
Abstract
Accumulating evidence has indicated that sirtuins are key components of diverse physiological processes, including metabolism and aging. Sirtuins confer protection from a wide array of metabolic and age-related diseases, such as cancer, cardiovascular and neurodegenerative diseases. Recent studies have also suggested that sirtuins regulate autophagy, a protective cellular process for homeostatic maintenance in response to environmental stresses. Here, we describe various biological and pathophysiological processes regulated by sirtuin-mediated autophagy, focusing on cancer, heart, and liver diseases, as well as stem cell biology. This review also emphasizes key molecular mechanisms by which sirtuins regulate autophagy. Finally, we discuss novel insights into how new therapeutics targeting sirtuin and autophagy may potentially lead to effective strategies to combat aging and aging-related diseases.
Similar content being viewed by others

Introduction
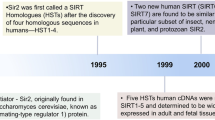
Sirtuins act predominantly as NAD+-dependent deacetylases for a wide range of target proteins, which are crucial for various biological processes. Sir2 (silent information regulator 2) was the first sirtuin to be discovered, and numerous studies have demonstrated that Sir2 is critical for transcriptional silencing in budding yeast1. The increased expression of yeast Sir2 and its invertebrate orthologs delay aging, increase lifespan and may mediate the effects of dietary restriction2.
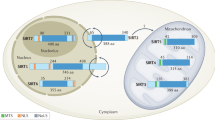
Mammals have seven sirtuin proteins, denoted as SIRT1 through SIRT7, each of which exhibits distinct subcellular localization and rather specific functions. These seven mammalian sirtuins regulate diverse physiological processes ranging from metabolism to epigenetic modification in many tissues3,4,5,6. SIRT1 and SIRT2 are localized to both the nucleus and cytoplasm7. SIRT1, SIRT6, and SIRT7, which are mainly localized to the nucleus, deacetylate intracellular signaling proteins, including histones, and regulate gene expression5,8. SIRT2 can act as a nuclear protein and regulate the cell cycle, although SIRT2 is mainly localized to the cytoplasm5,9,10. SIRT3, SIRT4, and SIRT5 are exclusively localized to the mitochondria and regulate energy metabolism in response to mitochondrial stress4,7,11. For instance, SIRT3 deacetylates various proteins to regulate amino acid metabolism, fatty acid oxidation, the tricarboxylic acid (TCA) cycle, electron transport chain (ETC) activity, mtDNA replication, transcription, and translation. Many target proteins of SIRT4 overlap with proteins that are regulated by SIRT3. SIRT5 specifically interacts with several enzymes crucial for fuel utilization and energy production4. Overall, mitochondrial sirtuins regulate mitochondrial protein networks, orchestrate mitochondrial function, and allow cells to adapt to metabolic stresses. In addition, emerging evidence indicates that sirtuins regulate yet another important cellular process, autophagy.
Autophagy is a process by which damaged proteins and organelles, including damaged mitochondria, endoplasmic reticulum (ER), and peroxisomes, are targeted for lysosomal-mediated degradation. This autophagy-mediated degradation process is useful for removing damaged cellular components and for providing metabolic intermediates necessary for protein synthesis and metabolism. When cells are stressed, autophagosome formation from the phagophore is induced. This de novo formation of autophagosomes likely derives from existing organelles such as the ER or mitochondria12,13. Autophagosomes then elongate, and the double membranous autophagosome encapsulates its damaged cargo, eventually fusing with the lysosome. This cellular process is, as mentioned above, induced by a number of stresses, including nutrient withdrawal, a classic inducer of autophagy. Nonetheless, autophagy is also essential for basal homeostatic maintenance, as shown in animal models in which autophagy is disrupted in a tissue-specific fashion, leading rapidly to the massive accumulation of damaged proteins and organelles within autophagy-deficient tissues14,15.
In this review, we focus on recent progress regarding sirtuin-mediated autophagy and its physiological importance in human biology.
The regulation of autophagy by various sirtuins
The regulation of autophagy by SIRT1
Both autophagy and sirtuins appear to protect cells from environmental stresses, including nutrient stress14,16 (Fig. 1). Essentially, cells induce autophagy in response to nutrient withdrawal. Starvation increases the levels of SIRT1 in mammalian cells, as well as in certain mouse tissues. This SIRT1 transcriptional induction requires the forkhead transcription factor FOXO3a, which forms a complex with p53 in a nutrient-sensitive manner17. In addition, calorie restriction protects mammalian cells from apoptosis18. Calorie restriction increases the level of SIRT118, which can deacetylate targets, including the DNA repair factor Ku70 and p5318. The deacetylation of p53 by SIRT1 suppresses the activity of p53 in essential physiological roles such as apoptosis and induces autophagy19,20 (Fig. 1). Cytoplasmic p53 inhibits autophagy in a transcription-independent manner19,21,22(Fig. 1), but nutrient depletion, a physiological inducer of autophagy, decreases p53 expression by activating the ubiquitin E3 ligase Mdm2 and activates autophagy23. SIRT1 overexpression also decreases p53 expression and protects cells from apoptosis20. However, p53 has dual roles in autophagy depending on its subcellular localization. For example, nuclear p53 induces autophagy21,22,24, and the molecular mechanisms by which p53 regulates autophagy need to be further clarified.

The level of nutrients (e.g., glucose, lipids, and amino acids) is detected by energy sensors, namely, AMPK, SIRT1, or mTOR17,97,104. Calorie restriction activates AMPK or SIRT1 but inhibits mTOR activity. AMPK and SIRT1 upregulate the expression of Atgs through the activation of forkhead box transcription factors (FOXOs)97,105. In addition, SIRT1 also forms a molecular complex with Atg proteins, including Atg7 and Atg8 (LC3). SIRT1 deacetylates the core autophagy machinery in nutrient restricted conditions and thereby increases autophagy. Calorie restriction also potentiates the expression of SIRT1 and increases its deacetylase activity17,18. SIRT1 deacetylates p5318, which downregulates p53 and increases autophagy19,20. However, another well-known negative regulator of autophagy, mTOR, is downregulated under low nutrient conditions
Under cellular stress, including nutrient depletion, SIRT1 activity is increased, autophagy proteins are deacetylated, and subsequently autophagy is induced25 (Fig. 1). While the increase in sirtuin activity is likely multifactorial, the relationship between caloric restriction and a rise in NAD+ levels is clearly one attractive hypothesis that connects nutrient deprivation, sirtuin activation and autophagy induction18,25,26,27. Several autophagy proteins (Atgs), including Atg7 and Atg8, directly interact and are deacetylated by SIRT125. Furthermore, Sirt1-deficient mice display increased basal acetylation of essential autophagic proteins and decreased autophagy function. This defective autophagy is particularly striking given the apparent accumulation of damaged mitochondria in Sirt1-deficient heart tissues25.
Another hallmark of mammalian autophagy is an increase in the number of LC3 (autophagy-related protein 8, Atg8) puncta, as measured by using a fusion protein consisting of green fluorescent protein (GFP) and LC3 (GFP-LC3). This fusion protein is often used as a marker of autophagosome formation. Starvation increases the translocation of GFP-LC3 from the nucleus to the cytoplasm in wild-type mouse embryonic fibroblasts (MEFs), indicating the stimulation of autophagy under nutrient-depleted conditions25. However, in Sirt1-deficient cells, GFP-LC3 puncta formation is highly reduced. The expression of wild-type SIRT1 in Sirt1-deficient MEFs restores autophagy in these cells, while a deacetylase-inactive mutant of SIRT1 is unable to restore autophagic activity25. Thus, the deacetylase activity of SIRT1 is required for the stimulation of autophagy in nutrient-depleted cells. In addition to these loss-of-function experiments, many lines of evidence suggest that the increased expression of SIRT1 is sufficient to induce autophagy by promoting the conversion of LC3-I to LC3-II through proteolytic cleavage and lipidation25.
How does SIRT1 regulate autophagy? SIRT1 forms molecular complexes with essential components of the autophagy machinery, including Atg5, Atg7 and LC3. SIRT1 deacetylates these proteins in cultured cells and neonatal tissues25. As mentioned above, the genetic inhibition of SIRT1 markedly increases the acetylation of endogenous Atg7 in various tissues, including the heart, brain and liver. The level of Atg7 acetylation is also elevated by treatment with nicotinamide, a pharmacological sirtuin inhibitor. Moreover, purified wild-type SIRT1, but not a catalytically inactive point mutant, can deacetylate Atg7 in an NAD-dependent manner in vitro25.
These initial studies have established that starvation activates SIRT1, which in turn deacetylates the autophagic components to augment autophagy. Subsequent studies have delineated additional aspects of this process. In particular, the deacetylation of nuclear LC3 drives autophagy initiation under starvation28. Upon nutrient depletion, SIRT1 deacetylates nuclear LC3 at lysine residues K49 and K51, which leads to the interaction between LC3 and the nuclear protein DOR (diabetes- and obesity-regulated nuclear factor), which is required for the translocation of both LC3 and DOR to the cytoplasm. LC3 then binds to Atg7 and other autophagy factors and subsequently undergoes phosphatidylethanolamine conjugation to form a preautophagic membrane structure28.
SIRT1-mediated autophagy is inhibited by REGγ, a proteasome activator, which regulates the stability of SIRT1 under starvation conditions29. Under normal nutrient conditions, REGγ mediates the ubiquitin-independent degradation of SIRT1, which inhibits autophagy by preventing SIRT1 from binding and deacetylating the autophagy complex components. Thus, REGγ deficiency causes dramatic autophagy induction. Similarly, REGγ dissociates from SIRT1 in response to energy depletion (GD, glucose deprivation), and this contributes to the induction of autophagy because SIRT1 can then interact with the autophagic machinery. GD reduces the interaction between REGγ and SIRT1, at least in part through the phosphorylation of SIRT1 at a specific threonine residue (T530). Evidence suggests that this phosphorylation is mediated by AMP-activated protein kinase (AMPK)29. Treatment with compound C, an AMPK inhibitor, reduces GD-induced SIRT1 phosphorylation and inhibits the dissociation of SIRT1 from REGγ. Conversely, treatment with AICAR, an AMPK activator, decreases the interaction between REGγ and SIRT1 and in turn activates autophagy29.
The regulation of autophagy by SIRT2
Recent reports have suggested that SIRT2 inhibits autophagy under basal conditions30,31. Specifically, SIRT2 binds and deacetylates FOXO1 under basal conditions. However, FOXO1 is highly acetylated when autophagy is induced in response to serum starvation or oxidative stress. This is because cytosolic FOXO1-SIRT2 complexes dissociate in response to oxidative stress or serum starvation, leading to an increase in the acetylation of FOXO1. Under SIRT2 deficiency, acetylated FOXO1 interacts with Atg7, which appears to be required for the induction of autophagy30. These observations stand in contrast to the results described for SIRT1, for which the levels of Atg protein acetylation under SIRT1 deficiency25 are negatively correlated with autophagic flux25,32.
In addition, other observations have suggested that SIRT2 regulates mitochondrial protein deacetylation, mitophagy, and mitochondrial function33,34,35. The depletion of Sirt2 results in an increase in the acetylation of mitochondrial proteins, morphological changes to mitochondria, increases in oxidative stress and decreases in ATP production. Dysfunctional mitochondria need to be cleared by mitophagy, a selective type of autophagy that specifically removes damaged mitochondria. The loss of Sirt2 appears to cause defects in mitophagy, as increased levels of p62, PINK1/Parkin, and ubiquitin, are detected in the brains of mice that lack Sirt2. In addition, autophagy is impaired in Sirt2-deficient MEFs in response to external stimuli. Moreover, the chemical uncoupler carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) induces mitophagy in wild-type MEFs but not in Sirt2-deficient MEFs33.
The regulation of autophagy by SIRT3
SIRT3 is a mitochondrial sirtuin and is responsible for the deacetylation of various mitochondrial proteins, thereby regulating ATP generation, beta oxidation, and urea cycle activity36,37. SIRT3 also regulates hepatic autophagy in a manner that is distinct from that of SIRT136. In particular, SIRT3 acts as a negative regulator of autophagy with respect to lipid overload. Indeed, the levels of SIRT3 correlate with the susceptibility of hepatocytes to lipotoxicity induced by saturated fatty acids (SFAs). This finding is of particular interest because these conditions are thought to contribute to nonalcoholic fatty liver disease (NAFLD)38. Hepatic SIRT3 expression is significantly elevated in SFA-rich (palm oil) high-fat diet (HFD) conditions. SIRT3 overexpression strongly inhibits hepatic autophagy by downregulating AMPK via MnSOD, a known SIRT3 target. In addition, SIRT3 overexpression activates mammalian target of rapamycin complex 1 (mTORC1), which inhibits autophagy. The increased expression of SIRT3 promotes the susceptibility of palmitic acid/palm oil-induced hepatic injury. In contrast, SIRT3 deficiency induces autophagy and protects hepatocytes from lipotoxicity38.
Curiously, in other settings, SIRT3 can also act as a positive regulator of autophagy and mitophagy37,39,40,41. SIRT3 activates essential regulators of mitochondrial function, including peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α) and AMPK36,37. For instance, SIRT3 appears to protect neurons from ischemia, at least in part by inducing autophagy. SIRT3 decreases neuronal apoptosis in primary cultures of cortical neurons exposed to oxygen glucose deprivation (OGD) to mimic neuronal ischemia. SIRT3 increases AMPK activity and potentiates the induction of autophagy in OGD-treated neuronal cells. In addition, the overexpression of SIRT3 partially rescues mitochondrial function, including the generation of ATP and the mitochondrial membrane potential39. SIRT3-mediated mitophagy has also been demonstrated to protect tumor cells from apoptosis under hypoxic conditions40.
The regulation of autophagy by SIRT6
SIRT6, a predominantly nuclear protein, deacetylates various proteins, including the histones H3K9 and H3K5642. Recent reports have demonstrated a protective role for SIRT6 against cardiovascular diseases43,44,45. SIRT6 reduces the formation of foam cells associated with early atherosclerosis (AS) in an autophagy-dependent manner43. The oxidized form of low density lipoprotein-cholesterol (ox-LDL) plays a role in the initiation and progression of AS46. Human monocytic THP1 cells treated with ox-LDL display increased lipid droplets in the cytoplasm. Treatment with ox-LDL reduces the level of SIRT6, which results in the inhibition of autophagy and decreases the levels of LC3 and Atg5. Foam cell formation induced by ox-LDL is ameliorated by the overexpression of wild-type SIRT6 but not by the expression of a catalytically inactive SIRT6 mutant (SIRT6 H133Y). In addition, SIRT6 overexpression preserves autophagic flux in THP1 cells treated with ox-LDL. This occurs in part because SIRT6 overexpression decreases the expression of miR-33, which increases autophagy and the expression of ABCA1 and ABCG1. This upregulation of ABCA1 and ABCG promotes cholesterol efflux and prevents the formation of macrophage foam cells43. As such, SIRT6-mediated autophagy plays a protective role in preventing atherosclerosis.
SIRT6 also suppresses isoproterenol (ISO)-induced cardiac hypertrophy through the activation of autophagy. ISO reduces autophagy and induces cardiac hypertrophy in primary neonatal rat cardiomyocytes (NRCMs) and Sprague-Dawley (SD) rats. SIRT6-mediated autophagy attenuates ISO-stimulated cardiomyocyte hypertrophy. SIRT6 also regulates cardiac autophagy in a FOXO3-dependent manner through the suppression of Akt signaling44. Moreover, autophagy induction by SIRT6 decreases the senescence of human bronchial epithelial cells (HBECs) induced by cigarette smoke extract (CSE). These effects appear to result from a SIRT6-dependent attenuation of the insulin-like growth factor-Akt-mTOR signaling axis47.
The role of sirtuin-induced autophagy in diseases
The role of sirtuin-mediated autophagy in cancer biology
Sirtuin-mediated autophagy is a potential therapeutic target for the treatment of cancers (Table 1). The regulation of autophagy by SIRT1 seems to play an important role in the development of gastric cancer. The high expression of SIRT1 and Beclin-1 (Atg6) is associated with shorter overall survival and reduced relapse-free survival in gastric cancer patients. In tumor gastric tumor samples, over 80% of malignant cells demonstrate increased autophagosome levels when compared to those of cells from adjacent normal tissue. Thus, the expression levels of SIRT1 and/or Beclin-1 are potential prognostic indicators for gastric cancer and are potential therapeutic targets for the disease48,49 (Table 1).
Interestingly, sirtuin-dependent autophagy can also play a role in tumor suppression. As noted, SIRT2 deacetylates the transcription factor FOXO1, which regulates autophagy in human cancer cell lines in response to oxidative stress or serum starvation. In response to cellular stresses, the acetylation of FOXO1 is increased by dissociation from SIRT2. Furthermore, acetylated FOXO1 interacts with Atg7 to potentiate autophagic cell death. This FOXO1-dependent cell death appears to act as an important tumor suppressor mechanism in a human colon xenograft model30 (Table 1). In addition, SIRT2 suppresses basal autophagy, leading to mitotic catastrophe, an essential cell death mechanism that kills cancer cells in the setting of microtubule inhibitors31 (Table 1).
SIRT3 also mediates autophagy, including mitophagy, in response to hypoxia. Hypoxia increases the interaction between VDAC and Parkin. In cells subjected to hypoxia, SIRT3 deficiency results in reduced mitophagy, leading to further decreases in the mitochondrial membrane potential. This event subsequently increases the generation of reactive oxygen species (ROS), stimulating the degradation of the anti-apoptotic proteins Mcl-1 and survivin. Thus, the inhibition of SIRT3 appears to promote apoptosis and enhances the sensitivity of cancer cells to hypoxia40 (Table 1).
Dysfunctional mitophagy and autophagy disrupt mitochondrial homeostasis. Mitochondrial dysfunction also causes metabolic reprogramming in response to environmental stresses. In addition, these bioenergetic changes associated with mitochondrial dysfunction alter stemness and cell fate decisions, which in turn impacts diseases such as cancer50.
The role of sirtuin-mediated autophagy in kidney diseases
SIRT1-dependent autophagic induction in response to environmental stress is essential for the maintenance of renal function. Dysfunctional autophagy in the kidneys, including in cells such as podocytes, mesangial cells, endothelial cells, and tubular cells, contributes to the pathogenesis of diabetic nephropathy51,52,53 (Table 1). SIRT1, a positive regulator of autophagy, is also downregulated in diabetic kidneys54. In addition, numerous studies have suggested that SIRT1 protects against oxidative stress, reduces fibrosis and inflammation, and inhibits apoptosis in the kidneys54,55,56 (Table 1).
Protein acetylation plays a crucial role in the regulation of autophagy and can potentially be exploited as a potential new therapeutic target for the treatment of diabetic nephropathy52,53,56,57,58. In addition, hypoxia-mediated mitochondrial and renal damage in aged kidneys are prevented in a SIRT1-dependent autophagic process59. As noted, calorie restriction (CR) increases the level and activity of SIRT117 and induces autophagy. Adult-onset and long-term CR in mice elevates SIRT1 expression in aged kidneys and attenuates hypoxia-associated mitochondrial and renal damage, at least in part by enhancing BCL2/adenovirus E1B 19-kDa-interacting protein 3 (Bnip3)-dependent autophagy. The deacetylation of FOXO3 by SIRT1 in CR increases the expression of Bnip-3 as well as that of p27, which results in increased autophagy and improved survival of primary renal proximal tubular cells (PTCs) under hypoxic conditions59.
The role of sirtuin-mediated autophagy in neuronal diseases
Sirtuin-induced autophagy also has protective roles in the brain. Mammalian SIRT1 maintains brain integrity and protects against various neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease60,61,62,63 (Table 1). SIRT1 promotes the growth and survival of neurons in the central nervous system by negatively regulating mTOR signaling, which in turn is a negative regulator of autophagy64. SIRT1 overexpression promotes neurite outgrowth in primary neurons and potentiates cell viability under nutrient stress conditions64. SIRT1-overexpressing neurons display increased tolerance to the accumulation of neurotoxic molecules such as β-amyloid (Aβ). SIRT1 overexpression decreases mTOR signaling by reducing the phosphorylation of mTOR at S2448 and the phosphorylation of p70S6K kinase at T289. In line with such findings, Sirt1-deficient mouse primary neurons display increased mTOR activity, which results in impaired cell survival and neurite outgrowth64. Consistent with these observations, the mTOR inhibitor rapamycin markedly improves neuronal cell survival in response to nutrient deprivation and significantly enhances neurite outgrowth in wild-type mouse neurons64 (Table 1).
SIRT1 also regulates the transcriptional activities as well as the acetylation status of essential neuroprotective proteins such as p53, nuclear factor kappa B, peroxisome proliferator-activated receptor γ (PPARγ), PPARγ coactivator-1α (PGC-1α), liver X receptor, and forkhead box O (FoxO)61,62,65. For instance, the overexpression of SIRT1 in the hippocampus of p25 transgenic mice, which exhibit massive degeneration of the forebrain with features of Alzheimer’s disease, significantly protects the mouse brain from neurodegeneration61. In addition, the treatment of these mice with resveratrol, an established SIRT1 activator, attenuates neurodegeneration in the hippocampus and prevents learning impairment by decreasing the acetylation of PGC-1α and p5361,66 (Table 1). The activation of SIRT1 in neurons by treatment with resveratrol deacetylates p53, and this reduces p53- and FOXO-dependent apoptosis. In addition, SIRT1 inhibits NF-κB signaling in microglia and astrocytes and protects AD neurons against Aβ-induced toxicity62. In Huntington’s disease, the mutant huntingtin protein inhibits the expression of PGC-1α, leading to the impairment of mitochondrial function. SIRT1 activates PGC-1α via deacetylation, which can act to reduce HD-associated mitochondrial impairment62,67,68 (Table 1).
The connection between sirtuins and autophagy might also be important for Parkinson’s disease. Resveratrol, a SIRT1 activator, induces autophagy and potentiates the degradation of α-synucleins in α-synuclein-expressing PC12 cell lines. The inhibition of AMPK decreases SIRT1 activity and reduces the beneficial effects of resveratrol on rotenone-induced apoptosis. Thus, AMPK and SIRT1 are required for resveratrol-mediated autophagy induction and for augmenting neuronal survival. In 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-treated mice (a model of Parkinson’s disease), resveratrol increases the activity of SIRT1 and thereby enhances the autophagic degradation of α-synuclein. In these MPTP-treated mice, resveratrol increases the survival of dopaminergic neurons, augments dopamine levels, and ameliorates behavioral impairments69. In simple model organisms such as yeast, the regulation of Atg32 by Sir2 prevents α-synuclein-induced toxicity during aging70,71,72. In addition, the regulation of autophagy by SIRT1 plays a protective role against prion-induced neuronal cell death and related neurodegenerative disorders73 (Table 1).
In sporadic Parkinson’s disease, unusual microtubule-dependent transport, mitochondrial dysfunction, and impaired autophagy have been reported. Changes in NAD+ metabolism in cells derived from sporadic Parkinson’s disease patients activate SIRT2, which in turn deacetylates α-tubulin, a SIRT2 target. The pharmacological or genetic inhibition of SIRT2 increases the level of acetylated α-tubulins. SIRT2 deficiency facilitates trafficking by preventing microtubule network disruption and clearing misfolded proteins or protein aggregates74,75,76. Therefore, the negative regulation of autophagy by SIRT2 has the potential to be a therapeutic target for neurodegenerative diseases, including sporadic Parkinson’s disease75,77,78 (Table 1).
SIRT3 induces autophagy during neuronal ischemia. SIRT3 regulates mitochondrial energy metabolism, including ATP generation, and has protective roles in neuronal survival following oxidative stress. SIRT3 is upregulated during oxygen and glucose deprivation (OGD), a neuronal ischemia mimetic. The ectopic expression of SIRT3 markedly reduces OGD-induced lactate dehydrogenase release and neuronal apoptosis. These cells also display a decrease in hydrogen peroxide (H2O2) production, increases in ATP generation, and the maintenance of mitochondrial membrane potential (MMP). The overexpression of SIRT3 in cortical neurons dramatically increases the phosphorylation of AMPK and the subsequent induction of autophagy. Moreover, phosphorylated mTOR levels are decreased, providing another stimulus for autophagic induction. This suggests that SIRT3-induced autophagy is regulated by an AMPK-mTOR signaling axis, which represents a potential therapeutic target for neuronal protection39 (Table 1).
The role of sirtuin-mediated autophagy in heart diseases
The regulation of autophagy by the sirtuin family appears to play a beneficial role in protection against cardiovascular diseases (Table 1). For instance, SIRT1 deficiency increases oxidative stress, inflammation, and foam cell formation and augments the senescence of endothelial cells, vascular smooth muscle cells, and monocytes/macrophages. Autophagy deficiency also contributes to vascular aging and atherosclerosis79 (Table 1). The overexpression of SIRT1 protects mouse heart tissues against ischemia-reperfusion (I/R) injury. In these mice, autophagy is stimulated, FOXO3 signaling is activated, nitric oxide synthase is hyperphosphorylated, and NF-kB is deacetylated80 (Table 1).
SIRT3-dependent autophagy also appears to play a role in cardiac hypertrophy. The induction of autophagy as well as the deacetylation of FOXO1 by SIRT3 reduces myocardial hypertrophy following treatment with angiotensin II. For instance, the overexpression of SIRT3 in the cardiomyocyte H9C2 cell line increases autophagy and attenuates cardiomyocyte hypertrophy induced by angiotensin II81. However, Sirt3 knockout mice display impaired autophagy and increased angiotensin II-induced myocardial hypertrophy81 (Table 1).
SIRT3 also has a pivotal cardioprotective role in the setting of diabetic cardiomyopathy in part by activating Parkin-mediated mitophagy82 (Table 1). Streptozotocin (STZ) exposure causes diabetes mellitus-induced myocardial damage and interstitial fibrosis in mice. In addition, STZ-treated mice display increased apoptotic cardiomyocyte death and mitochondrial injury with increases in the number of abnormally shaped mitochondria. The effect of STZ is more pronounced in Sirt3-deficient mice than in wild-type mice. In Sirt3 knockout mice, deacetylated FOXO3A levels and Parkin expression levels are decreased. SIRT3 overexpression in neonatal mouse cardiomyocytes increases autophagy and protects against mitochondrial injury and cardiomyocyte apoptosis. As such, a SIRT3-FOXO3A-Parkin-mediated mitophagy pathway appears to play key protective roles in moderating diabetic cardiomyopathy82 (Table 1).
SIRT6 is another important mammalian sirtuin protein that appears to protect against cardiac hypertrophy. Cardiac hypertrophy induced by isoproterenol significantly decreases autophagy, which is regulated in part by SIRT644. SIRT6 overexpression increases autophagy in primary neonatal rat cardiomyocytes and in rat hearts. Thus, the cardioprotective effect of SIRT6 in an isoproterenol (ISO)-stimulated cardiac hypertrophy model appears to be dependent on the induction of autophagy. SIRT6 dramatically increases the nuclear accumulation of the FOXO3 transcription factor possibly via attenuating Akt signaling. The positive regulation of autophagy by SIRT6 in isolated cardiomyocytes and intact rat hearts protects against ISO-induced cardiac hypertrophy44 (Table 1).
The role of sirtuin-mediated autophagy in liver diseases
Autophagy regulation by SIRT1 contributes to protecting against liver damage by maintaining basic hepatic function83,84(Table 1). Mortality after liver surgery is largely dependent on ischemia/reperfusion (I/R) injury. After I/R injury in the liver, SIRT1 expression is highly reduced. The ectopic expression or increased activity of SIRT1 dramatically restores defective autophagy, reduces the onset of the mitochondrial permeability transition, and mitigates hepatocyte death after I/R injury. Mechanistically, SIRT1 interacts with mitofusin-2, a target of the SIRT1 deacetylase. Furthermore, SIRT1 overexpression moderately induces autophagy in wild-type cells but not in MFN2-deficient cells. Thus, Sirt1 deficiency results in a series of sequential events, including defective autophagy, mitochondrial dysfunction, and hepatocyte death after I/R84 injury (Table 1).
The physiological roles of sirtuin-induced autophagy
The effect of sirtuin-induced autophagy on lifespan
Sirtuins and autophagy generally have lifespan-extending effects6,85,86. Dietary-restricted model organisms, such as yeast, worms, flies, and mice, live longer due to an increase in SIRT1 activity as well as the induction of autophagy. In addition, the overexpression of SIRT1 or specific autophagy genes in model organisms, including mice, and the administration of autophagy-activating or sirtuin-regulatory small molecules such as rapamycin, resveratrol, or spermidine increase lifespan87,88,89.
Mitophagy contributes to a wide range of physiological processes, including aging and lifespan. Patients with ataxia telangiectasia have defects in ATM kinase, a master regulator of DNA damage and a facilitator of DNA double-strand break repair. In mice and worms, ATM-deficient neurons display significantly increased PARylation, low NAD+ levels, and mitochondrial dysfunction. Augmenting intracellular NAD+ levels reduces the severity of A–T neuropathology, normalizes neuromuscular function, delays memory loss, and extends lifespan in both worms and mice. Increased intracellular NAD+ levels also lead to neuronal DNA repair and improve mitochondrial quality via mitophagy90, although the precise role of NAD-dependent sirtuins in this process is incompletely understood.
The effect of sirtuin-induced autophagy on inflammation and senescence
SIRT1-dependent autophagy blocks endothelial inflammation. Resveratrol, a SIRT1 activator, alleviates tumor necrosis factor (TNF)-induced inflammation by increasing autophagy in human umbilical vein endothelial cells (HUVECs)91. The genetic inhibition of autophagy using siRNAs targeting ATG5 or BECN1 suppresses the downregulation of inflammatory factors, including ICAM1, PTGS2, and MMP9, through treatment with resveratrol. Again, autophagy is required for the suppression of TNF-induced inflammation in a SIRT1-dependent manner and protects vascular endothelial cells against inflammation. Resveratrol also increases cyclic AMP content and the expression of PKA and SIRT1 as well as the activity of AMPK. Overall, resveratrol reduces endothelial inflammation by inducing autophagy, which is regulated by the activation of a cAMP-PKA-AMPK-SIRT1 signaling pathway91.
The activation of autophagy by sirtuins also attenuates cellular senescence. The expression levels of SIRT6 are decreased in lung homogenates from chronic obstructive pulmonary disease patients47. Cigarette smoke extract (CSE) reduces SIRT6 expression in human bronchial epithelial cells (HBECs). The CSE-induced senescence of human bronchial epithelial cells is inhibited by SIRT6 overexpression, and SIRT6 knockdown and deacetylase-defective mutant SIRT6 (H133Y) enhance the senescence of HBECs. SIRT6 overexpression induces autophagy via the attenuation of IGF-Akt-mTOR signaling. SIRT6-induced autophagy prevents senescence by CSE47. SIRT6 is involved in CSE-induced HBEC senescence via autophagy regulation, which contributes to the attenuation of IGF-Akt-mTOR signaling. In addition, the negative regulation of SIRT1-dependent autophagy, which is targeted by miR-212, augments cellular senescence92.
The effect of sirtuin-induced autophagy on stem cell functions
Recent reports have indicated that SIRT1 and autophagy regulate stem cell fate decisions and protect stem cells from diverse stresses, such as hypoxia, serum deprivation, and oxidative stress93,94,95. Oxidative stress further increases the level of apoptosis in Sirt1−/− murine embryonic stem cells (ESCs) compared to that in wild-type cells. In both Sirt1-deficient murine ESCs and SIRT1 knockdown human ESCs, autophagy is markedly decreased in response to oxidative stress, as measured by the decreased conversion of LC3-I to LC-II, a low level of Beclin-1 protein, and reduced LC3 puncta. In addition, the class III PI3K/Beclin-1 and mTOR pathways are partially required for the protective roles of SIRT1-dependent autophagy in ESCs under oxidative stress conditions95.
Sirt1 deficiency also causes impaired mitochondrial dynamics in ESCs. The generation of intracellular ROS by high concentrations of exogenous H2O2, in combination with the autophagy inhibitor 3-methyladenine (3-MA), further increases apoptosis in Sirt1−/− murine ESCs compared with wild-type ESCs. Cellular oxidative stress leads to the disruption of the mitochondrial membrane potential. Treatment with H2O2, in conjunction with 3-MA treatment, results in mitochondrial depolarization in wild-type cells. However, the combinatorial treatment of 3-MA with H2O2 in Sirt1−/− cells does not cause an increased loss of membrane potential because Sirt1−/− leads to impaired autophagy. Thus, autophagy appears to regulate the mitochondrial membrane potential in response to H2O2 in a Sirt1-dependent manner95.
Concluding remarks and future directions
Although the mechanisms remain incompletely understood, as outlined above, the functional links between autophagy and sirtuins play essential roles in diverse cellular processes, including aging and various age-related diseases. Both sirtuins and autophagy function as sensors and effectors of nutrient depletion and appear to integrate this information into a coordinated response to maintain cellular homeostasis. The question therefore arises as to why this relationship between the sirtuin family and autophagy evolved? While no definitive answer for this question exists, it is tempting to speculate that autophagy arose, in part, to allow organisms to resist metabolic stresses, including starvation. After all, starvation is a classic stimulus of autophagic induction. Nonetheless, the enzymatic core machinery of autophagy (i.e., Atg proteins such as Atg5, Atg7, etc.) has little intrinsic capacity to monitor metabolic conditions. In contrast, sirtuins, the enzymatic activity of which is dependent on the NAD/NADH ratio and the enzymatic reaction of which involves acetyl-CoA, are perfectly positioned to act as energy rheostats. Under nutrient limiting conditions, NAD+ levels rise26,27, activating sirtuin proteins, which in turn can deacetylate the autophagy machinery. The observation that the autophagy machinery is highly acetylated under basal conditions might reflect high acetyl-CoA conditions, which occur when nutrients are available. Under this scenario, starvation induces sirtuin activity in an NAD+-dependent manner, triggering the deacetylation of the core autophagy machinery (Fig. 2). This deacetylation results in two independent events that help the organism withstand nutrient stress, namely, unlocking the enzymatic activity of the autophagy machinery and liberating acetyl-CoA. Although speculative, this scenario is consistent with the growing appreciation that cytosolic acetyl-CoA levels play an important role in autophagy96.

Nutrient limitation increases the level of the essential cellular metabolite NAD+, which activates sirtuins by inducing autophagy within cells and tissues. In contrast, the activity of sirtuins and autophagy decreases during normal aging, as well as due to some diseases26, and correlates with the known reduction in levels of tissue NAD+
The link between sirtuins and autophagy may explain one other important aspect of regulation that was intimated in this review. It is generally believed that autophagic flux slows down as organisms age86,97,98. Recent evidence has suggested that this is also true for more specialized forms of autophagy, such as mitophagy99. The molecular basis for this phenomenon is not understood. Again, it is tempting to hypothesize that the well-known age-dependent decline in NAD+ levels might provide a basis for this effect. Under this scenario, a primary fall in tissue levels of NAD+ would limit sirtuin activity and thereby limit autophagic flux (Fig. 2). Given the growing interest in NAD biology6,26,27,100, it will be important in the future to determine whether NAD supplementation exerts its beneficial effects through sirtuin-dependent autophagy induction. In addition, since direct sirtuin-activating compounds (STACs), including resveratrol and SRT2104, and indirect activators, such as nicotinamide riboside, are currently in clinical trials for the treatment of cardiovascular diseases, cancer, diabetes, and neuropathy26,101, it is critical to more fully understand how these agents work. Again, it is tempting to hypothesize that their clinical benefits might result from the link between sirtuin activity and autophagic flux. Finally, the small molecule activation of sirtuins and hence the indirect activation of autophagy might be advantageous over the direct and constitutive activation of autophagy. This supposition is based on the growing literature that autophagy might provide a survival advantage for tumor cells102,103. In such a scenario, there may be safety concerns for the long-term administration of direct autophagy inducers. In contrast, therapeutic strategies that exploit the interaction between sirtuins and the autophagy machinery might provide for a more regulated and physiological approach. Undoubtedly, a fuller understanding of the cross-regulation between sirtuins and autophagy will likely provide important insights and potentially novel therapeutic avenues to combat a host of age-related diseases, as well as potentially a strategy to slow aging itself.
References
Imai, S.-I., Armstrong, C. M., Kaeberlein, M. & Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000).
Sinclair, D. A. & Guarente, L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell 91, 1033–1042 (1997).
Finkel, T., Deng, C.-X. & Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 460, 587–591 (2009).
van de Ven, R. A., Santos, D. & Haigis, M. C. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol. Med. 23, 320–331 (2017).
Chang, H.-C. & Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrin Met. 25, 138–145 (2014).
Imai, S.-i & Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2, 16017 (2016).
Choi, J.-E. & Mostoslavsky, R. Sirtuins, metabolism, and DNA repair. Curr. Opin. Genet Dev. 26, 24–32 (2014).
Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 27, 2072–2085 (2013).
Dryden, S. C., Nahhas, F. A., Nowak, J. E., Goustin, A.-S. & Tainsky, M. A. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell Biol. 23, 3173–3185 (2003).
Serrano, L. et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 27, 639–653 (2013).
Yang, W. et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 167, 985–1000. e1021 (2016).
Antonioli, M., Di Rienzo, M., Piacentini, M. & Fimia, G. M. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 42, 28–41 (2017).
Hurley, J. H. & Young, L. N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 86, 225–244 (2017).
Kroemer, G., Mariño, G. & Levine, B. Autophagy and the integrated stress response. Mol. Cell 40, 280–293 (2010).
Boya, P., Reggiori, F. & Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 15, 713–720 (2013).
Kim, J. et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303 (2013).
Nemoto, S., Fergusson, M. M. & Finkel, T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306, 2105–2108 (2004).
Cohen, H. Y. et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392 (2004).
Green, D. R. & Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 458, 1127–1130 (2009).
Liu, T. et al. SIRT1 reverses senescence via enhancing autophagy and attenuates oxidative stress-induced apoptosis through promoting p53 degradation. Int J. Biol. Macromol. 117, 225–234 (2018).
Maiuri, M. C. et al. Autophagy regulation by p53. Curr. Opin. Cell Biol. 22, 181–185 (2010).
Tang, J., Di, J., Cao, H., Bai, J. & Zheng, J. p53-mediated autophagic regulation: a prospective strategy for cancer therapy. Cancer Lett. 363, 101–107 (2015).
Tasdemir, E. et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 10, 676–687 (2008).
White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med 6, a026120 (2016).
Lee, I. H. et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl Acad. Sci. USA 105, 3374–3379 (2008).
Bonkowski, M. S. & Sinclair, D. A. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 17, 679–690 (2016).
Canto, C., Menzies, K. J. & Auwerx, J. NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53 (2015).
Huang, R. et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell 57, 456–466 (2015).
Dong, S. et al. The REGγ proteasome regulates hepatic lipid metabolism through inhibition of autophagy. Cell Meta. 18, 380–391 (2013).
Zhao, Y. et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell biol. 12, 665–675 (2010).
Inoue, T. et al. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J. 281, 2623–2637 (2014).
Lee, I. H. & Finkel, T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 284, 6322–6328 (2009).
Liu, G. et al. Loss of NAD-dependent protein deacetylase sirtuin-2 alters mitochondrial protein acetylation and dysregulates mitophagy. Antioxid. Redox Signal. 26, 849–863 (2017).
Lemos, V. et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum. Mol. Genet. 26, 4105–4117 (2017).
Silva, D., Esteves, A., Oliveira, C. & Cardoso, S. Mitochondrial metabolism power SIRT2-dependent deficient traffic causing Alzheimer’s-disease related pathology. Mol. Neurobiol. 54, 4021–4040 (2017).
Cho, C. S., Lombard, D. B. & Lee, J. H. SIRT3 as a regulator of hepatic autophagy. Hepatology 66, 700–702 (2017).
Lombard, D. B. & Zwaans, B. M. SIRT3: as simple as it seems? Gerontology 60, 56–64 (2014).
Li, S. et al. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 66, 936–952 (2017).
Dai, S.-H. et al. Sirt3 confers protection against neuronal ischemia by inducing autophagy: involvement of the AMPK-mTOR pathway. Free Radic. Biol. Med. 108, 345–353 (2017).
Qiao, A. et al. Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget 7, 43390–43400 (2016).
Duan, W.-J. et al. A SIRT3/AMPK/autophagy network orchestrates the protective effects of trans-resveratrol in stressed peritoneal macrophages and RAW 264.7 macrophages. Free Radic. Biol. Med. 95, 230–242 (2016).
Sosnowska, B. et al. The sirtuin family members SIRT1, SIRT3 and SIRT6: their role in vascular biology and atherogenesis. Atherosclerosis 265, 275–282 (2017).
He, J. et al. SIRT 6 reduces macrophage foam cell formation by inducing autophagy and cholesterol efflux under ox‐LDL condition. FEBS J. 284, 1324–1337 (2017).
Lu, J. et al. SIRT6 suppresses isoproterenol-induced cardiac hypertrophy through activation of autophagy. Trans. Res. 172, 96–112. e116 (2016).
D’Onofrio, N., Servillo, L. & Balestrieri, M. L. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid. Redox Signal. 28, 711–732 (2018).
Mitra, S., Deshmukh, A., Sachdeva, R., Lu, J. & Mehta, J. L. Oxidized low-density lipoprotein and atherosclerosis implications in antioxidant therapy. Am. J. Med. Sci. 342, 135–142 (2011).
Takasaka, N. et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 192, 958–968 (2014).
Qiu, G. et al. The prognostic role of SIRT1-autophagy axis in gastric cancer. Dis. Markers 2016, 1–11 (2016).
Qiu, G. et al. SIRT1 is a regulator of autophagy: implications in gastric cancer progression and treatment. FEBS Lett. 589, 2034–2042 (2015).
Drake, L. E., Springer, M. Z., Poole, L. P., Kim, C. J. & Macleod, K. F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 47, 110–124 (2017).
Kitada, M., Ogura, Y., Monno, I. & Koya, D. Regulating autophagy as a therapeutic target for diabetic nephropathy. Curr. Diab Rep. 17, 53 (2017).
Kume, S., Thomas, M. C. & Koya, D. Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes 61, 23–29 (2012).
Lenoir, O., Tharaux, P.-L. & Huber, T. B. Autophagy in kidney disease and aging: lessons from rodent models. Kidney Int. 90, 950–964 (2016).
Kong, L. et al. Sirtuin 1: a target for kidney diseases. Mol. Med. 21, 87–97 (2015).
Polak-Jonkisz, D. et al. Nephroprotective action of sirtuin 1 (SIRT1). J. Physiol. Biochem. 69, 957–961 (2013).
Kitada, M., Kume, S., Takeda-Watanabe, A., Kanasaki, K. & Koya, D. Sirtuins and renal diseases: relationship with aging and diabetic nephropathy. Clin. Sci. 124, 153–164 (2013).
Khan, S., Bhat, Z. R. & Jena, G. Role of autophagy and histone deacetylases in diabetic nephropathy: current status and future perspectives. Genes Dis. 3, 211–219 (2016).
Kume, S., Koya, D., Uzu, T. & Maegawa, H. Role of nutrient-sensing signals in the pathogenesis of diabetic nephropathy. Biomed. Res Int. 2014, 1–9 (2014).
Kume, S. et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Invest. 120, 1043–1055 (2010).
Tang, B. L. Sirtuins as modifiers of Parkinson’s disease pathology. J. Neurosci. Res. 95, 930–942 (2017).
Donmez, G. & Outeiro, T. F. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol. Med. 5, 344–352 (2013).
Paraíso, A. F., Mendes, K. L. & Santos, S. H. S. Brain activation of SIRT1: role in neuropathology. Mol. Neurobiol. 48, 681–689 (2013).
Zhang, A., Wang, H., Qin, X., Pang, S. & Yan, B. Genetic analysis of SIRT1 gene promoter in sporadic Parkinson’s disease. Biochem. Biophys. Res. Commun. 422, 693–696 (2012).
Guo, W. et al. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J. Neurosci. Res. 89, 1723–1736 (2011).
Pallàs, M., Porquet, D., Vicente, A. & Sanfeliu, C. Resveratrol: new avenues for a natural compound in neuroprotection. Curr. Pharm. Des. 19, 6726–6731 (2013).
Kim, D. et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179 (2007).
Gan, L. & Mucke, L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron 58, 10–14 (2008).
Cui, L. et al. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69 (2006).
Guo, Y. J. et al. Resveratrol alleviates MPTP‐induced motor impairments and pathological changes by autophagic degradation of α‐synuclein via SIRT1‐deacetylated LC3. Mol. Nutr. Food Res. 60, 2161–2175 (2016).
Wu, Y. et al. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 19, 163–174 (2011).
Sampaio-Marques, B. et al. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on Sir2-mediated mitophagy. Autophagy 8, 1494–1509 (2012).
Ham III, P. B. & Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 157, 92–116 (2017).
Jeong, J.-K., Moon, M.-H., Lee, Y.-J., Seol, J.-W. & Park, S.-Y. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol. Aging 34, 146–156 (2013).
Reed, N. A. et al. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16, 2166–2172 (2006).
Esteves, A. R. et al. Mitochondrial metabolism regulates microtubule acetylome and autophagy trough Sirtuin-2: impact for Parkinson’s disease. Mol. Neurobiol. 55, 1440–1462 (2018).
Xie, R., Nguyen, S., McKeehan, W. L. & Liu, L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 11, 89 (2010).
Gal, J., Bang, Y. & Choi, H. J. SIRT2 interferes with autophagy-mediated degradation of protein aggregates in neuronal cells under proteasome inhibition. Neurochem Int. 61, 992–1000 (2012).
Esteves, A. R. et al. Acetylation as a major determinant to microtubule-dependent autophagy: relevance to Alzheimer’s and Parkinson disease pathology. Biochim Biophys. Acta Mol. Basis Dis. 1865, 2008–2023 (2018).
Kitada, M., Ogura, Y. & Koya, D. The protective role of Sirt1 in vascular tissue: its relationship to vascular aging and atherosclerosis. Aging (Albany NY) 8, 2290–2307 (2016).
Nadtochiy, S. M. et al. SIRT1-mediated acute cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 301, H1506–H1512 (2011).
Li, J. et al. Mouse Sirt3 promotes autophagy in AngII-induced myocardial hypertrophy through the deacetylation of FoxO1. Oncotarget 7, 86648–86659 (2016).
Yu, W. et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys. Acta Mol. Basis Dis. 1863, 1973–1983 (2017).
Ueno, T. & Komatsu, M. Autophagy in the liver: functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 14, 170–184 (2017).
Biel, T. G. et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ. 23, 279–290 (2016).
Arensman, M. D. & Eng, C. H. Self-digestion for lifespan extension: enhanced autophagy delays aging. Mol. Cell. 71, 485–486 (2018).
Hansen, M., Rubinsztein, D. C. & Walker, D. W. Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol. 19, 579–593 (2018).
Cuervo, A. M. Autophagy and aging-when” all you can eat” is yourself. Sci. Aging Knowl. Environ. 2003, pe25 (2003).
He, L.-Q, Lu, J.-H & Yue, Z.-Y. Autophagy in ageing and ageing-associated diseases. Acta Pharm. Sin. 34, 605–611 (2013).
Morselli, E. et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 1, e10 (2011).
Fang, E. F. et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 24, 566–581 (2016).
Chen, M.-l et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 9, 2033–2045 (2013).
Ramalinga, M. et al. MicroRNA-212 negatively regulates starvation induced autophagy in prostate cancer cells by inhibiting SIRT1 and is a modulator of angiogenesis and cellular senescence. Oncotarget 6, 34446–34457 (2015).
Phadwal, K., Watson, A. S. & Simon, A. K. Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell. Mol. Life Sci. 70, 89–103 (2013).
Matsui, K. et al. NAD-dependent histone deacetylase, SIRT1, plays essential roles in the maintenance of hematopoietic stem cells. Biochem. Biophys. Res. Commun. 418, 811–817 (2012).
Ou, X., Lee, M. R., Huang, X., Messina‐Graham, S. & Broxmeyer, H. E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 32, 1183–1194 (2014).
Marino, G. et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell. 53, 710–725 (2014).
Escobar, K. A., Cole, N. H., Mermier, C. M. & VanDusseldorp, T. A. Autophagy and aging: maintaining the proteome through exercise and caloric restriction. Aging Cell 18, e12876 (2018).
Leidal, A. M., Levine, B. & Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 20, 1338–1348 (2018).
Sun, N. et al. Measuring in vivo mitophagy. Mol. Cell. 60, 685–696 (2015).
Imai, S.-I. & Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471 (2014).
Dai, H., Sinclair, D. A., Ellis, J. L. & Steegborn, C. Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharm. Ther. 188, 140–154 (2018).
Nazio, F., Bordi, M., Cianfanelli, V., Locatelli, F. & Cecconi, F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 26, 690–702 (2019).
Dikic, I. & Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364 (2018).
Finkel, T. The metabolic regulation of aging. Nat. Med. 21, 1416–1423 (2015).
Rubinsztein, D. C., Marino, G. & Kroemer, G. Autophagy and aging. Cell 146, 682–695 (2011).
Acknowledgements
I apologize to the scientists whose work was not cited due to space limitations. I appreciate Pf. Toren Finkel for their helpful discussions. I also thank Pf. Seungjae V. Lee, Jiyun Kwon and Seon Young Kim for technical support. This work was supported by grants from the National Research Foundation of Korea (NRF-2016R1D1A1B04934603, NRF-2014R1A1A3051320, and NRF-2014M3A9D8034459)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that the author has no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp Mol Med 51, 1–11 (2019). https://doi.org/10.1038/s12276-019-0302-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-019-0302-7
This article is cited by
-
Revealing the role of epigenetic and post-translational modulations of autophagy proteins in the regulation of autophagy and cancer: a therapeutic approach
Molecular Biology Reports (2024)
-
Research and study of 2-((4,6 dimethyl pyrimidine-2-yle) thio)-N-phenyl acetamide derivatives as inhibitors of sirtuin 2 protein for the treatment of cancer using QSAR, molecular docking and molecular dynamic simulation
Journal of Molecular Modeling (2022)
-
Human M1 macrophages express unique innate immune response genes after mycobacterial infection to defend against tuberculosis
Communications Biology (2022)
-
Epigenetic targeting of the ACE2 and NRP1 viral receptors limits SARS-CoV-2 infectivity
Clinical Epigenetics (2021)
-
Build-UPS and break-downs: metabolism impacts on proteostasis and aging
Cell Death & Differentiation (2021)
