
Abstract
Numerous lines of evidence indicate an association between sustained inflammation and Parkinson’s disease, but whether increased inflammation is a cause or consequence of Parkinson’s disease remains highly contested. Extensive efforts have been made to characterize microglial function in Parkinson’s disease, but the role of peripheral immune cells is less understood. Natural killer cells are innate effector lymphocytes that primarily target and kill malignant cells. Recent scientific discoveries have unveiled numerous novel functions of natural killer cells, such as resolving inflammation, forming immunological memory, and modulating antigen-presenting cell function. Furthermore, natural killer cells are capable of homing to the central nervous system in neurological disorders that exhibit exacerbated inflammation and inhibit hyperactivated microglia. Recently, a study demonstrated that natural killer cells scavenge alpha-synuclein aggregates, the primary component of Lewy bodies, and systemic depletion of natural killer cells results in exacerbated neuropathology in a mouse model of alpha-synucleinopathy, making them a highly relevant cell type in Parkinson’s disease. However, the exact role of natural killer cells in Parkinson’s disease remains elusive. In this review, we introduce the systemic inflammatory process seen in Parkinson’s disease, with a particular focus on the direct and indirect modulatory capacity of natural killer cells in the context of Parkinson’s disease.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease that affects over one million people in the United States, with predictable healthcare-associated costs of nearly $52 billion per year1. PD is pathologically characterized by the misfolding and aggregation of the alpha-synuclein (α-syn) protein into insoluble inclusions known as Lewy bodies (LBs) and Lewy neurites2. Monomeric α-syn protein is highly expressed in presynaptic terminals3 and the nucleus4 of cells. However, α-syn can acquire toxic properties through the pathogenic process of misfolding into α-syn fibrils that comprise the major component of LBs. LBs are present in PD, dementia with Lewy bodies (DLB), and Alzheimer’s disease (AD) brains; however, each disease shows distinct regional specificity, with PD patients displaying LBs most prominently in the substantia nigra (SN)5. Furthermore, in human sporadic PD cases and murine models of PD, LB formation is present in numerous cell types other than dopaminergic (DA) neurons such as astrocytes6,7. The exact cause of α-syn aggregation is debated, but the importance of this protein in PD is highlighted through genetic studies revealing missense, duplication, or triplication mutations in the SNCA gene encoding α-syn leading to early-onset PD with prominent dementia8. Importantly, human patient samples revealed aberrant α-syn distribution peripherally within blood plasma9, interstitial fluid10,11, and the enteric nervous system (ENS)12, implicating prion-like replication and spreading of α-syn. The accumulation of α-syn aggregates can promote sustained activation of pattern-recognition receptors (PRRs), leading to the production of proinflammatory cytokines in microglia13,14. These inflammatory mediators from microglia and chemoattractant molecules from damaged neurons possibly play a role in infiltrating peripheral cells into the central nervous system (CNS)15. Peripheral inflammation has been shown to exacerbate DA neurodegeneration in numerous animal models of PD16,17. Extracellular α-syn aggregates may elicit a self-amplifying cycle of immune responses in the CNS14,18 and periphery19 through an overproduction of inflammatory mediators, thus providing the tertiary hit required for PD-associated dysfunction to spread to neighboring neurons in the CNS20,21 and the periphery22. Therefore, immunomodulatory-based approaches aimed at halting the propagation and burden of extracellular α-syn, and in turn diminishing the inflammatory response, are currently being tested as a therapeutic for PD and related synucleinopathies (reviewed in23). Recently, it was discovered that natural killer (NK) cells efficiently internalize and degrade α-syn aggregates via the endosomal/lysosomal pathway, a novel and highly relevant function of NK cells in synucleinopathies24. The number of circulating NK cells in PD patients is increased compared to non-PD controls25. Furthermore, NK cells are present in the human postmortem PD SN24 and PD mouse brain26, and depletion of NK cells in a mouse model of synucleinopathies is associated with increased neuropathology24. In this review, we review α-syn-associated immune responses, the potential role of NK cells, and the mechanisms by which NK cells exert their effects on PD pathogenesis.
Implications of α-syn-associated immune responses in PD
Although the etiology of PD is considered multifactorial in nature, genetic studies have identified several loci involved in inflammation, including the human leukocyte antigen-DR isotype (HLA-DR), that lead to an increased risk of PD27. It is hypothesized that misfolded α-syn may potentiate the observed increase in inflammation seen in PD, as excessive aggregated α-syn can hyperactivate microglia18 and promote neurotoxic effects by enhancing the release of tumor necrosis factor-alpha (TNF-α), interleukin (IL) 1-beta (IL-1β), IL-6, nitric oxide (NO), inducible nitric oxide synthase (iNOS), and reactive oxygen species (ROS). Microglia phagocytose extracellular α-syn28 via various receptors, including Toll-like receptors13,14. As microglia clear α-syn aggregates, they become activated independent of internalization and phagocytosis29,30. The distinct mechanisms of α-syn-induced activation seem to be α-syn species-specific28. For example, monomeric and mutant α-syn can bind to CD36 on microglia, leading to the production of TNF-α, oxidative stress, and Erk phosphorylation31,32, while oligomeric α-syn interacts with CD11b to induce nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and ROS production21. The α-syn-induced cascade of proinflammatory cytokines and oxygenating mediators from microglia is sufficient to induce neurodegeneration33.
Neuronal expression of α-syn also induces reactive astrogliosis26,34,35,36. Furthermore, α-syn-containing inclusions are present astrocytes in sporadic PD cases7 and transgenic mouse models of synucleinopathy6. α-Syn is efficiently transferred between astrocytes37,38 and from neurons to astrocytes37 in vitro and in vivo. Astrocytic transfer of α-syn is conducted via direct contact and tunneling nanotubes (TNTs)38. α-Syn is localized to the lysosomal compartment of astrocytes and neurons; however, a progressive increase in cleaved and full-length α-syn was displayed in neurons over time, while astrocytes displayed efficient degradation of α-syn37. However, failure of the lysosomal digestion of excess α-syn oligomers in astrocytes results in α-syn deposits in the trans-Golgi network followed by endoplasmic reticulum swelling and mitochondrial disturbances38. Moreover, exogenous α-syn induced increased mRNA expression of proinflammatory cytokines (IL-1β, TNF-α), iNOS, and cyclooxygenase-2 (COX-2) from primary mouse astrocytes in a TLR4-dependent manner39.
Peripheral inflammation has been deemed a significant contributing factor in PD pathogenesis. Serum levels of IL-1β, IL-2, IL-10, interferon-gamma (IFN-γ), and TNF-α have been correlated with the severity of PD symptoms40,41 and rate of disease progression42. Serum autoimmune antibodies against α-syn are significantly elevated in PD patients43. Autoantibody titers, α-syn monomers, and oligomers plus fibrils in 72%, 56%, and 17% of PD patients are elevated, respectively, within a 5-year disease duration44, which implicates systemic adaptive immune responses against different α-syn species. PD patients also have a T helper cell 1 (Th1) bias in peripheral blood with naïve CD4+ T cells from patients preferentially differentiating to a Th1 proinflammatory lineage and showing augmented production of IFN-γ and TNF-α45. Moreover, peripheral blood mononuclear cell (PBMC)-derived CD4+ T cells from PD patients specifically react to antigenic major histocompatibility class two (MHC-II) epitopes derived from α-syn46, revealing T cell specificity to the α-syn antigen. Substantial increases in inflammatory cytokines (IL-1β, TNF-α, IL-6)47 and intestinal permeability48,49,50 are correlated with the presence of α-syn aggregates within the gut51. Increased intestinal permeability is thought to result from reductions in barrier-promoting proteins and disruptions of tight-junction networks49, a phenotype consistent with low-grade inflammation52,53.
Properties of NK cells
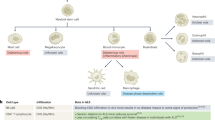
NK cells are bone marrow (BM)-derived hematopoietic cells54 that represent 10–15% of total circulating lymphocytes55 and are widely located throughout lymphoid and nonlymphoid tissues56,57. NK cells primarily target and destroy malignant cells through germline-encoded activating and inhibitory receptors58, perforin and granzyme production59,60,61 following immune synapse formation with a target cell, and death receptor pathways Fas Ligand and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)62,63. Immunological synapse formation leads to polarization of NK cells towards their target synapse, followed by lysosomal docking at the plasma membrane and finally the fusion and release of contents61. NK cell function extends beyond malignant cell lysis with various roles, such as antimicrobial defense64,65, formation of immunological memory66, resolution of inflammation67,68,69,70, and endocytosis of extracellular proteins24,71.
NK cells are capable of recognizing self- and nonself-molecules through the expression of a variety of activating and inhibitory receptors that regulate NK cell activity (summarized in Fig. 1). NK cell activating and inhibitory receptors contain immunoreceptor tyrosine activating motifs (ITAMs) and immunoreceptor tyrosine-based inhibitory motifs (ITIMs), respectively58,72,73. NK cells express killer immunoglobulin-like receptors (KIRs) in humans for molecules known as human leukocyte antigen DR isotype (HLA-DR) and Ly49 receptors in rodents for major histocompatibility complex class I (MHC-I)72,74,75. All nucleated cells express HLA-DR/MHC-I molecules, and therefore, the binding of these NK cell receptors to these molecules is the prominent mechanism for ‘tolerance’ of self-cells76,77,78. Other pivotal inhibitory receptors found on NK cells are CD85 for HLA-A, CD94 for HLA-E in humans and NKG2A for Qa-1b and CD244 for CD48 in mice79 (Fig. 1). Cells undergoing malignant transformation often downregulate the expression of MHC class I molecules62, which permit the activation of NK cells. Activating receptors found on the surface of NK cells in humans include CD16 for immune complexes, CD122 for IL-2 and IL-15, CD266 (DNAM-1) for Nectin-2, and KIR2S for HLA-1 (Fig. 1). Activating receptors on NK cells found in mice include CD16 for immune complexes, NKG2C for Qa-1b, NKG2D for MHC class I polypeptide-related sequence A (MICA), and CD28 for CD80 (Fig. 1) (summarized in79).

NK cell function is mediated by the integration of activating and inhibitory signals. The combinatorial nature of signal integration involves ITAM (immunoreceptor tyrosine-based activation motif)-bearing molecules, ITIMs (immunoreceptor tyrosine-based inhibitory motifs), and other stimulatory receptors for cytokines and adhesion molecules. *Activated by multiple ligands (viral hemagglutinins, unknown tumor ligands).
In humans, NK cells are primarily divided into CD3- CD16+ CD56dim and CD3- CD16+ CD56bright subsets56. Approximately 90% of peripheral blood and splenic NK cells are CD3-CD16+ CD56dim, which have cytotoxic effector functions, including the production of perforin and IFN-γ, upon interaction with tumor cells80. CD3- CD16+ CD56bright NK cell subsets play an immunoregulatory role through cytokine production that can directly and indirectly modulate innate and adaptive immune responses81. In the CNS, the majority of NK cells in cerebrospinal fluid (CSF) are CD56bright NK cells82. Both CD3- CD16+ CD56dim and CD3- CD16+ CD56bright NK cell subsets can take on highly specific effector functions through dynamic and combinatorial alterations in receptor expression under pathophysiological conditions such as viral infections and autoimmune diseases83,84. In mice, NK cells are generally defined as CD3-NK1.1+ or CD3-NKCR1/NKp46+ cells that typically express integrin alpha 2/CD49b, integrin alpha M/CD11b, CD27, T-bet, and Eomes and lack expression of CD127/IL-7R alpha79,85. Three major subsets of mouse NK cells have been characterized based on the differential expression of integrin alpha M/CD11b and CD27, including CD11bdim CD27bright NK cells, CD11bbrightCD27dim NK cells, and CD11bbrightCD27bright NK cells. CD27dim NK cells have lower cytotoxic potential and produce lower levels of cytokines than CD27bright NK cells86. However, strain differences have been observed, as C57BL/6J mouse NK cells are identified by the presence of NK1.1 (NKR-P1C) and NCR1 (NKp46/CD335), while CD49b (DX5, Integrin VLA-2α) is a common NK cell marker in other mouse backgrounds86.
The presence of NK cells in the CNS
The existence of brain residential NK cells has been strongly supported by transcriptomic analysis of brain myeloid cells, revealing that NK cells are identified as small but distinct and biologically meaningful clusters of cells in the brain parenchyma87,88. Peripheral NK cells are recruited to the CNS during high levels of inflammation by chemokines such as CX3C ligand 1 (CX3CL1) produced by neurons89 or C-C motif chemokine ligand 2 (CCL2) and CXC ligand 10 (CXCL10) produced by microglia, astrocytes, and other inflammatory cells57. NK cells found in the CNS display various functions in neurological disorders. NK cells migrate to the mouse brain in a CX3CL1-dependent manner in the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS)69 and attenuate inflammation in the early stages of EAE, suggesting anti-inflammatory effects57, while others reported neurotoxic effects in EAE90. NK cells can inhibit microglial transactivation of T helper 17 cell (Th17) signature transcription factors on pathogenic T cells and in turn decrease neuroinflammation69. Furthermore, NK cells display cytotoxicity against astrocytes91 during infection through astrocytic expression of the inducible NKp44 ligand92. NK cells were found in the motor cortex and spinal cord in a CCL2-dependent manner and modulate the progression of motor neuron degeneration in amyotrophic lateral sclerosis (ALS)93. In neurological autoimmune conditions, NK cells have been shown to reciprocally interact with neural stem cells (NSCs) to regulate neural repair during the chronic stage of disease progression94, demonstrating the capacity to engage with neural cells.
Implications of NK cells in PD pathologies
In PD patients, NK cell numbers are increased in the blood compared to controls25,95,96. Mihara et al reported that NKG2A expression on circulating NK cells is lower than that of non-PD controls with no changes in NKG2D expression95. Another study reported that the percentages of NKG2D-positive NK cells were higher in PD patients25. The functional activity of NK cells against K562 leukemia cells is positively correlated with disease duration in PD patients, suggesting that NK activity increases as the disease advances95. However, these studies must be further evaluated, as K562 cells express high levels of ligands for NKG2D but very low levels of ligands for NKG2A (HLA-E).97. Although these findings are purely correlational, they do suggest a consistent deviation in NK cell number and receptor expression patterns during pathogenesis96,98,99. In addition, analysis of subsets of NK cells and their function associated with PD pathologies needs to be conducted to further understand their function in PD. The presence of NK cells in human brains with synucleinopathies was recently reported24. Immunohistochemical analysis of postmortem brain tissue containing abnormal α-syn aggregates, including PD, DLB, Parkinson’s disease dementia (PDD), and progressive supranuclear palsy (PSS) cases, demonstrated that NK cells were found in close proximity to α-syn aggregates24. Moreover, NK cells are also present in the brain parenchyma of mouse models of PD24,26. While the presence of NK cells in the CNS parenchyma is undoubtedly significant, the role NK cells play in PD pathogenesis has remained elusive until a recent study suggested a neuroprotective role of NK cells in PD24. The study demonstrated that NK cells reduce synuclein burden in vitro, and systemic depletion of NK cells in a preclinical mouse model of PD results in increased pathological α-syn burden in numerous brain regions, including the striatum, SNpc, and brainstem24. The study also demonstrated that increased α-syn pathology is positively correlated with gross motor deficits and mortality24. Furthermore, NK cell-deficient animals displayed increased inflammation in the CNS, as shown through glial fibrillary acidic protein (GFAP) and ionized calcium binding adaptor molecule-1 (Iba-1) immunoreactivity24. While this study provides a critical foundation for the argument that NK cells play a protective role in PD and potentially other synucleinopathies, the precise mechanism by which NK cells exert protection requires further investigation. The possible mechanisms by which NK cells exert effects during PD pathogenesis (α-syn deposition, augmented inflammation, DA neurodegeneration, and motor dysfunction) are as follows (summarized in Fig. 2).

The proposed mechanisms of NK cells in the CNS are as follows: 1) NK cells efficiently scavenge α-syn aggregates via receptor-mediated endocytosis; 2) NK cells modulate CNS inflammation by directly interacting with neurons or glia. NK cells have been involved in the cytotoxicity to immature or damaged neurons. Neurons and microglia may change the surface expression of ligands for the inhibitory receptor of NK cells or ligands for activating receptors that affect the activation status of NK cells; 3) Lastly, NK cells may attenuate neuroinflammation by modulating CNS-infiltrated leukocytes.
NK cells as direct scavengers of α-syn species
α-Syn deposition has been shown to promote the release of senescence-associated secretory (SASP) profiles from cells100, which converts senescent cells into continuous sources of proinflammatory mediators, reactive oxygen species and metalloproteinases101,102. NK cells can recognize and clear senescent cells through a mechanism involving perforin and granzyme-containing granule exocytosis and production of IFN-γ following senescent cell interaction103 Therefore, NK cells may target senescent cells burdened with α-syn for lysis. Furthermore, human NK cells efficiently scavenge various forms of α-syn species24, making it plausible that NK cells can reduce the α-syn burden seen in PD patients. Although NK cells are not classically defined as phagocytic cells, they possess efficient endocytosis machinery, as receptor trafficking through endocytic pathways has been well documented in NK cells71. α-Syn aggregates are degraded within NK cells, and cytoplasmic α-syn is colocalized with endosomal and lysosomal protein markers24. Importantly, α-syn aggregates attenuate NK cell release of IFN-γ24. This finding is highly significant, as α-syn augments the release of proinflammatory cytokines and oxidative species in microglia during the phagocytosis of α-syn aggregates33. It is also conceivable that NK cells will preferentially home to areas of increased synuclein burden.
Interaction of NK cells and CNS-resident cells in PD
NK cells have previously been shown to be selectively involved in neurotoxicity in immature or injured neurons via an NKG2D-retinoic acid early inducible gene 1 (RAE-1)-mediated mechanism following peripheral nerve injury, leading to the resolution of painful neuropathies104,105. NKG2D ligands comprise several MHC class I-like molecules106. From analysis of mRNA-seq data by Dumitriu et al.107, the levels of stress-inducible glycoproteins MICA and MICB were significantly higher in PD brains than in healthy brains, which could induce interaction with NK cells within the CNS. Thus, NK cells may selectively target neurons with α-syn inclusions via NKG2D ligands. Another potential mechanism by which NK cells exert protection is through a microglia-NK cell interaction. This interaction can induce decreased expression of the MHC class I molecule Qa1 on activated microglia, which in turn triggers NK cell-mediated cytotoxicity towards hyperactive microglia69. As microglia are known to become aberrantly activated in the presence of sustained α-syn burden, targeting microglial activation states by suppressing their deleterious proinflammatory neurotoxicity may be a valid therapeutic approach for PD treatment23. Therefore, NK cells could exert protection by mitigating microglial toxic effects within the CNS.
Interaction of NK cells and immune cells and their implications in PD
Systemic NK cell depletion in vivo in EAE led to disease enhancement associated with increased autoreactive T cell proliferation and a proinflammatory response108. NK cells display enhanced cytotoxicity towards the myelin proteolipid protein (PLP) autoreactive T cell line in comparison to naïve splenic T cells in vitro90, indicating that NK cells exert protection in EAE by killing myelin antigen-specific T cells. SN DA neurons display MHC-I in response to microglia stimulated with α-syn and take on antigen-presenting cell (APC) functions such as loading and displaying of antigen46. Neuronal antigen-loaded MHC-I was competent to trigger cytotoxic T cell (CTL)-mediated neuronal death in vitro46. It seems plausible that T cells are autoreactive to α-syn, as peptides derived from α-syn elicit in vitro responses from CD8+ and CD4+ T cells in PD patients but not healthy controls109. Therefore, NK cells could exert effects through direct and indirect interactions with T cells. NK cells can negatively regulate the T cell response directly by their ability to recognize and kill activated T cells110,111,112. In vitro experiments show that activated CD4+ and CD8+ T cells are susceptible to NK cell-mediated lysis through perforin-dependent mechanisms110 and increased expression of NKG2D ligands112. Of high relevance, in vivo activated murine T cells become targets for NK cells under LPS treatment, which mimics an inflammatory condition110.
NK cells are a major source of IFN-γ and their potential role in PD
Many studies have demonstrated a relationship between PD and IFN-γ levels, with recent evidence showing elevated blood plasma levels of IFN-γ in PD patients113. NK cells are major producers of IFN-γ114, and NK cell depletion in a murine model of synucleinopathy led to a six-fold reduction in IFN-γ serum concentration24. IFN-γ directly or indirectly affects APCs by increasing the expression of subunits of MHC class I and II molecules, transporters associated with antigen processing (TAP1/2), invariant chain, and the expression and activity of the proteasome115. IFN-γ contributes to macrophage activation by increasing phagocytosis and priming the production of proinflammatory mediators115. Furthermore, Th1 development is heavily influenced by IFN-γ produced by NK cells115.
In the CNS, IFN-γ ameliorated autoimmune disease symptoms with EAE116,117, implicating a neuroprotective effect. Furthermore, a study showed the potential role of α-syn in modulating the expression of IFN genes, as IFN genes were downregulated in the CNS following brain infection in α-syn knockout mice118. Whether IFN-γ potentiates or attenuates the phagocytic activity of microglial α-syn aggregates needs to be investigated. α-Syn-containing inclusions are also present in the lysosomal compartments of astrocytes in sporadic PD cases7 and transgenic mouse models of α-synucleinopathy6. Microarray analysis of IFN-γ response genes in astrocytes showed increased expression of genes associated with protein degradation (Ubiquitin D) and proteasome degradation (proteasome subunit β9)119, implicating that IFN-γ may promote the lysosomal digestion of excess α-syn and reduce organelle dysfunction. As the amount of IFN-γ produced by NK cells decreases in older adults120,121, it could have deleterious effects on the ability of APCs to manage pathogens such as α-syn aggregates centrally and peripherally.
Role of NK cells in the gut and their potential role in PD pathologies
Peripherally, gut-resident NK cells are found as intraepithelial lymphocytes (IEPs) and in the lamina propria122. The cytokines produced by NK cells, including IFN-γ and TNF, have a primary role in gut homeostasis and damage (reviewed in ref. 122). In addition, NK cells play key roles against gut infections of Salmonella, Listeria, Citrobacter rodentium, and other enteric pathogen injections in mice123,124,125,126,127. NK cell activity in the gut with autoimmune inflammatory bowel diseases (IBDs), such as Crohn’s disease and ulcerative colitis, is substantially dampened (reviewed in ref. 122). Macrophages within the myenteric plexus are present at sites of aggregated α-syn deposition and are able to phagocytose deposits, as evidenced by inclusions present in the cytoplasm of macrophages51. As α-syn inclusions are found within the ENS in PD, they may mobilize resident macrophage populations to clear this protein and induce an inflammatory milieu within the gut. NK cell-macrophage interactions have been characterized via LPS stimulation of macrophages, inducing NKG2D upregulation and subsequent lysis of activated macrophages by NK cells128. Therefore, gut-resident NK cells, which come into contact with a multitude of antigens and adaptive immune cells (macrophages, dendritic cells, T cells)122, may have the capacity to mitigate the peripheral macrophage proinflammatory response to the α-syn antigen.
NK cell immunosenescence and its potential role in PD pathologies
NK cell immunosenescence can impair crosstalk between the innate and adaptive immune systems129, which may have substantial implications for the aging population. Human studies have shown significant increases in the frequency and total number of circulating NK cell populations with age129,130,131, while there are significant reductions in both the numbers and percentage of splenic NK cells in aged mice (unpublished observation). Since human studies are restricted to circulating mononuclear cells (PBMCs), the profiles of lymphocytes with aging in local tissues may explain the observed differences between species. There have been conflicting reports regarding the functionality of aging NK cells, as human NK cells are hyporesponsive or hyperresponsive to IL-2 compared to young NK cells as measured by IFN-γ production120,121,132. As IFN-γ function is so diverse, the potential implications for dysregulated release of this proinflammatory mediator with age are immense.
Conclusion and future directions
The mechanisms by which NK cells can exert protection in synucleinopathies are abundant. NK cells finely orchestrate immune responses and modulate inflammation, making them a highly relevant cell type to study in inflammatory conditions. NK cells may play a key role in alleviating a sustained, unmitigated immune response to α-syn systemically. Furthermore, NK cells can internalize and degrade α-syn aggregates and subsequently reduce the α-syn burden. However, additional studies must be conducted to elucidate the precise mechanisms by which NK cells exert effects in PD. In addition, NK cell immunosenescence may result in the accumulation of α-syn aggregates and the deceleration of the resolution of immune responses. As a future direction, age-related alterations in NK cell phenotypes would provide insight into the relationship between NK cell immunosenescence and age-related neurological disorders. In addition, PD has an increased prevalence in males compared to females; therefore, interrogating the effects of sex on NK cell phenotype would further expand our knowledge on how NK cells contribute to age-related neurodegenerative diseases that display sexual dimorphic patterns.
References
Marras, C. et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis. 4, 21 (2018).
Lees, A. J., Hardy, J. & Revesz, T. Parkinson’s disease. Lancet 373, 2055–2066 (2009).
Burre, J. The synaptic function of alpha-synuclein. J. Parkinsons Dis. 5, 699–713 (2015).
Burre, J. et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 (2010).
Kim, W. S., Kagedal, K. & Halliday, G. M. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res Ther. 6, 73 (2014).
Lau, A. et al. alpha-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. https://doi.org/10.1038/s41593-019-0541-x (2019).
Wakabayashi, K., Hayashi, S., Yoshimoto, M., Kudo, H. & Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 99, 14–20 (2000).
Poewe, W. et al. Parkinson disease. Nat. Rev. Dis. Prim. 3, 17013 (2017).
Foulds, P. G. et al. A longitudinal study on alpha-synuclein in blood plasma as a biomarker for Parkinson’s disease. Sci. Rep. 3, 2540 (2013).
El-Agnaf, O. M. et al. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 20, 419–425 (2006).
Malek, N. et al. Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease - a systematic review. Acta Neurol. Scand. 130, 59–72 (2014).
Braak, H., de Vos, R. A., Bohl, J. & Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 396, 67–72 (2006).
Fellner, L. et al. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 61, 349–360 (2013).
Kim, C. et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562 (2013).
Ramesh, G., MacLean, A. G. & Philipp, M. T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013, 480739 (2013).
Cunningham, C., Wilcockson, D. C., Campion, S., Lunnon, K. & Perry, V. H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 25, 9275–9284 (2005).
Hernandez-Romero, M. C. et al. Peripheral inflammation increases the deleterious effect of CNS inflammation on the nigrostriatal dopaminergic system. Neurotoxicology 33, 347–360 (2012).
Beraud, D. et al. Microglial activation and antioxidant responses induced by the Parkinson’s disease protein alpha-synuclein. J. Neuroimmune Pharm. 8, 94–117 (2013).
Allen Reish, H. E. & Standaert, D. G. Role of alpha-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinsons Dis. 5, 1–19 (2015).
Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 30, 244–250 (2007).
Zhang, W. et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542 (2005).
Braak, H., Rub, U., Gai, W. P. & Del Tredici, K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 110, 517–536 (2003).
Zella, M. A. S. et al. Novel immunotherapeutic approaches to target alpha-synuclein and related neuroinflammation in Parkinson’s disease. Cells 8, https://doi.org/10.3390/cells8020105 (2019).
Earls, R. H. et al. NK cells clear alpha-synuclein and the depletion of NK cells exacerbates synuclein pathology in a mouse model of alpha-synucleinopathy. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.1909110117 (2020).
Niwa, F., Kuriyama, N., Nakagawa, M. & Imanishi, J. Effects of peripheral lymphocyte subpopulations and the clinical correlation with Parkinson’s disease. Geriatr. Gerontol. Int 12, 102–107 (2012).
Earls, R. H. et al. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J. Neuroinflammation 16, 250 (2019).
Hamza, T. H. et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 42, 781–785 (2010).
Lee, H. J., Suk, J. E., Bae, E. J. & Lee, S. J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 372, 423–428 (2008).
Zhang, W. et al. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia 55, 1178–1188 (2007).
Ferreira, S. A. & Romero-Ramos, M. Microglia response during Parkinson’s disease: alpha-synuclein intervention. Front. Cell Neurosci. 12, 247 (2018).
Su, X. et al. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 29, 1690–1701 (2008).
Su, X., Federoff, H. J. & Maguire-Zeiss, K. A. Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox. Res 16, 238–254 (2009).
Zhang, Q. S., Heng, Y., Yuan, Y. H. & Chen, N. H. Pathological alpha-synuclein exacerbates the progression of Parkinson’s disease through microglial activation. Toxicol. Lett. 265, 30–37 (2017).
Lee, M. K. et al. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 -> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl Acad. Sci. USA 99, 8968–8973 (2002).
Masliah, E. et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269 (2000).
Richfield, E. K. et al. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp. Neurol. 175, 35–48 (2002).
Loria, F. et al. alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 134, 789–808 (2017).
Rostami, J. et al. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 37, 11835–11853 (2017).
Rannikko, E. H., Weber, S. S. & Kahle, P. J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 16, 57 (2015).
Gao, H. M. & Hong, J. S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365 (2008).
White, A. J. et al. The peripheral inflammatory response to alpha-synuclein and endotoxin in Parkinson’s disease. Front Neurol. 9, 946 (2018).
Williams-Gray, C. H. et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 31, 995–1003 (2016).
Shalash, A. et al. Elevated serum alpha-synuclein autoantibodies in patients with Parkinson’s disease relative to Alzheimer’s disease and controls. Front. Neurol. 8, 720 (2017).
Gruden, M. A. et al. Immunoprotection against toxic biomarkers is retained during Parkinson’s disease progression. J. Neuroimmunol. 233, 221–227 (2011).
Kustrimovic, N. et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflammation 15, 205 (2018).
Cebrian, C. et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 5, 3633 (2014).
Devos, D. et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 50, 42–48 (2013).
Forsyth, C. B. et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE 6, e28032 (2011).
Clairembault, T. et al. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol. Commun. 3, 12 (2015).
Salat-Foix, D., Tran, K., Ranawaya, R., Meddings, J. & Suchowersky, O. Increased intestinal permeability and Parkinson disease patients: chicken or egg? Can. J. Neurol. Sci. 39, 185–188 (2012).
Phillips, R. J., Billingsley, C. N. & Powley, T. L. Macrophages are unsuccessful in clearing aggregated alpha-synuclein from the gastrointestinal tract of healthy aged Fischer 344 rats. Anat. Rec. 296, 654–669 (2013).
Houser, M. C. & Tansey, M. G. The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? Npj Parkinsons Dis. 3, https://doi.org/10.1038/s41531-016-0002-0 (2017).
Edelblum, K. L. & Turner, J. R. The tight junction in inflammatory disease: communication breakdown. Curr. Opin. Pharm. 9, 715–720 (2009).
Hazenberg, M. D. & Spits, H. Human innate lymphoid cells. Blood 124, 700–709 (2014).
Cheent, K. & Khakoo, S. I. Natural killer cells: integrating diversity with function. Immunology 126, 449–457 (2009).
Vivier, E., Tomasello, E., Baratin, M., Walzer, T. & Ugolini, S. Functions of natural killer cells. Nat. Immunol. 9, 503–510 (2008).
Shi, F. D., Ljunggren, H. G., La Cava, A. & Van Kaer, L. Organ-specific features of natural killer cells. Nat. Rev. Immunol. 11, 658–671 (2011).
Lanier, L. L., Corliss, B. & Phillips, J. H. Arousal and inhibition of human NK cells. Immunol. Rev. 155, 145–154 (1997).
Burkhardt, J. K., Hester, S., Lapham, C. K. & Argon, Y. The lytic granules of natural killer cells are dual-function organelles combining secretory and pre-lysosomal compartments. J. Cell Biol. 111, 2327–2340 (1990).
Lettau, M., Schmidt, H., Kabelitz, D. & Janssen, O. Secretory lysosomes and their cargo in T and NK cells. Immunol. Lett. 108, 10–19 (2007).
Topham, N. J. & Hewitt, E. W. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology 128, 7–15 (2009).
Guillerey, C., Huntington, N. D. & Smyth, M. J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 17, 1025–1036 (2016).
Romee, R., Leong, J. W. & Fehniger, T. A. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Science 2014, 205796 (2014).
Small, C. L. et al. NK cells play a critical protective role in host defense against acute extracellular Staphylococcus aureus bacterial infection in the lung. J. Immunol. 180, 5558–5568 (2008).
Schmidt, R. L., Filak, H. C., Lemon, J. D., Potter, T. A. & Lenz, L. L. A LysM and SH3-domain containing region of the Listeria monocytogenes p60 protein stimulates accessory cells to promote activation of host NK cells. PLoS Pathog. 7, e1002368 (2011).
Nikzad, R. et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci. Immunol. 4, https://doi.org/10.1126/sciimmunol.aat8116 (2019).
Thoren, F. B. et al. Human NK Cells induce neutrophil apoptosis via an NKp46- and Fas-dependent mechanism. J. Immunol. 188, 1668–1674 (2012).
Waggoner, S. N. & Kumar, V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front. Immunol. 3, 377 (2012).
Hao, J. et al. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J. Exp. Med 207, 1907–1921 (2010).
Jiang, W. et al. Acetylcholine-producing NK cells attenuate CNS inflammation via modulation of infiltrating monocytes/macrophages. Proc. Natl Acad. Sci. USA 114, E6202–E6211 (2017).
Masilamani, M., Peruzzi, G., Borrego, F. & Coligan, J. E. Endocytosis and intracellular trafficking of human natural killer cell receptors. Traffic 10, 1735–1744 (2009).
Cerwenka, A. & Lanier, L. L. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 1, 41–49 (2001).
Moretta, A. et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 19, 197–223 (2001).
Karre, K., Ljunggren, H. G., Piontek, G. & Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678 (1986).
Ravetch, J. V. & Lanier, L. L. Immune inhibitory receptors. Science 290, 84–89 (2000).
Raulet, D. H. Missing self recognition and self tolerance of natural killer (NK) cells. Semin. Immunol. 18, 145–150 (2006).
Raulet, D. H. & Vance, R. E. Self-tolerance of natural killer cells. Nat. Rev. Immunol. 6, 520–531 (2006).
Gasser, S. & Raulet, D. H. Activation and self-tolerance of natural killer cells. Immunol. Rev. 214, 130–142 (2006).
Di Santo, J. P. Natural killer cell developmental pathways: a question of balance. Annu Rev. Immunol. 24, 257–286 (2006).
Anfossi, N. et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 25, 331–342 (2006).
Poli, A. et al. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 126, 458–465 (2009).
Han, S. et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J. Immunol. 192, 2551–2563 (2014).
Marcenaro, E., Notarangelo, L. D., Orange, J. S. & Vivier, E. Editorial: NK cell subsets in health and disease: new developments. Front. Immunol. 8, 1363 (2017).
Mikulak, J., Di Vito, C., Zaghi, E. & Mavilio, D. Host immune responses in HIV-1 infection: the emerging pathogenic role of siglecs and their clinical correlates. Front Immunol. 8, 314 (2017).
Zamora, A. E., Grossenbacher, S. K., Aguilar, E. G. & Murphy, W. J. Models to study NK cell biology and possible clinical application. Curr. Protoc. Immunol. 110, 11–14 (2015). 14 37.
Abel, A. M., Yang, C., Thakar, M. S. & Malarkannan, S. Natural killer cells: development, maturation, and clinical utilization. Front. Immunol. 9, 1869 (2018).
Li, Q. et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101, 207–223 e210 (2019).
Van Hove, H. et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 22, 1021–1035 (2019).
Huang, D. et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 20, 896–905 (2006).
Xu, W., Fazekas, G., Hara, H. & Tabira, T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 163, 24–30 (2005).
Morse, R. H., Seguin, R., McCrea, E. L. & Antel, J. P. NK cell-mediated lysis of autologous human oligodendrocytes. J. Neuroimmunol. 116, 107–115 (2001).
Bowen, K. E., Mathew, S. O., Borgmann, K., Ghorpade, A. & Mathew, P. A. A novel ligand on astrocytes interacts with natural cytotoxicity receptor NKp44 regulating immune response mediated by NK cells. PLoS ONE 13, e0193008 (2018).
Garofalo, S. et al. Natural killer cells modulate motor neuron-immune cell cross talk in models of amyotrophic lateral sclerosis. Nat. Commun. 11, 1773 (2020).
Liu, Q. et al. Neural stem cells sustain natural killer cells that dictate recovery from brain inflammation. Nat. Neurosci. 19, 243–252 (2016).
Mihara, T. et al. Natural killer cells of Parkinson’s disease patients are set up for activation: a possible role for innate immunity in the pathogenesis of this disease. Parkinsonism Relat. Disord. 14, 46–51 (2008).
Cen, L. et al. Peripheral lymphocyte subsets as a marker of Parkinson’s disease in a Chinese population. Neurosci. Bull. 33, 493–500 (2017).
Tremblay-McLean, A., Coenraads, S., Kiani, Z., Dupuy, F. P. & Bernard, N. F. Expression of ligands for activating natural killer cell receptors on cell lines commonly used to assess natural killer cell function. BMC Immunol. 20, 8 (2019).
Bokor, M., Farago, A., Garam, T., Malatinszky, G. & Schnabel, R. Antibody-dependent cell-mediated cytotoxicity (ADCC) in Parkinson’s disease. J. Neurol. Sci. 115, 47–50 (1993).
Jiang, S., Gao, H., Luo, Q., Wang, P. F. & Yang, X. L. The correlation of lymphocyte subsets, natural killer cell, and Parkinson’s disease: a meta-analysis. Neurological Sci. 38, 1373–1380 (2017).
Angelova, D. M. & Brown, D. R. Model senescent microglia induce disease related changes in alpha-synuclein expression and activity. Biomolecules 8, https://doi.org/10.3390/biom8030067 (2018).
Coppe, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev. Pathol. 5, 99–118 (2010).
Kritsilis, M. et al. Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 19, https://doi.org/10.3390/ijms19102937 (2018).
Antonangeli, F., Zingoni, A., Soriani, A. & Santoni, A. Senescent cells: living or dying is a matter of NK cells. J. Leukoc. Biol. 105, 1275–1283 (2019).
Backstrom, E., Chambers, B. J., Kristensson, K. & Ljunggren, H. G. Direct NK cell-mediated lysis of syngenic dorsal root ganglia neurons in vitro. J. Immunol. 165, 4895–4900 (2000).
Davies, A. J. et al. Natural killer cells degenerate intact sensory afferents following nerve injury. Cell 176, 716–728 e718 (2019).
Carapito, R. & Bahram, S. Genetics, genomics, and evolutionary biology of NKG2D ligands. Immunol. Rev. 267, 88–116 (2015).
Dumitriu, A. et al. Integrative analyses of proteomics and RNA transcriptomics implicate mitochondrial processes, protein folding pathways and GWAS loci in Parkinson disease. BMC Med. Genomics 9, 5 (2016).
Zhang, B., Yamamura, T., Kondo, T., Fujiwara, M. & Tabira, T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J. Exp. Med. 186, 1677–1687 (1997).
Sulzer, D. et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546, 656–661 (2017).
Crouse, J., Xu, H. C., Lang, P. A. & Oxenius, A. NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol. 36, 49–58 (2015).
Rabinovich, B. A. et al. Activated, but not resting, T cells can be recognized and killed by syngeneic NK cells. J. Immunol. 170, 3572–3576 (2003).
Cerboni, C. et al. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK- cell lysis. Blood 110, 606–615 (2007).
Mount, M. P. et al. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 27, 3328–3337 (2007).
Paolini, R., Bernardini, G., Molfetta, R. & Santoni, A. NK cells and interferons. Cytokine Growth Factor Rev. 26, 113–120 (2015).
Schoenborn, J. R. & Wilson, C. B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 96, 41–101 (2007).
Voorthuis, J. A. et al. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-gamma in Lewis rats. Clin. Exp. Immunol. 81, 183–188 (1990).
Ottum, P. A., Arellano, G., Reyes, L. I., Iruretagoyena, M. & Naves, R. Opposing roles of interferon-gamma on cells of the central nervous system in autoimmune neuroinflammation. Front. Immunol. 6, 539 (2015).
Massey, A. R. et al. Alpha-synuclein expression supports interferon stimulated gene expression in neurons. bioRxiv, https://doi.org/10.1101/2020.04.25.061762 (2020).
Halonen, S. K., Woods, T., McInnerney, K. & Weiss, L. M. Microarray analysis of IFN-gamma response genes in astrocytes. J. Neuroimmunol. 175, 19–30 (2006).
Di Lorenzo, G. et al. Granulocyte and natural killer activity in the elderly. Mech. Ageing Dev. 108, 25–38 (1999).
Hazeldine, J., Hampson, P. & Lord, J. M. Reduced release and binding of perforin at the immunological synapse underlies the age-related decline in natural killer cell cytotoxicity. Aging Cell 11, 751–759 (2012).
Poggi, A. et al. Human gut-associated natural killer cells in health and disease. Front Immunol. 10, 961 (2019).
Bregenholt, S., Berche, P., Brombacher, F. & Di Santo, J. P. Conventional alpha beta T cells are sufficient for innate and adaptive immunity against enteric Listeria monocytogenes. J. Immunol. 166, 1871–1876 (2001).
Naper, C., Shegarfi, H., Inngjerdingen, M. & Rolstad, B. The role of natural killer cells in the defense against Listeria monocytogenes lessons from a rat model. J. Innate Immun. 3, 289–297 (2011).
Ashkar, A. A., Reid, S., Verdu, E. F., Zhang, K. & Coombes, B. K. Interleukin-15 and NK1.1+ cells provide innate protection against acute Salmonella enterica serovar Typhimurium infection in the gut and in systemic tissues. Infect. Immun. 77, 214–222 (2009).
Hall, L. J. et al. Natural killer cells protect against mucosal and systemic infection with the enteric pathogen Citrobacter rodentium. Infect. Immun. 81, 460–469 (2013).
Sotolongo, J. et al. Host innate recognition of an intestinal bacterial pathogen induces TRIF-dependent protective immunity. J. Exp. Med. 208, 2705–2716 (2011).
Nedvetzki, S. et al. Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood 109, 3776–3785 (2007).
Hazeldine, J. & Lord, J. M. The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Res. Rev. 12, 1069–1078 (2013).
Lutz, C. T., Moore, M. B., Bradley, S., Shelton, B. J. & Lutgendorf, S. K. Reciprocal age related change in natural killer cell receptors for MHC class I. Mech. Ageing Dev. 126, 722–731 (2005).
Lutz, C. T. et al. Human NK cells proliferate and die in vivo more rapidly than T cells in healthy young and elderly adults. J. Immunol. 186, 4590–4598 (2011).
Krishnaraj, R. & Bhooma, T. Cytokine sensitivity of human NK cells during immunosenescence. 2. IL2-induced interferon gamma secretion. Immunol. Lett. 50, 59–63 (1996).
Acknowledgements
This work was supported by startup funds from the Department of Physiology and Pharmacology at the University of Georgia and the Michael J. Fox Foundation Target Advancement Grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Earls, R.H., Lee, JK. The role of natural killer cells in Parkinson’s disease. Exp Mol Med 52, 1517–1525 (2020). https://doi.org/10.1038/s12276-020-00505-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-00505-7
This article is cited by
-
The role of CD56bright NK cells in neurodegenerative disorders
Journal of Neuroinflammation (2024)
-
Dynamic molecular network analysis of iPSC-Purkinje cells differentiation delineates roles of ISG15 in SCA1 at the earliest stage
Communications Biology (2024)
-
Immunological shifts during early-stage Parkinson’s disease identified with DNA methylation data on longitudinally collected blood samples
npj Parkinson's Disease (2024)
-
Advancements in Single-Cell RNA Sequencing Research for Neurological Diseases
Molecular Neurobiology (2024)
-
Role of the immune system in amyotrophic lateral sclerosis. Analysis of the natural killer cells and other circulating lymphocytes in a cohort of ALS patients
BMC Neurology (2023)
