
Abstract
T cells are the central mediators of both humoral and cellular adaptive immune responses. Highly specific receptor-mediated clonal selection and expansion of T cells assure antigen-specific immunity. In addition, encounters with cognate antigens generate immunological memory, the capacity for long-term, antigen-specific immunity against previously encountered pathogens. However, T-cell receptor (TCR)-independent activation, termed “bystander activation”, has also been found. Bystander-activated T cells can respond rapidly and secrete effector cytokines even in the absence of antigen stimulation. Recent studies have rehighlighted the importance of antigen-independent bystander activation of CD4+ T cells in infection clearance and autoimmune pathogenesis, suggesting the existence of a distinct innate-like immunological function performed by conventional T cells. In this review, we discuss the inflammatory mediators that activate bystander CD4+ T cells and the potential physiological roles of these cells during infection, autoimmunity, and cancer.
Similar content being viewed by others

Introduction
The immune system is classically divided into the innate and adaptive arms1. Innate immune cells express invariant antigen receptors, such as pattern recognition receptors (PRRs) that recognize conserved molecular patterns of pathogens. Adaptive immunity is mediated by cells known as lymphocytes, which have evolved to recognize pathogens through highly precise and diverse antigen-specific receptors generated by gene rearrangement. While innate immunity generates less specific but rapid inflammatory responses, adaptive immunity provides long-lasting, highly specific defense and protection against pathogens by generating immunological memory2. This dichotomy, supported by the clonal selection theory and the concept of antigen processing and presentation, has been accepted for decades3,4. However, the recent discovery of innate lymphoid cells (ILCs) has drawn special attention to the importance of the innate-like function of lymphocytes. ILCs are derived from common lymphoid progenitors, which also give rise to T lymphocytes. ILC subsets, including ILC1, ILC2, and ILC3, mirror the transcriptional and cytokine profiles of the effector CD4+ T-helper TH1, TH2, and TH17 subsets5. As ILCs are activated by cytokines rather than antigen receptors, this correlation suggests the possibility of T cells having an innate-like capacity. Of note, the concept of antigen-independent “bystander activation” of conventional T cells has been previously reported. An increasing body of evidence suggests that effector/memory T cells can be activated in the absence of antigen stimulation by pro-inflammatory mediators6. The antigen-independent activation of bystander effector/memory CD8+ T cells has been shown to play an important role in immune responses in viral infection, cancer, and autoimmunity7,8,9. Although the bystander activation of CD8+ T cells has been recently reviewed6,10, the key concept of T-cell receptor (TCR)-independent CD4+ T-cell activation has not been well characterized. In this review, we focus on the understanding of the TCR-independent bystander activation of CD4+ T cells and the importance of bystander activation in potential immunological roles and therapeutic approaches during infection, autoimmunity, or cancer.
Overview of bystander CD4+ T-cell activation
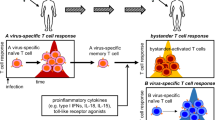
Traditionally, the activation and differentiation of naïve CD4+ T cells into effector T-helper cells require THREE distinct signals: TCR engagement of antigen peptides presented by major histocompatibility complex class II molecules (signal 1) and the interaction of costimulatory molecules (signal 2) initiate T-cell activation. These activated naïve CD4+ T cells further differentiate into distinct subsets of helper T cells, including TH1, TH2, TH17, Treg, and TFH cells, in different cytokine milieus (signal 3), as defined by their pattern of effector cytokine production and immunological function11,12. Compared to conventional T-cell activation, bystander T-cell activation is independent of TCR signaling6,10 (Fig. 1). Bystander CD4+ T cells were first reported in lymphocytic choriomeningitis virus infection, where TCR-independent proliferation of unrelated CD4+ T cells was observed13. Bystander proliferation has also been demonstrated with direct injection of lipopolysaccharide (LPS) or with cytokine stimulation14,15. Interestingly, the TCR-independent activation of CD4+ T cells has primarily been observed in memory (CD44high) cells, while naïve (CD44low) cells exhibit lower reactivity13,14,15. Thus, it appears that cytokines and innate receptors, such as Toll-like receptors (TLRs), can play important roles in TCR-independent bystander activation and that effector/memory CD4+ T cells have a lower threshold than naïve CD4+ T cells, which indicates that effector/memory CD4+ T cells have a higher probability of undergoing this bystander activation.

a Antigen-specific T-cell activation requires three distinct signals. Signal 1 is antigen-specific signaling mediated by T-cell receptor (TCR) engagement of pathogenic peptides presented by major histocompatibility complex (MHC) molecules. Signal 2 is costimulatory signaling, which is mainly mediated by the interaction of CD28 with one of the B7 molecules (CD80 and CD86). Signal 3 is polarizing signaling mediated by various cytokine milieus produced by dendritic cells. b In contrast, bystander T-cell activation is the concept of T-cell activation independent of antigen stimulation. Bystander-activated T cells can respond rapidly to inflammatory mediators (cytokine and TLR signaling) in a TCR-independent manner. TLR2 Toll-like receptor 2, TLR4 Toll-like receptor 4.
Bystander CD4+ T-cell activation: IL-1 family cytokines and STAT activators
Cytokines are the central mediators that regulate innate and adaptive immune responses. Previous studies have revealed that pro-inflammatory cytokines can directly induce the effector function of T cells6,10,16. Of note, Interleukin-1 (IL-1) family cytokines (IL-1, Interleukin-18 (IL-18), and Interleukin-33 (IL-33)) and signal transducer and activator of transcription (STAT) activators (Interleukin-2 (IL-2), Interleukin-12 (IL-12), Interleukin-23 (IL-23), and Interleukin-27 (IL-27)) appear to be potent activators of antigen-independent bystander activation of CD4+ T cells (Fig. 2).

Effector/memory CD4+ T cells (TH1, TH2, and TH17) can undergo bystander activation by directly responding to inflammatory cytokines and TLR agonists. These signals induce effector cytokine production that promotes important pathological responses in autoimmunity and pathogen infections. Naïve CD4+ T cells can also be activated in a TCR-independent manner under the influence of cytokines such as IL-2, IL-18, and IL-27. Bystander activation of naïve CD4+ T cells can promote immunosuppressive functions that regulate autoimmune pathogenesis. IL interleukin, IFN-γ interferon-γ, GM-CSF granulocyte-macrophage colony-stimulating factor, PD-L1 programmed death-ligand 1, LPS lipopolysaccharide, TH1 T-helper 1, TH2 T-helper 2, TH17 T-helper 17, TN naïve CD4+ T cell.
Interleukin-1 (IL-1)
IL-1, the first identified interleukin, is a central mediator of innate and adaptive immune responses17. IL-1β has been shown to contribute to the early differentiation and maintenance of TH17 cells by regulating the expression of IFN regulatory factor (IRF4) and retinoid acid-related orphan receptor (ROR)γt18. Importantly, IL-1β can potently induce cytokine production by effector TH17 cells in the absence of TCR engagement18,19,20. As differentiated TH17 cells upregulate the expression of IL-23 and IL-12 receptors, IL-1β acts synergistically with IL-23 and IL-1219,21,22. In the presence of IL-1β, TH17 cells can produce IL-17 and IFN-γ in a TCR-independent manner when stimulated with IL-2318,19,20,21 or IL-1221, respectively. The IL-1β- and IL-23-mediated mechanisms underlying the bystander activation of TH17 cells are dependent on nuclear factor (NF)-κB and p38 mitogen-activated protein kinase (MAPK) signaling20. We and others have shown that both murine and human memory CD4+ T cells express high levels of interleukin receptor type 1 (IL-1R1)23,24. Memory but not naïve CD4+ T cells primarily respond to IL-1β in the absence of TCR engagement. This bystander activation synergizes with IL-23 to induce pathogenic TH17 signature genes (e.g., Csf2, Il23r, Bhlhe40, Ccr6, and Rorc) while downregulating the expression of nonpathogenic TH17 signature genes (e.g., Foxp3, Il10, and IL6st). Murine models of multiple sclerosis (MS) (experimental autoimmune encephalomyelitis, EAE) have revealed the potential pathogenicity of antigen-nonrelated memory-like TH17 cells responding to IL-1β and IL-23. The recruitment of bystander T cells into the central nervous system (CNS) and their pathogenic function were found to be directly mediated by IL-1R1 signaling24.
Interleukin-18 (IL-18)
IL-18, a member of the IL-1 cytokine family, is a pleiotropic cytokine potentially capable of inducing bystander activation of CD4+ T cells in both mice and humans17. The TCR-independent function of IL-18 has been shown in various T-helper subsets including TH1, TH2, and TH17 cells19,21,25. IL-18 overexpression in the murine lungs spontaneously induced effector CD4+ T cells expressing IFN-γ, IL-13, and IL-17A without exogenous antigen challenge26. Effector TH1 cells extensively express IL-18 receptor α (IL-18Rα), and signaling through this receptor increases the expression levels of IFN-γ and IL-18Rα in direct response to IL-1819. In addition, IL-18 promotes IL-17A production in TH17 cells generated in vitro or in vivo in the absence of TCR engagement21. IL-18 has also been demonstrated to contribute to the bystander activation of both murine and human naïve and memory CD4+ T cells to produce high levels of IFN-γ27,28. Recent studies have identified a major subset of human mucosal memory CD4+ T cells expressing IL-18Rα. IL-18Rα+ memory CD4+ T cells respond to IL-18 by producing IFN-γ, TNF-α, IL-6, IL-5, IL-13, GM-CSF, and IL-22 in an antigen-independent manner29. IL-18 has been shown to have synergistic effects with various combinations of cytokines, including IL-2, IL-12, IL-23, and IL-15. Of note, these “cytokine synergies” were mostly dependent on IL-18 signaling29, thus implicating IL-18 as an important initial mediator of the bystander activation of antigen-nonrelated CD4+ T cells during host defense.
Interleukin-33 (IL-33)
IL-33, an epithelial-derived cytokine in the IL-1 family, is a particularly potent activator of ILC2s that induces the production of the effector cytokines IL-5 and IL-1317,30. TH2 cells, which selectively express the IL-33 receptor (ST2), are another important effector cell type responsive to IL-33. Previous studies have reported that ST2 is expressed on TH2 cells but not on TH1, TH17, or Treg cells19,31,32. Because of this ST2 expression pattern, TH2 cells are considered to directly respond to IL-3319,33,34. In vitro studies have revealed that TH2 cells can produce IL-13 and IL-5 but not IL-4 in response to IL-33 without TCR engagement. The antigen-independent activation of TH2 cells responding to IL-33 was dependent on NFκB and p38 but independent of nuclear factor of activated T cells19. Notably, memory TH2 cells express ST2 at higher levels than effector TH2 cells35,36. A murine infection model demonstrated the innate immunological function of bystander memory TH2 cells in allergic inflammation and protection against early helminth infection. This bystander activation of airway memory TH2 cells was dependent on IL-33 but not on TCR stimulation33. Furthermore, lung-resident ST2+ memory CD4+ T cells have been reported to be involved in IL-33-induced lung inflammation. Compared with ILC2s, ST2+ memory CD4+ T cells were the major contributors to the pathogenicity of eosinophilic inflammation and functioned by producing IL-5 and IL-1334. Thus, collectively, these results suggested a distinct innate mechanism of bystander-activated effector/memory TH2 cells in type-2 inflammation that correlates with ILC2s.
Interleukin-2 (IL-2)
IL-2, originally called T-cell growth factor, is a member of the common γ-chain family37. IL-2 induces STAT5 signaling, which has important roles in the regulation of T-cell proliferation and immune responses38. Both in vitro and in vivo studies have revealed that IL-2 drives the expansion and proliferation of bystander CD4+ T cells39,40. However, the direct effect of IL-2 on bystander-activated CD4+ T cells is unknown. When naïve murine CD4+ T cells are treated with high doses of IL-2, they can respond to IL-12 and IL-18 to produce IFN-γ in the absence of TCR engagement27. This result suggests that high-dose IL-2 stimulation can replace TCR signaling to activate naïve CD4+ T cells. Furthermore, IL-2-mediated STAT5 signaling has been shown to be important in the TCR-independent activation of TH2 cells. IL-2 is directly involved in GATA3 and ST2 upregulation in TH2 cells, which has a synergistic effect with IL-33 to drive the production of the effector cytokine IL-1319. Further studies may be necessary to establish the potential roles of IL-2 in the effector functions of bystander-activated CD4+ T cells.
Interleukin-12 (IL-12)
IL-12 is an important cytokine that induces IFN-γ production and TH1 differentiation from naïve CD4+ T cells through STAT4 signaling. Previous studies have shown that IL-12 is one of the key factors in the induction of TCR-independent IFN-γ production by CD4+ T cells in both mice and humans27,28. Without antigen stimulation, IL-12 can directly upregulate T-bet and IL-18Rα expression in TH1 cells19. Together with IL-18, IL-12 induces bystander activation of effector TH1 and TH17 cells to induce IFN-γ production19,21. Studies of in vivo mouse models indicate that IL-12 signaling generates and maintains T-bethigh memory-phenotype (MP) CD4+ T cells. These MP cells rapidly produce IFN-γ in response to IL-12, which provides host resistance against Toxoplasma gondii infection in the absence of cognate antigen recognition41. Thus, IL-12 appears to be an important inflammatory mediator of bystander T-cell responses in type-1 immunity.
Interleukin-23 (IL-23)
IL-23, a pro-inflammatory cytokine belonging to the IL-12 family, is essential for the generation of pathogenic TH17 cells42,43,44,45. IL-23 upregulates the expression of transcription factors, such as RORγt and T-bet, and induces effector molecules, including IL-17A, IFN-γ, GM-CSF, and IL-22, by activating the JAK2/STAT3 signaling pathway43,44,45,46,47. We and others have shown that IL-23 alone cannot induce TCR-independent activation of effector/memory CD4+ T cells19,21,24. However, in the presence of IL-1β, IL-23 increases the expression and production of the effector cytokines IL-17A, IFN-γ, GM-CSF, and IL-22. IL-1β appears to be an important factor in the initial upregulation of IL-23R expression, and compared with either of these two cytokines alone, cotreatment with IL-1β and IL-23 significantly increases the expression levels of both IL-1R1 and IL-23R, indicating the existence of positive feedback between IL-1 and IL-2324. Further studies are necessary to reveal the distinct mechanisms by which IL-1β and IL-23 transduce antigen-independent signals to effector/memory CD4+ T cells.
Interleukin-27 (IL-27)
IL-27 is a member of the IL-12 family of heterodimeric cytokines that have immunosuppressive functions. IL-27 can inhibit pro-inflammatory cytokine production by effector T cells, including TH1, TH2, and TH17 cells48. Of note, naïve CD4+ T cells are reported to express IL-27R, whose expression is downregulated in the presence of TCR signaling. IL-27 can directly upregulate the expression of CD274, which encodes PD-L1, an immune checkpoint inhibitor that binds to PD-1, on naïve CD4+ T cells in a STAT1-dependent manner49. IL-27-primed naïve T cells inhibit TH17 differentiation by expressing PD-L1, which ameliorates the development of autoimmune encephalomyelitis50. Hence, IL-27 can induce bystander activation of naïve CD4+ T cells, leading to acquisition of antigen-independent inhibitory function. This suggests a potent role for the antigen-independent regulatory function of T cells during inflammation, which merits further investigation.
Induction of bystander CD4+ T-cell activation through TLR signaling
TLRs are one of the key participants in innate immunity. The discovery and functional characterization of TLRs furthered our understanding of how innate immune cells rapidly respond to microbial pathogen invasion51. Recent studies, however, demonstrate that T cells can express a variety of TLRs52. Direct activation of TLR signaling in effector/memory CD4+ T cells induces innate-like effector function (Fig. 2).
TLR2
TLR2 is an important PRR for the recognition of bacterial lipoteichoic acid, lipoproteins, and peptidoglycans53. Previous studies have demonstrated the functional role of TLR2 in effector/memory CD4+ T cells54,55,56,57,58,59,60,61,62. TLR2 agonists can act directly on effector TH1 cells to induce production of IFN-γ in the absence of TCR stimulation. This TLR2-induced IFN-γ production can be greatly enhanced by the combination of the cytokines IL-2 and IL-12, which induce the activation of MAPK signaling58. In addition, TH17 cells have been linked to the expression of TLRs, including TLR2 and TLR4. TLR2 signaling, which has a synergistic effect with IL-23 signaling, directly induces the TCR-independent activation and proliferation of TH17 cells, resulting in the secretion of the effector cytokines IL-17A and IL-22. A murine model of EAE revealed that TH17 cell-intrinsic TLR2 activation is important for the pathogenesis of autoimmune neuroinflammation57. ΤLR2 stimulation has been shown to impair the suppressive function of regulatory T cells61,63,64. Although TLR2 signaling drives Treg cell glycolysis and proliferation, it reduces the suppressive capacity in an mTORC1-dependent manner. Thus, these findings demonstrate the potential roles of TLRs in regulating Treg metabolism to balance proliferation and suppressive function61. In humans, TLR2 is expressed in memory and activated CD4+ T cells54. In the absence of antigen stimulation, human memory CD4+ T cells have been reported to respond to TLR2 agonists with higher IFN-γ production than that achieved by naïve CD4+ T cells54,62.
TLR4
TLR4 is a receptor that recognizes for gram-negative bacterial LPS. LPS stimulation has been reported to enhance the proliferation and survival of CD4+ T cells. Loss of TLR4 in CD4+ T cells abrogated disease symptoms in a murine model of EAE by substantially reducing TH17 and TH1 cell numbers in the CNS. Thus, TLR4 expression in CD4+ T cells is essential in EAE pathogenesis65. In contrast, TLR4 signaling in CD4+ T cells seems to ameliorate spontaneous colitis. A T-cell transfer model of colitis showed that LPS stimulation inhibited ERK1/2 activation and TH1 responses66. Regulatory T cells also selectively express TLR4. LPS treatment directly increases the survival/proliferation and suppressor efficiency of Treg cells in the absence of TCR engagement67. In human CD4+ T cells, LPS stimulation directly increases adherence to fibronectin in a PKC- and p38-dependent manner, indicating that TLR4 signaling can regulate T-cell behavior in inflammation68. Thus, TLR signaling is important not only for phagocytic cell functions and inflammation but also direct stimulation of effector T cells, which induces activation of antigen-nonrelated T cells during inflammatory responses.
Bystander CD4+ T-cell activation in immunity and disease
Infection
The published mechanistic studies on infection are mainly focused on antigen-specific T-cell responses69,70. However, the importance of bystander CD8+ T cells in viral infections has been recently reviewed10, indicating an unexpected role for antigen-nonrelated T cells during infection. Notably, previous studies have revealed the potential role of antigen-nonrelated CD4+ T cells in various infectious diseases33,41,59,71,72,73,74. Bystander activation of CD4+ T cells has been reported during herpes simplex virus (HSV) infection. Ocular infection with HSV can cause an immunopathological disease called herpetic stromal keratitis (HSK) in the corneal stroma75. HSV-unrelated CD4+ T cells were shown to contribute to establishing ocular lesions in the absence of antigen specificity71,72, thus indicating a significant role for virus-unrelated CD4+ T cells in HSK pathogenesis. Moreover, recent studies have shown evidence for bystander activation of CD4+ T cells in parasitic infections33,41,59. Mouse models of Borrelia burgdorferi infection have demonstrated expansion and activation of Borrelia-unrelated TLR2+CD4+ T cells in the synovial joint. TLR2 expression on bystander T cells contributed to IFN-γ production and arthritis pathogenesis59, indicating the existence of a microbe-induced innate mechanism during infection. Recently, TH2 cells generated in response to the helminth Ascaris suum were shown to contribute to the clearance of unrelated Nippostrongylus brasiliensis. OVA-induced airway TH2 cells also were found to participate in antigen-nonspecific protection against helminth infection. Such bystander activation of TH2 cells was dependent on IL-33 but not on TCR33. Furthermore, in Toxoplasma gondii infection, compared with pathogen-specific effector T cells, MP CD4+ T cells induced rapid IFN-γ production. MP CD4+ T cells provided nonspecific IL-12-dependent host resistance against infection41. Taken together, these findings indicate that antigen-unrelated CD4+ T cells can undergo bystander activation in the absence of TCR stimulation during infection, which could be implicated in both protective and pathogenic immune responses.
Autoimmunity
MS, one of the most common autoimmune diseases, is an inflammatory demyelinating human autoimmune disease of the CNS76. Previous studies on EAE, a model of MS, have reported that myelin-specific TH1 and TH17 cells mediate the pathogenesis of autoimmune neuroinflammation77,78,79. However, we and other groups have shown that the majority of CNS-infiltrating effector CD4+ T cells in the spinal cord of EAE mice are not specific for myelin oligodendrocyte glycoprotein (MOG)24,80,81,82. These T cells unrelated to MOG expressed high levels of effector cytokines (IL-17A, IFN-γ, and GM-CSF)24. Murine models of EAE have suggested that effector/memory CD4+ T cells can invade the CNS without antigen specificity24,83,84,85,86. One study reported that T cells unrelated to myelin contributed to EAE development by stimulating the function of antigen-presenting cells85. Furthermore, we recently confirmed that memory-like CD4+ T cells unrelated to myelin contribute to EAE pathogenicity by amplifying the production of the effector cytokines IL-17A, IFN-γ, and GM-CSF24 (Fig. 3), thereby indicating a potential role for bystander CD4+ T cells in the pathogenesis of EAE development. GM-CSF, which is directly regulated by the transcription factor RORγt87, is a well-defined pathogenic cytokine in TH17-related diseases, including EAE45,87 and MS88. Interestingly, overexpression of GM-CSF in CD4+ T cells has been reported to induce spontaneous neuroinflammation regardless of antigen specificity. The transcription factor Bhlhe40, whose expression is a characteristic of encephalitic TH17 cells, was primarily expressed in CNS-infiltrating effector T cells unrelated to MOG82,89. As further studies have revealed that IL-1β is one of the key cytokines that directly induces GM-CSF and Bhlhe40 expression45,82,90, Bhlhe40 and GM-CSF may be potent pathogenic mediators of the bystander activation of CD4+ T cells responding to IL-1β. In humans, the CD4+ T cells of patients with MS have been reported to significantly express the cytokine receptor IL-1R1 and TLRs, including TLR2 and TLR4, indicating the potential of CD4+ T cells to exert innate-like function in MS55,91. TLR agonists directly induced IL-6, IFN-γ, IL-17, and GM-CSF production by CD4+ T cells in patients with MS55. Several groups have also recently reported the protective role of bystander CD4+ T cells in the pathology of CNS inflammation50,92. T-cell-mediated neuroprotection after CNS injury has been reported to occur in the absence of TCR engagement. This neuroprotective bystander T-cell response was dependent on IL-4, which attenuated axonal damage92. In a study of an EAE murine model, naïve CD4+ T cells primed with IL-27 induced PD-L1, which inhibited severe autoimmune encephalomyelitis50. Together, these findings suggest that bystander CD4+ T cells can contribute to the development of and protection against autoimmune encephalomyelitis. Further work is necessary to reveal the precise pathological mechanisms involving bystander CD4+ T cells in various autoimmune diseases.

IL-1β and IL-23 induce innate-like pathogenic function in memory-like CD4+ T cells that are not specific for myelin. Along with myelin-specific effector T cells (TH1 and TH17), bystander memory-like CD4+ T cells contribute to the development of autoimmune pathogenesis by increasing IL-17A, IFN-γ, and GM-CSF levels in the CNS. Thus, bystander-activated memory-like CD4+ T cells responding to IL-1β and IL-23 perform a pathogenic role in an antigen-independent manner in autoimmune encephalomyelitis. IL interleukin, IFN-γ interferon-γ, GM-CSF granulocyte-macrophage colony-stimulation factor, IL-1R1 interleukin-1 receptor type 1, IL-23R interleukin-23 receptor, RORγt RAR-related orphan receptor gamma, CCR6 chemokine receptor 6, TH1 T-helper 1, TH17 T-helper 17, CNS central nervous system.
Cancer
The role of bystander T cells in anticancer immunity remains an open question. However, a recent study revealed that in human lung and colorectal cancers, tumor-infiltrating CD8+ TILs can be antigen nonspecific. These bystander CD8+ TILs, which lacked expression of CD39, were specific for unrelated viruses including Epstein–Barr virus, human cytomegalovirus, and influenza virus. The absence of CD39 expression, which indicates chronic antigen stimulation, suggested a potent biomarker for bystander T cells in antitumor immunity8. Interestingly, murine models of cancer have revealed that CD4+ T cells can also infiltrate tumors independent of antigen specificity. TCR-transgenic murine models have demonstrated nonspecific T-cell migration and accumulation in tumors. This bystander activation of CD4+ T cells required an effector/memory phenotype93. In mice treated with dual costimulation of CD134 (OX40) and CD137 (4-1BB), bystander activation of effector CD4+ T cells unrelated to the tumor contributed to the antitumor response, although the precise mechanism is unknown94. Collectively, these findings demonstrate the potent role of bystander CD4+ T cells in antitumor immunity and immunotherapy. Further investigation of the distinct mechanism involving bystander T cells in tumor pathogenesis may provide a new platform for understanding cancer immunopathology.
Concluding remarks
Bystander activation of CD4+ T cells occurs when T cells are stimulated by inflammatory mediators such as cytokines or TLR signaling molecules in the absence of antigen-specific TCR stimulation. In addition to providing rapid effector cytokine production, these bystander-activated CD4+ T cells significantly contribute to disease pathology, including that of infection, autoimmunity, and cancer, via their innate-like capacity. In reality, it may be more than just the few antigen-specific T cells that are capable of responding to pathogen invasion or related inflammation. Therefore, the dichotomy of rapidly responding innate immune cells of low specificity and highly antigen-specific responsive immune memory cells may need to be reconsidered. Concerning the importance of antigen-independent bystander T-cell activation, numerous questions remain unanswered. Further studies dissecting the molecular mechanisms driving bystander T-cell activation and the precise roles of these cells in immunity will provide new insights to improve the understanding of the pathological mechanisms of immune-mediated diseases. In addition, the characterization of specific T-cell subsets undergoing bystander activation needs to be completed. Recently, tissue-resident memory T cells have emerged as key components of immunological memory95. They are the dominant T-cell subsets residing in human tissues (the skin, lungs, gastrointestinal tract, etc.) that form a front-line defense against reinfection96. Being situated within peripheral tissues, TRM cells may be ideal subset candidates capable of responding to local inflammatory cytokines. Furthermore, identifying distinct and reproducible biomarkers of bystander T cells is necessary for generating new therapeutic approaches. Taken together, these studies will provide a foundation for a better understanding of the pivotal roles of bystander T-cell activation, thereby suggesting new directions for the treatment of immune-mediated diseases.
Change history
08 June 2023
A Correction to this paper has been published: https://doi.org/10.1038/s12276-023-01032-x
References
Chaplin D. D. Overview of the immune response. J. Allergy Clin. Immunol. 125, S3–S23 (2010).
Schenten, D. & Medzhitov, R. The control of adaptive immune responses by the innate immune system. Adv. Immunol. 109, 87–124 (2011).
Burnet F. M. A modification of Jerne’s theory of antibody production using the concept of clonal selection. CA Cancer J. Clin. 26, 119–121 (1976).
Neefjes, J., Jongsma, M. L., Paul, P. & Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11, 823–836 (2011).
Walker, J. A., Barlow, J. L. & McKenzie, A. N. Innate lymphoid cells-how did we miss them. Nat. Rev. Immunol. 13, 75–87 (2013).
Whiteside, S. K., Snook, J. P., Williams, M. A. & Weis, J. J. Bystander T cells: a balancing act of friends and foes. Trends Immunol. 39, 1021–1035 (2018).
Kim J. et al. Innate-like cytotoxic function of bystander-activated CD8(+) T cells is associated with liver injury in acute hepatitis A. Immunity 48, 161–173 (2018).
Simoni Y. et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018).
Christoffersson, G., Chodaczek, G., Ratliff, S. S., Coppieters, K. & von Herrath, M. G. Suppression of diabetes by accumulation of non-islet-specific CD8+ effector T cells in pancreatic islets. Sci. Immunol. 3(21), (2018).
Kim, T. S. & Shin, E. C. The activation of bystander CD8+ T cells and their roles in viral infection. Exp. Mol. Med. 51, 1–9 (2019).
Zhu, J., Yamane, H. & Paul, W. E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28, 445–489 (2010).
Crotty S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 (2011).
Tough, D. F., Borrow, P. & Sprent, J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 272, 1947–1950 (1996).
Tough, D. F., Sun, S. & Sprent, J. T cell stimulation in vivo by lipopolysaccharide (LPS). J. Exp. Med. 185, 2089–2094 (1997).
Eberl, G., Brawand, P. & MacDonald, H. R. Selective bystander proliferation of memory CD4+ and CD8+ T cells upon NK T or T cell activation. J. Immunol. 165, 4305–4311 (2000).
McGinty, J. W. & von Moltke, J. A three course menu for ILC and bystander T cell activation. Curr. Opin. Immunol. 62, 15–21 (2020).
Sims, J. E. & Smith, D. E. The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10, 89–102 (2010).
Chung Y. et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30, 576–587 (2009).
Guo L. et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc. Natl Acad. Sci. U. S. A. 106, 13463–13468 (2009).
Sutton, C., Brereton, C., Keogh, B., Mills, K. H. & Lavelle, E. C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med 203, 1685–1691 (2006).
Lee Y. K. et al. TCR-independent functions of Th17 cells mediated by the synergistic actions of cytokines of the IL-12 and IL-1 families. PLoS One 12, e0186351 (2017).
Lee Y. K. et al. Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107 (2009).
Lee W. W. et al. Regulating human Th17 cells via differential expression of IL-1 receptor. Blood 115, 530–540 (2010).
Lee H. G. et al. Pathogenic function of bystander-activated memory-like CD4+ T cells in autoimmune encephalomyelitis. Nat. Commun. 10, 709 (2019).
Hand T. W. et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science 337, 1553–1556 (2012).
Kang M. J. et al. IL-18 induces emphysema and airway and vascular remodeling via IFN-γ, IL-17A, and IL-13. Am. J. Respir. Crit. Care Med. 185, 1205–1217 (2012).
Chakir, H., Lam, D. K., Lemay, A. M. & Webb, J. R. “Bystander polarization” of CD4+ T cells: activation with high-dose IL-2 renders naive T cells responsive to IL-12 and/or IL-18 in the absence of TCR ligation. Eur. J. Immunol. 33, 1788–1798 (2003).
Munk R. B. et al. Antigen-independent IFN-γ production by human naïve CD4 T cells activated by IL-12 plus IL-18. PLoS One 6, e18553 (2011).
Holmkvist P. et al. A major population of mucosal memory CD4+ T cells, coexpressing IL-18Rα and DR3, display innate lymphocyte functionality. Mucosal Immunol. 8, 545–558 (2015).
Moro K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Xu D. et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J. Exp. Med. 187, 787–794 (1998).
Löhning M. et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc. Natl Acad. Sci. U. S. A. 95, 6930–6935 (1998).
Guo L. et al. Innate immunological function of TH2 cells in vivo. Nat. Immunol. 16, 1051–1059 (2015).
Mato N. et al. Memory-type ST2+CD4+ T cells participate in the steroid-resistant pathology of eosinophilic pneumonia. Sci. Rep. 7, 6805 (2017).
Endo Y. et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity 42, 294–308 (2015).
Morimoto Y. et al. Amphiregulin-producing pathogenic memory T helper 2 cells instruct eosinophils to secrete osteopontin and facilitate airway fibrosis. Immunity 49, 134–150.e6 (2018).
Malek T. R. The biology of interleukin-2. Annu. Rev. Immunol. 26, 453–479 (2008).
Liao, W., Lin, J. X. & Leonard, W. J. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol. 23, 598–604 (2011).
Unutmaz, D., Pileri, P. & Abrignani, S. Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J. Exp. Med. 180, 1159–1164 (1994).
Di Genova, G., Savelyeva, N., Suchacki, A., Thirdborough, S. M. & Stevenson, F. K. Bystander stimulation of activated CD4+ T cells of unrelated specificity following a booster vaccination with tetanus toxoid. Eur. J. Immunol. 40, 976–985 (2010).
Kawabe T. et al. Memory-phenotype CD4+ T cells spontaneously generated under steady-state conditions exert innate TH1-like effector function. Sci. Immunol. 2(12), (2017).
Vignali, D. A. & Kuchroo, V. K. IL-12 family cytokines: immunological playmakers. Nat. Immunol. 13, 722–728 (2012).
Lee Y. et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999 (2012).
Ghoreschi K. et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467, 967–971 (2010).
El-Behi M. et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 12, 568–575 (2011).
Hirota K. et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263 (2011).
McGeachy M. J. et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol. 10, 314–324 (2009).
Yoshida, H. & Hunter, C. A. The immunobiology of interleukin-27. Annu. Rev. Immunol. 33, 417–443 (2015).
Sun, C., Mezzadra, R. & Schumacher, T. N. Regulation and function of the PD-L1 checkpoint. Immunity 48, 434–452 (2018).
Hirahara K. et al. Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity 36, 1017–1030 (2012).
Medzhitov R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1, 135–145 (2001).
Reynolds, J. M. & Dong, C. Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol. 34, 511–519 (2013).
Oliveira-Nascimento, L., Massari, P. & Wetzler, L. M. The role of TLR2 in infection and immunity. Front. Immunol. 3, 79 (2012).
Komai-Koma, M., Jones, L., Ogg, G. S., Xu, D. & Liew, F. Y. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc. Natl Acad. Sci. U. S. A. 101, 3029–3034 (2004).
Ferreira T. B. et al. Different interleukin-17-secreting Toll-like receptor+ T-cell subsets are associated with disease activity in multiple sclerosis. Immunology 154, 239–252 (2018).
Sobek V. et al. Direct Toll-like receptor 2 mediated co-stimulation of T cells in the mouse system as a basis for chronic inflammatory joint disease. Arthritis Res. Ther. 6, R433–R446 (2004).
Reynolds J. M. et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 32, 692–702 (2010).
Imanishi T. et al. Cutting edge: TLR2 directly triggers Th1 effector functions. J. Immunol. 178, 6715–6719 (2007).
Whiteside S. K. et al. IL-10 Deficiency reveals a role for TLR2-dependent bystander activation of T cells in lyme arthritis. J. Immunol. 200, 1457–1470 (2018).
Sinnott, B. D., Park, B., Boer, M. C., Lewinsohn, D. A. & Lancioni, C. L. Direct TLR-2 costimulation unmasks the proinflammatory potential of neonatal CD4+ T cells. J. Immunol. 197, 68–77 (2016).
Gerriets V. A. et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 17, 1459–1466 (2016).
Caron G. et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J. Immunol. 175, 1551–1557 (2005).
Nyirenda M. H. et al. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: a novel mechanism of reduced regulatory T cell function in multiple sclerosis. J. Immunol. 194, 5761–5774 (2015).
Nyirenda M. H. et al. TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J. Immunol. 187, 2278–2290 (2011).
Reynolds, J. M., Martinez, G. J., Chung, Y. & Dong, C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc. Natl Acad. Sci. U. S. A. 109, 13064–13069 (2012).
González-Navajas J. M. et al. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J. Clin. Invest. 120, 570–581 (2010).
Caramalho I. et al. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J. Exp. Med. 197, 403–411 (2003).
Zanin-Zhorov A. et al. Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J. Immunol. 179, 41–44 (2007).
Jenkins M. K. et al. In vivo activation of antigen-specific CD4 T cells. Annu. Rev. Immunol. 19, 23–45 (2001).
Doherty, P. C. & Christensen, J. P. Accessing complexity: the dynamics of virus-specific T cell responses. Annu. Rev. Immunol. 18, 561–592 (2000).
Gangappa, S., Deshpande, S. P. & Rouse, B. T. Bystander activation of CD4(+) T cells can represent an exclusive means of immunopathology in a virus infection. Eur. J. Immunol. 29, 3674–3682 (1999).
Gangappa, S., Deshpande, S. P. & Rouse, B. T. Bystander activation of CD4+ T cells accounts for herpetic ocular lesions. Invest. Ophthalmol. Vis. Sci. 41, 453–459 (2000).
Suwannasaen, D., Romphruk, A., Leelayuwat, C. & Lertmemongkolchai, G. Bystander T cells in human immune responses to dengue antigens. BMC Immunol. 11, 47 (2010).
Polley, R., Zubairi, S. & Kaye, P. M. The fate of heterologous CD4+ T cells during Leishmania donovani infection. Eur. J. Immunol. 35, 498–504 (2005).
Tullo A. Pathogenesis and management of herpes simplex virus keratitis. Eye 17, 919–922 (2003).
Trapp, B. D., Ransohoff, R. & Rudick, R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr. Opin. Neurol. 12, 295–302 (1999).
Ivanov I. I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006).
Harris T. J. et al. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 179, 4313–4317 (2007).
Goverman J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 9, 393–407 (2009).
Korn T. et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 13, 423–431 (2007).
Sabatino, J. J., Shires, J., Altman, J. D., Ford, M. L. & Evavold, B. D. Loss of IFN-gamma enables the expansion of autoreactive CD4+ T cells to induce experimental autoimmune encephalomyelitis by a nonencephalitogenic myelin variant antigen. J. Immunol. 180, 4451–4457 (2008).
Lin C. C. et al. IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. J. Exp. Med. 213, 251–271 (2016).
Hickey, W. F., Hsu, B. L. & Kimura, H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 28, 254–260 (1991).
Ludowyk, P. A., Willenborg, D. O. & Parish, C. R. Selective localisation of neuro-specific T lymphocytes in the central nervous system. J. Neuroimmunol. 37, 237–250 (1992).
Jones, R. E., Kay, T., Keller, T. & Bourdette, D. Nonmyelin-specific T cells accelerate development of central nervous system APC and increase susceptibility to experimental autoimmune encephalomyelitis. J. Immunol. 170, 831–837 (2003).
Lees, J. R., Sim, J. & Russell, J. H. Encephalitogenic T-cells increase numbers of CNS T-cells regardless of antigen specificity by both increasing T-cell entry and preventing egress. J. Neuroimmunol. 220, 10–16 (2010).
Codarri L. et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 12, 560–567 (2011).
Hartmann F. J. et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat. Commun. 5, 5056 (2014).
Lin C. C. et al. Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nat. Commun. 5, 3551 (2014).
Komuczki J. et al. Fate-mapping of GM-CSF expression identifies a discrete subset of inflammation-driving T helper cells regulated by cytokines IL-23 and IL-1β. Immunity 50, 1289–1304.e6 (2019).
Sha, Y. & Markovic-Plese, S. Activated IL-1RI signaling pathway induces Th17 cell differentiation via interferon regulatory factor 4 signaling in patients with relapsing-remitting multiple sclerosis. Front. Immunol. 7, 543 (2016).
Walsh J. T. et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J. Clin. Invest. 125, 2547 (2015).
Joncker N. T. et al. Antigen-independent accumulation of activated effector/memory T lymphocytes into human and murine tumors. Int. J. Cancer 118, 1205–1214 (2006).
Mittal P. et al. Tumor-unrelated CD4 T cell help augments CD134 plus CD137 dual costimulation tumor therapy. J. Immunol. 195, 5816–5826 (2015).
Schenkel, J. M. & Masopust, D. Tissue-resident memory T cells. Immunity 41, 886–897 (2014).
Mueller, S. N. & Mackay, L. K. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol. 16, 79–89 (2016).
Acknowledgements
This research was supported by the Basic Science Research Program (NRF-2019R1A2C3006155) of the National Research Foundation funded by the Korean government to J-.M.C.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, HG., Cho, MJ. & Choi, JM. Bystander CD4+ T cells: crossroads between innate and adaptive immunity. Exp Mol Med 52, 1255–1263 (2020). https://doi.org/10.1038/s12276-020-00486-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-00486-7
This article is cited by
-
Protective effect of low‐intensity pulsed ultrasound on immune checkpoint inhibitor-related myocarditis via fine-tuning CD4+ T-cell differentiation
Cancer Immunology, Immunotherapy (2024)
-
Liver metastasis from colorectal cancer: pathogenetic development, immune landscape of the tumour microenvironment and therapeutic approaches
Journal of Experimental & Clinical Cancer Research (2023)
-
Steady-state memory-phenotype conventional CD4+ T cells exacerbate autoimmune neuroinflammation in a bystander manner via the Bhlhe40/GM-CSF axis
Experimental & Molecular Medicine (2023)
-
Immune Escape Mechanism of Cancer
Current Molecular Biology Reports (2023)
-
Soluble factors from TLR4- or TCR-activated cells contribute to stability of the resting phenotype and increase the expression of CXCR4 of human memory CD4 T cells
Immunologic Research (2023)
