations show robust results in vitro and high potential to develop into treatment in vivo.
By fully understanding the metabolic flexibility and diversity of amino acid usage in cancer cells, it is possible to provide further insight into metabolic dependences and liabilities that can be exploited therapeutically.
References
Warburg, O., Wind, F. & Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927).
Hensley, C. T., Wasti, A. T. & DeBerardinis, R. J. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Investig. 123, 3678–3684 (2013).
Green, C. R. et al. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 12, 15–21 (2016).
Moffatt, B. A. & Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arabidopsis Book 1, e0018 (2002).
Zhang, Y., Morar, M. & Ealick, S. E. Structural biology of the purine biosynthetic pathway. Cell Mol. Life Sci. 65, 3699–3724 (2008).
Locasale, J. W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583 (2013).
Shuvalov, O. et al. One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 8, 23955–23977 (2017).
Lu, S., Chen, G. L., Ren, C., Kwabi-Addo, B. & Epner, D. E. Methionine restriction selectively targets thymidylate synthase in prostate cancer cells. Biochem. Pharm. 66, 791–800 (2003).
Pegg, A. E. Mammalian polyamine metabolism and function. IUBMB Life 61, 880–894 (2009).
Greene, L. I. et al. A role for tryptophan-2,3-dioxygenase in CD8 T-cell suppression and evidence of tryptophan catabolism in breast cancer patient plasma. Mol. Cancer Res. 17, 131–139 (2019).
Fallarino, F. et al. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 4, 1206–1212 (2003).
DiNatale, B. C. et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 115, 89–97 (2010).
Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 (2011).
Nguyen, N. T. et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl Acad. Sci. USA 107, 19961–19966 (2010).
Mezrich, J. D. et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198 (2010).
Vaughn, A. E. & Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 10, 1477–1483 (2008).
Chung, W. J. et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 25, 7101–7110 (2005).
Lo, M., Ling, V., Wang, Y. Z. & Gout, P. W. The xc- cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br. J. Cancer 99, 464–472 (2008).
Fan, J. et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302 (2014).
Ulanovskaya, O. A., Zuhl, A. M. & Cravatt, B. F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 9, 300–306 (2013).
Maddocks, O. D. K. et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376 (2017).
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F. & Kroemer, G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 (2015).
Son, S. M. et al. Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab. 29, 192–201.e197 (2019).
Wu, Z. et al. TPO-induced metabolic reprogramming drives liver metastasis of colorectal cancer CD110+ tumor-initiating cells. Cell Stem Cell 17, 47–59 (2015).
Son, J. et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013).
De Marchi, T. et al. Phosphoserine aminotransferase 1 is associated to poor outcome on tamoxifen therapy in recurrent breast cancer. Sci. Rep. 7, 2099 (2017).
Gupta, N. et al. Upregulation of the amino acid transporter ATB0,+ (SLC6A14) in colorectal cancer and metastasis in humans. Biochimica et. Biophysica Acta 1741, 215–223 (2005).
Gupta, N. et al. Up-regulation of the amino acid transporter ATB(0,+) (SLC6A14) in carcinoma of the cervix. Gynecol. Oncol. 100, 8–13 (2006).
Karunakaran, S. et al. Interaction of tryptophan derivatives with SLC6A14 (ATB0,+) reveals the potential of the transporter as a drug target for cancer chemotherapy. Biochem. J. 414, 343–355 (2008).
Lukey, M. J., Katt, W. P. & Cerione, R. A. Targeting amino acid metabolism for cancer therapy. Drug Disco. Today 22, 796–804 (2017).
Abuchowski, A. et al. Cancer therapy with chemically modified enzymes. I. Antitumor properties of polyethylene glycol-asparaginase conjugates. Cancer Biochem. Biophys. 7, 175–186 (1984).
Wetzler, M. et al. Effective asparagine depletion with pegylated asparaginase results in improved outcomes in adult acute lymphoblastic leukemia: Cancer and Leukemia Group B Study 9511. Blood 109, 4164–4167 (2007).
Leu, S. Y. & Wang, S. R. Clinical significance of arginase in colorectal cancer. Cancer 70, 733–736 (1992).
Savoca, K. V., Davis, F. F., van Es, T., McCoy, J. R. & Palczuk, N. C. Cancer therapy with chemically modified enzymes. II. The therapeutic effectiveness of arginase, and arginase modified by the covalent attachment of polyethylene glycol, on the taper liver tumor and the L5178Y murine leukemia. Cancer Biochem. Biophys. 7, 261–268 (1984).
Schulte, M. L. et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 24, 194–202 (2018).
Timmerman, L. A. et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465 (2013).
Possemato, R. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 (2011).
Pacold, M. E. et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 12, 452–458 (2016).
Cheong, J. E. & Sun, L. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy—challenges and opportunities. Trends Pharm. Sci. 39, 307–325 (2018).
Eggermont, A. M. M., Crittenden, M. & Wargo, J. Combination Immunotherapy Development in Melanoma. Am. Soc. Clin. Oncol. Educ. Book 38, 197–207 (2018).
Keenan, M. M. & Chi, J.-T. Alternative fuels cancer cells. Cancer J. 21, 49–55 (2015).
Curthoys, N. P. & Watford, M. Regulation of glutaminase activity and glutamine metabolism. Annu. Rev. Nutr. 15, 133–159 (1995).
Yang, L., Venneti, S. & Nagrath, D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev. Biomed. Eng. 19, 163–194 (2017).
Choi, B. H. & Coloff, J. L. The diverse functions of non-essential amino acids in cancer. Cancers 11, https://doi.org/10.3390/cancers11050675 (2019).
Le, A. et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 (2012).
Metallo, C. M. et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2011).
Mullen, A. R. et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 (2012).
Zhang, J. et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 56, 205–218 (2014).
Pavlova, N. N. et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 27, 428–438.e425 (2018).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Mayers, J. R. et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 20, 1193–1198 (2014).
Commisso, C. et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013).
Zeitouni, D., Pylayeva-Gupta, Y., Der, C. J. & Bryant, K. L. KRAS mutant pancreatic cancer: no lone path to an effective treatment. Cancers 8, https://doi.org/10.3390/cancers8040045 (2016).
Sheen, J. H., Zoncu, R., Kim, D. & Sabatini, D. M. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell 19, 613–628 (2011).
Wang, J. et al. Dependence of mouse embryonic stem cells on threonine catabolism. Science 325, 435–439 (2009).
Majumdar, R. et al. Glutamate, ornithine, arginine, proline, and polyamine metabolic interactions: the pathway is regulated at the post-transcriptional level. Front. Plant Sci. 7, https://doi.org/10.3389/fpls.2016.00078 (2016).
Delage, B. et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 126, 2762–2772 (2010).
Amelio, I., Cutruzzola, F., Antonov, A., Agostini, M. & Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 39, 191–198 (2014).
Ducker, G. S. & Rabinowitz, J. D. One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017).
Krall, A. S., Xu, S., Graeber, T. G., Braas, D. & Christofk, H. R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7, 11457 (2016).
Cory, J. G. & Cory, A. H. Critical roles of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis: asparaginase treatment in childhood acute lymphoblastic leukemia. Vivo 20, 587–589 (2006).
Wang, Y. et al. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 10, 201 (2019).
Yoneshiro, T. et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 572, 614–619 (2019).
Li, S. et al. Essential amino acid ratios and mTOR affect lipogenic gene networks and miRNA expression in bovine mammary epithelial cells. J. Anim. Sci. Biotechnol. 7, 44 (2016).
Gerner, E. W. & Meyskens, F. L. Jr Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer 4, 781–792 (2004).
Shirahata, A. & Pegg, A. E. Increased content of mRNA for a precursor of S-adenosylmethionine decarboxylase in rat prostate after treatment with 2-difluoromethylornithine. J. Biol. Chem. 261, 13833–13837 (1986).
Holtta, E. Oxidation of spermidine and spermine in rat liver: purification and properties of polyamine oxidase. Biochemistry 16, 91–100 (1977).
Liu, R. et al. Plasma N-acetylputrescine, cadaverine and 1,3-diaminopropane: potential biomarkers of lung cancer used to evaluate the efficacy of anticancer drugs. Oncotarget 8, 88575–88585 (2017).
Xu, H. et al. Polyamine metabolites profiling for characterization of lung and liver cancer using an LC-tandem MS method with multiple statistical data mining strategies: discovering potential cancer biomarkers in human plasma and urine. Molecules 21, https://doi.org/10.3390/molecules21081040 (2016).
van Dam, L., Korolev, N. & Nordenskiold, L. Polyamine-nucleic acid interactions and the effects on structure in oriented DNA fibers. Nucleic Acids Res. 30, 419–428 (2002).
Drew, H. R. & Dickerson, R. E. Structure of a B-DNA dodecamer. III. Geometry of hydration. J. Mol. Biol. 151, 535–556 (1981).
Panagiotidis, C. A., Artandi, S., Calame, K. & Silverstein, S. J. Polyamines alter sequence-specific DNA-protein interactions. Nucleic Acids Res. 23, 1800–1809 (1995).
Nakanishi, S. & Cleveland, J. L. Targeting the polyamine-hypusine circuit for the prevention and treatment of cancer. Amino Acids 48, 2353–2362 (2016).
Wang, Z., Jiang, J., Qin, T., Xiao, Y. & Han, L. EIF5A regulates proliferation and chemoresistance in pancreatic cancer through the sHH signalling pathway. J. Cell Mol. Med. 23, 2678–2688 (2019).
Guan, X. Y. et al. Oncogenic role of eIF-5A2 in the development of ovarian cancer. Cancer Res. 64, 4197–4200 (2004).
Mathews, M. B. & Hershey, J. W. The translation factor eIF5A and human cancer. Biochimica et. Biophysica Acta 1849, 836–844 (2015).
Miyamoto, S., Kashiwagi, K., Ito, K., Watanabe, S. & Igarashi, K. Estimation of polyamine distribution and polyamine stimulation of protein synthesis in Escherichia coli. Arch. Biochem. Biophys. 300, 63–68 (1993).
Choudhari, S. K., Chaudhary, M., Bagde, S., Gadbail, A. R. & Joshi, V. Nitric oxide and cancer: a review. World J. Surg. Oncol. 11, 118 (2013).
Badawy, A. A. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int. J. Tryptophan Res. 10, 1178646917691938 (2017).
Masaki, A. et al. Clinical significance of tryptophan catabolism in Hodgkin lymphoma. Cancer Sci. 109, 74–83 (2018).
Sperner-Unterweger, B. et al. Enhanced tryptophan degradation in patients with ovarian carcinoma correlates with several serum soluble immune activation markers. Immunobiology 216, 296–301 (2011).
Suzuki, Y. et al. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer 67, 361–365 (2010).
Barbul, A. Proline precursors to sustain mammalian collagen synthesis. J. Nutr. 138, 2021s–2024s (2008).
Tang, L. et al. Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma. Clin. Cancer Res. 24, 474–485 (2018).
Arrick, B. A. & Nathan, C. F. Glutathione metabolism as a determinant of therapeutic efficacy: a review. Cancer Res. 44, 4224–4232 (1984).
Doxsee, D. W. et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate 67, 162–171 (2007).
Mandal, P. K. et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 285, 22244–22253 (2010).
Lu, S. C. Regulation of glutathione synthesis. Mol. Asp. Med. 30, 42–59 (2009).
Ye, J. et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4, 1406–1417 (2014).
Zhou, X. et al. Serine alleviates oxidative stress via supporting glutathione synthesis and methionine cycle in mice. Mol. Nutr. Food Res. 61, https://doi.org/10.1002/mnfr.201700262 (2017).
Maddocks, O. D. et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546 (2013).
DeNicola, G. M. et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 47, 1475–1481 (2015).
Nikkanen, J. et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 23, 635–648 (2016).
Egger, G., Liang, G., Aparicio, A. & Jones, P. A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463 (2004).
Feinberg, A. P. & Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 4, 143–153 (2004).
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487 (2016).
Edwards, J. R., Yarychkivska, O., Boulard, M. & Bestor, T. H. DNA methylation DNA methyltransferases. Epigenet. Chromatin 10, 23 (2017).
Kulis, M. & Esteller, M. DNA methylation and cancer. Adv. Genet. 70, 27–56 (2010).
Herman, J. G. Hypermethylation of tumor suppressor genes in cancer. Semin. Cancer Biol. 9, 359–367 (1999).
Esteller, M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 21, 5427–5440 (2002).
Hyun, K., Jeon, J., Park, K. & Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Amp; Mol. Med. 49, e324 (2017).
Ulrey, C. L., Liu, L., Andrews, L. G. & Tollefsbol, T. O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 14, R139–R147 (2005).
Williams, K. T. & Schalinske, K. L. New Insights into the regulation of methyl group and homocysteine metabolism. J. Nutr. 137, 311–314 (2007).
Borrego, S. L. et al. Metabolic changes associated with methionine stress sensitivity in MDA-MB-468 breast cancer cells. Cancer Metab. 4, 9 (2016).
Booher, K., Lin, D. W., Borrego, S. L. & Kaiser, P. Downregulation of Cdc6 and pre-replication complexes in response to methionine stress in breast cancer cells. Cell Cycle 11, 4414–4423 (2012).
Maddocks, O. D., Labuschagne, C. F., Adams, P. D. & Vousden, K. H. Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol. Cell 61, 210–221 (2016).
Shyh-Chang, N. et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226 (2013).
Yang, X. J. & Seto, E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310 (2007).
Lee, J. V. et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319 (2014).
Vermeulen, L. et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468–476 (2010).
Wise, D. R. et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl Acad. Sci. USA 105, 18782–18787 (2008).
Gao, P. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009).
Kang, Y. P. et al. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. eLife 8, https://doi.org/10.7554/eLife.45572 (2019).
Dunphy, M. P. S. et al. In vivo PET assay of tumor glutamine flux and metabolism: in-human trial of (18)F-(2S,4R)-4-fluoroglutamine. Radiology 287, 667–675 (2018).
Fuchs, B. C. & Bode, B. P. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin. Cancer Biol. 15, 254–266 (2005).
Kanai, Y. et al. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 273, 23629–23632 (1998).
Mastroberardino, L. et al. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature 395, 288–291 (1998).
Meier, C., Ristic, Z., Klauser, S. & Verrey, F. Activation of system L heterodimeric amino acid exchangers by intracellular substrates. EMBO J. 21, 580–589 (2002).
Yanagida, O. et al. Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines. Biochimica et. Biophysica Acta 1514, 291–302 (2001).
Elorza, A. et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 48, 681–691 (2012).
Miko, E. et al. miR-126 inhibits proliferation of small cell lung cancer cells by targeting SLC7A5. FEBS Lett. 585, 1191–1196 (2011).
Jewell, J. L. et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198 (2015).
Kimball, S. R., Shantz, L. M., Horetsky, R. L. & Jefferson, L. S. Leucine regulates translation of specific mRNAs in L6 myoblasts through mTOR-mediated changes in availability of eIF4E and phosphorylation of ribosomal protein S6. J. Biol. Chem. 274, 11647–11652 (1999).
Oda, K. et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 101, 173–179 (2010).
Yun, D. W. et al. JPH203, an L-type amino acid transporter 1-selective compound, induces apoptosis of YD-38 human oral cancer cells. J. Pharm. Sci. 124, 208–217 (2014).
Kanai, Y. et al. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol. Asp. Med. 34, 108–120 (2013).
Nicklin, P. et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534 (2009).
Wang, Q. et al. Androgen receptor and nutrient signaling pathways coordinate the demand for increased amino acid transport during prostate cancer progression. Cancer Res. 71, 7525–7536 (2011).
Reynolds, M. R. et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 33, 556–566 (2014).
Sontheimer, H. Malignant gliomas: perverting glutamate and ion homeostasis for selective advantage. Trends Neurosci. 26, 543–549 (2003).
Lyons, S. A., Chung, W. J., Weaver, A. K., Ogunrinu, T. & Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 67, 9463–9471 (2007).
Briggs, K. J. et al. Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell 166, 126–139 (2016).
Nabeyama, A. et al. xCT deficiency accelerates chemically induced tumorigenesis. Proc. Natl Acad. Sci. USA 107, 6436–6441 (2010).
Abdelmagid, S. A., Rickard, J. A., McDonald, W. J., Thomas, L. N. & Too, C. K. CAT-1-mediated arginine uptake and regulation of nitric oxide synthases for the survival of human breast cancer cell lines. J. Cell Biochem. 112, 1084–1092 (2011).
Sullivan, L. B. et al. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563 (2015).
Birsoy, K. et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015).
Beuster, G. et al. Inhibition of alanine aminotransferase in silico and in vivo promotes mitochondrial metabolism to impair malignant growth. J. Biol. Chem. 286, 22323–22330 (2011).
Wang, J. B. et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 (2010).
Gross, M. I. et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13, 890–901 (2014).
Cui, H. et al. Enhanced expression of asparagine synthetase under glucose-deprived conditions protects pancreatic cancer cells from apoptosis induced by glucose deprivation and cisplatin. Cancer Res. 67, 3345–3355 (2007).
Wyant, G. A. et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–654.e612 (2017).
Ananieva, E. Targeting amino acid metabolism in cancer growth and anti-tumor immune response. World J. Biol. Chem. 6, 281–289 (2015).
Shroff, E. H. et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl Acad. Sci. USA 112, 6539–6544 (2015).
Xiang, Y. et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 125, 2293–2306 (2015).
Yang, C. et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 56, 414–424 (2014).
Jin, L. et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 27, 257–270 (2015).
Jin, L. et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol. Cell 69, 87–99.e87 (2018).
Gout, P. W., Buckley, A. R., Simms, C. R. & Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640 (2001).
Cobler, L., Zhang, H., Suri, P., Park, C. & Timmerman, L. A. xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction. Oncotarget 9, 32280–32297 (2018).
Arensman, M. D. et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc. Natl Acad. Sci. USA 116, 9533–9542 (2019).
Wang, Q. et al. Rational design of selective allosteric inhibitors of PHGDH and serine synthesis with anti-tumor activity. Cell Chem. Biol. 24, 55–65 (2017).
Mullarky, E. et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl Acad. Sci. USA 113, 1778–1783 (2016).
Rohde, J. M. et al. Discovery and optimization of piperazine-1-thiourea-based human phosphoglycerate dehydrogenase inhibitors. Bioorg. Med. Chem. 26, 1727–1739 (2018).
Sullivan, M. R. et al. Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab. 29, 1410–1421.e1414 (2019).
Anglin, J. et al. Discovery and optimization of aspartate aminotransferase 1 inhibitors to target redox balance in pancreatic ductal adenocarcinoma. Bioorg. Med. Chem. Lett. 28, 2675–2678 (2018).
Tonjes, M. et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 19, 901–908 (2013).
Panosyan, E. H., Lin, H. J., Koster, J. & Lasky, J. L. 3rd In search of druggable targets for GBM amino acid metabolism. BMC Cancer 17, 162 (2017).
Zheng, Y. H. et al. BCAT1, a key prognostic predictor of hepatocellular carcinoma, promotes cell proliferation and induces chemoresistance to cisplatin. Liver Int. 36, 1836–1847 (2016).
Ananieva, E. A., Powell, J. D. & Hutson, S. M. Leucine metabolism in T cell activation: mTOR signaling and beyond. Adv. Nutr. 7, 798s–805s (2016).
Ananieva, E. A., Patel, C. H., Drake, C. H., Powell, J. D. & Hutson, S. M. Cytosolic branched chain aminotransferase (BCATc) regulates mTORC1 signaling and glycolytic metabolism in CD4+ T cells. J. Biol. Chem. 289, 18793–18804 (2014).
Mayers, J. R. et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165 (2016).
Kim, D. K. et al. System L-amino acid transporters are differently expressed in rat astrocyte and C6 glioma cells. Neurosci. Res. 50, 437–446 (2004).
Wempe, M. F. et al. Metabolism and pharmacokinetic studies of JPH203, an L-amino acid transporter 1 (LAT1) selective compound. Drug Metab. Pharmacokinet. 27, 155–161 (2012).
Hattori, A. et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 545, 500–504 (2017).
Nojiri, S., Fujiwara, K., Shinkai, N., Iio, E. & Joh, T. Effects of branched-chain amino acid supplementation after radiofrequency ablation for hepatocellular carcinoma: a randomized trial. Nutrition 33, 20–27 (2017).
Shiozawa, S. et al. Impact of branched-chain amino acid-enriched nutrient on liver cirrhosis with hepatocellular carcinoma undergoing transcatheter arterial chemoembolization in barcelona clinic liver cancer stage B: a prospective study. J. Nippon Med. Sch. 83, 248–256 (2016).
Park, J. G. et al. Effects of branched-chain amino acids (BCAAs) on the progression of advanced liver disease: A Korean nationwide, multicenter, retrospective, observational, cohort study. Medicine 96, e6580 (2017).
Erez, A. & DeBerardinis, R. J. Metabolic dysregulation in monogenic disorders and cancer - finding method in madness. Nat. Rev. Cancer 15, 440–448 (2015).
Chen, J. et al. Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget 4, 2502–2511 (2013).
Nilsson, L. M. et al. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 8, e1002573 (2012).
Singh, N. et al. Discovery of potent inhibitors for the large neutral amino acid transporter 1 (LAT1) by structure-based methods. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20010027 (2018).
Labadie, B. W., Bao, R. & Luke, J. J. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin. Cancer Res. 25, 1462–1471 (2019).
Acknowledgements
We are grateful to Paul M. Wehn for comments and editing of the review and to Nefertiti Muhammad for minor edits. J.K. is supported by the National Cancer Institute (1K22CA226676–01A1), the American Lung Association (LCD-614827) and the V Foundation (V2019–022).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lieu, E.L., Nguyen, T., Rhyne, S. et al. Amino acids in cancer. Exp Mol Med 52, 15–30 (2020). https://doi.org/10.1038/s12276-020-0375-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0375-3
This article is cited by
-
Unlocking the power of precision medicine: exploring the role of biomarkers in cancer management
Future Journal of Pharmaceutical Sciences (2024)
-
Cancer metabolism and carcinogenesis
Experimental Hematology & Oncology (2024)
-
Selective enhanced cytotoxicity of amino acid deprivation for cancer therapy using thermozyme functionalized nanocatalyst
Journal of Nanobiotechnology (2024)
-
Cancer immunometabolism: advent, challenges, and perspective
Molecular Cancer (2024)
-
Real-time detection of 20 amino acids and discrimination of pathologically relevant peptides with functionalized nanopore
Nature Methods (2024)
