
Abstract
During viral infection, virus-derived cytosolic nucleic acids are recognized by host intracellular specific sensors. The efficacy of this recognition system is crucial for triggering innate host defenses, which then stimulate more specific adaptive immune responses against the virus. Recent studies show that signal transduction pathways activated by sensing proteins are positively or negatively regulated by many modulators to maintain host immune homeostasis. However, viruses have evolved several strategies to counteract/evade host immune reactions. These systems involve viral proteins that interact with host sensor proteins and prevent them from detecting the viral genome or from initiating immune signaling. In this review, we discuss key regulators of cytosolic sensor proteins and viral proteins based on experimental evidence.
Similar content being viewed by others

Introduction
Viral infection is a major threat to human and animal health worldwide. Acute and chronic infections cause many economic and social problems. Over the past few decades, the field of molecular cell biology has contributed to our knowledge of both viruses and the host innate immune reactions that they trigger. In particular, we now understand how host cells recognize invading viruses and how the antiviral signaling cascade is regulated.
Host innate immunity is the first line of defense against viral infection. Efficient and rapid detection of invading viruses, coupled with mechanisms that distinguish viral components from host components, is a critical factor. Upon viral infection, virus-derived pathogen-associated molecular patterns (PAMPs), such as viral capsid proteins, surface glycoproteins, and the viral genome, are recognized by host pattern recognition receptors (PRRs). There are several types of PRRs, which are identified according to cellular localization and ligand specificity; these include Toll-like receptors, C-type lectin receptors, retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors, and cytosolic DNA sensors such as cyclic GMP-AMP synthetase1. Sensing of viral PAMPs by PRRs triggers signaling cascades via adapter proteins such as mitochondrial antiviral signaling protein (MAVS) or stimulator of interferon genes (STING), ultimately leading to the production of host defense molecules such as type I and III interferons (IFNs), proinflammatory cytokines, and chemokines2. Secreted IFNs and cytokines enhance innate immune responses via autocrine and paracrine mechanisms and induce expression of interferon-stimulated genes (ISGs) that inhibit viral replication and spread3. Secreted cytokines and chemokines are also critical for inducing effective adaptive and memory immune responses.
Nonetheless, excessive production of IFNs and prolonged inflammatory responses triggered by uncontrolled PRR signaling can have deleterious effects on the host by promoting the development of autoimmune disorders, allergies, and other immunopathologies4. In contrast, weak or ineffective PRR signal transduction exacerbates the severity of viral disease. Therefore, PRR-mediated signal transduction must be tightly regulated (either positively or negatively) to maintain host immune homeostasis5.
In addition, viruses have evolved several strategies to avoid detection of host antiviral immune responses; these range from interruption of viral sensors to manipulation of molecules within signaling cascades6. For example, the viral genome harbors structures that mask specific molecular motifs recognized by cytosolic sensors. Some viral proteins inhibit host sensor molecules by cleaving or mediating degradation of signaling molecules or by interfering with post-translational modifications (PTMs) of sensors6. From the perspective of the virus, these actions during the early phase of invasion are critical for successful infection.
Here, we summarize recent evidence regarding interactions between key intracellular sensors, viral RNA/DNA, and molecules that regulate efficient IFN responses or maintenance of host immune homeostasis. Furthermore, we describe recent advances in our knowledge about viral evasion of host cytosolic sensors, focusing on interactions between cytosolic sensors and specific viral proteins.
Host viral RNA sensors and viral evasion mechanisms
Upon viral infection, the viral genome is released into the cytoplasm to initiate viral protein biosynthesis. During this step, conserved molecular structures such as triphosphates and double-stranded (ds)RNA act as PAMPs that are recognized by sensors in the host cell cytosol (Table 1). The host innate immune system includes receptors, called PRRs, that distinguish the viral genome from the host genome. To achieve this, RLRs comprising RIG-I, melanoma differentiation-associated protein 5 (MDA5), laboratory of genetics and physiology 2 (LGP2), and other sensors such as NACHT, LRR, PYD domain-containing protein 3 (NLRP3), and nucleotide-binding oligomerization domain-containing protein 2, act as intracellular viral RNA sensors7. These proteins bind to viral RNA in the cell cytoplasm via RNA binding motifs, after which their signaling domain interacts with downstream adapter molecules, resulting in the activation of signaling cascades. The reactions are triggered as an immediate response to infection by RNA viruses and result in the production of type I IFNs, proinflammatory cytokines, and chemokines2,8. However, RNA viruses possess an arsenal of mechanisms to attenuate innate immune responses. Below, we describe the activation and regulation processes of major sensor molecules and mechanisms by which viruses evade them.
RIG-I
RIG-I, which belongs to the DExD/H box RNA helicase family, is an intracellular sensor of viral RNA. RIG-I recognizes 5′ tri- or di-phosphorylated dsRNA, the AU-rich 3′untranslated region (UTR), RNase L cleavage products, and circular viral RNA9,10. RIG-I detects the genomes of viruses such as vesicular stomatitis virus (VSV), influenza A virus (IAV), Sendai virus (SeV), Newcastle disease virus (NDV), respiratory syncytial virus (RSV), hepatitis C virus (HCV), and Japanese encephalitis virus (JEV)10,11,12. In addition, some DNA viruses such as vaccinia virus and Herpes simplex virus (HSV)9 and bacteria such as Listeria monocytogenes generate RNA that is then targeted by RIG-I13. Structurally, RIG-I comprises two N-terminal caspase activation and recruitment domains (CARDs), two helicase domains (Hel-1 and Hel-2), and a C-terminal repressor domain (RD)14. In the resting state, RIG-I is autoinhibited by its own RD. In response to virus invasion, RIG-I recognizes viral RNA via its two components: the RD and helicase domain. The RD facilitates viral RNA recognition through its strong affinity for the 5′ end triphosphate, and the positively charged pocket structure of the RD interacts with the 5′ end of viral RNA15,16. The helicase domain binds to dsRNA and mediates a conformational change that allows ATP binding to activate RIG-I15,16. This conformational change opens up the CARDs, which are essential for downstream signaling14,17. During this step, RIG-I is activated or inactivated by several regulators and/or PTMs (see below). Open CARDs interact with the CARD MAVS to activate downstream signaling cascades18. In addition, adapters such as TNF receptor associated factor (TRAF) 3 or TRAF6, serine/threonine-protein kinases, TANK-binding kinase (TBK1), and IκB kinase (IKK) are activated9,10. Consequently, transcription factors such as IRF3, IRF7, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) trigger production of type I IFNs and induce expression of antiviral molecules9,10.
RIG-I is essential for innate antiviral immunity; however, it is modulated by several regulatory molecules to protect against viral spread or the maintenance of host immune homeostasis (Fig. 1). First, RIG-I activation or inactivation is regulated by PTMs such as ubiquitination, phosphorylation, and acetylation5. During activation, RIG-I undergoes K63-linked ubiquitination by RING finger protein 135 (RNF135/Riplet), tripartite motif-containing protein (TRIM4), and TRIM2519,20,21,22,23. Importantly, K63-linked ubiquitination of the CARD at K172 is mediated by TRIM25, which induces RIG-I oligomerization22. Caspase 12 promotes K63-mediated ubiquitination of RIG-I via TRIM25 to promote RIG-mediated signaling, whereas linear ubiquitin chain assembly complex (LUBAC) negatively regulates TRIM25 via K48-linked ubiquitination to trigger proteasomal degradation24,25.

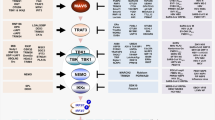
Schematic presentation of positive and negative regulators of RLRs (Top) and melanoma differentiation-associated protein-5 (MDA5) (Bottom) through PTMs or non-PTMs and immune invasion viral proteins interacting with RIG-I (Top) and MDA5 (Bottom). The RLR-MAVS pathway includes the key cytosolic sensors RIG-I and MDA5, which detect viral RNA. These sensors subsequently interact with the central antiviral signaling protein MAVS, which in turn activates the transcription factors NF-κB and IRF3/IRF7 via the cytosolic kinases IKK and TBK1/IKKε, respectively. Activated transcription factors NF-κB, IRF7 and IRF3 translocate to the nucleus and induce transcription of type I IFN and pro-inflammatory genes
Conversely, ubiquitin carboxyl-terminal hydrolase (USP) 15 mediates deubiquitination of K48-linked ubiquitination of TRIM2526. In addition, mex-3 RNA-binding family member C (MEX3C) mediates K63-linked ubiquitination of RIG-I to promote the formation of stress granules, which generate a platform complex for viral sensing and signaling27. In contrast to activation by K63-linked ubiquitination, removal of K63-linked polyubiquitin by the deubiquitinating enzyme CYLD negatively regulates RIG-I activity28. Two other deubiquitinases, USP3 and USP21, also negatively control RIG-I activity in the same way29,30. K63-linked ubiquitination of RIG-I by TRIM25, MEX3C, and TRIM4 and deubiquitination of RIG-I by CYLD, USP3, and USP21 occur in the CARDs, whereas K63-linked ubiquitination by the RNF135/Riplet occurs in the RD (K788), which has a positive effect on TRIM25-mediated K63-linked ubiquitination in the CARDs19,20,23,28,29,30. In contrast, K48-linked ubiquitination serves as a signal for proteasomal degradation of RIG-I. RNF122 and RNF125 mediate K48-linked ubiquitination, which inhibits RIG-I-mediated antiviral innate immune responses31,32. Sialic acid binding Ig-like lectin G (Siglec-G) recruits the E3 ligase c-cbl to RIG-I, resulting in degradation via K48-linked ubiquitination33. The deubiquitinase USP4 also serves as a negative regulator of ubiquitination34.
Phosphorylation and acetylation are also important PTMs involved in RIG-I regulation. In resting cells, for example, RIG-I is autoinhibited by phosphorylation and acetylation. Phosphorylation on the CARDs of RIG-I is maintained by protein kinase C (PKC) α/β and removed by protein phosphatase 1 (PP1) α/γ, which induces further K63-linked ubiquitination at this domain35,36. Casein kinase II (CK2) phosphorylates the RD of RIG-I, and removal of this phosphorylation allows for K63-linked ubiquitination via RNF13537. When RIG-I is inactivate, the RD domain is acetylated. In the presence of RNA ligands, histone deacetylase (HDAC) 6 deacetylates the RD and allows RIG-I to bind to viral RNA to promote oligomerization38,39. However, some regulators involved in RIG-I function do not act via PTMs. As a positive regulator, the shorter isoform of PARP-13 (ZAPS) associates with RIG-I and promotes its oligomerization, whereas IRF1 functions to increase expression of RIG-I40,41. The mitochondrial targeting chaperone protein 14-3-3ε interacts with RIG-I, thereby translocating it to the translocon42. As a negative regulator, RNF123 blocks RIG-I and inhibits signaling of MAVS without its E3 ligase function43. Caspase-8 is recruited to RIG-I upon viral infection, whereupon it cleaves the RIG-I signaling enhancer receptor-interacting protein (RIP) 144. Other molecules that negatively regulate RIG-I are listed in Table 2.
RIG-I is tightly controlled by a wide range of regulatory factors. For structural activation and initiation of innate immune responses, RIG-I is involved in many specific modification mechanisms with other regulatory factors. Since the discovery that TRIM25 activates RIG-I via K63 ubiquitination, a burgeoning number of other factors regulating RIG-1 activity, such as PTMs, oligomerization, RNA recognition, relocalization, and stabilization, have been identified. Moreover, beyond these processes directly affecting RIG-1 activity, some of the factors that regulate RIG-1 are controlled by other factors. Taken together, these findings highlight the complexity and delicate control of RIG-I-mediated innate immune signaling.
Viral evasion of RIG-I-mediated responses
In the early stage of viral infection, avoidance of innate immunity, including the interferon response, is important for successful viral infection. Because RIG-I is a key viral RNA sensor that initiates rapidly innate immune responses, it is targeted by diverse viral proteins.
Many viruses possess proteins that interfere directly with RIG-I. For example, HSV-1 tegument protein US11 interacts with RIG-I to block formation of the RIG-I-MAVS complex; porcine deltacoronavirus (PDCoV) accessory protein NS6 interferes with RIG-I binding to dsRNA45,46. The 3C protease of Enterovirus (EV) 71, Poliovirus, Echovirus, Rhinovirus type 16, Rhinovirus type 1A, and Encephalomyocarditis virus (EMCV) cleaves and inactivates RIG-I47. Latent membrane protein (LMP) 1 of Epstein–Barr virus (EBV) mediates proteasomal degradation of RIG-I, and the nonstructural (NS) protein of severe fever with thrombocytopenia syndrome virus (SFTSV) hijacks RIG-I and its signaling proteins in the cytoplasmic structure48,49. In addition, evidence suggests that human respiratory syncytial virus (HRSV) N and P proteins colocalize with RIG-I and that the influenza virus NS1 protein interacts with RIG-I directly50,51. Interaction between RIG-I and these viral proteins mainly leads to direct functional impairment of RIG-I. The processes used by these viral proteins include cleavage, degradation, suspension, and inhibition of RIG-I. Specific viral proteins also interfere with RIG-I activation. A number of viral proteins target TRIM25-mediated K63-linked ubiquitination of RIG-I. The NS1 protein of IAV targets TRIM25 to block K63-linked ubiquitination52, whereas Paramyxovirus V proteins, HRSV NS1, and the severe acute respiratory syndrome coronavirus (SARS-CoV) nucleocapsid protein target TRIM25 to inhibit activation of RIG-I53,54,55. Moreover, the human papillomavirus (HPV) E6 protein increases the activity of USP15 to promote proteasomal degradation of TRIM25, and Herpesvirus mediates autoubiquitination of TRIM25 to prevent K63-linked ubiquitination of RIG-I56,57. Furthermore, West Nile virus (WNV) NS1 interferes with the innate immune response by mediating proteasomal degradation of RIG-I and inhibiting K63-linked ubiquitination58. In addition, some viral proteins dephosphorylate RIG-I and inhibit its signaling, and measles virus (MV) activates the C-type lectin DC-SIGN and blocks PP1 activity to attenuate RIG-I in dendritic cells59. The viral proteins that interact with or affect RIG-I are listed in Table 3.
Taken together, results to date show that viruses express specific proteins that interfere with RIG-I function via diverse mechanisms and are essential for viral pathogenesis.
MDA5 and viral evasion
MDA5 is a major intracellular sensor that recognizes viral dsRNA, including the genomes of EMCV, Poliovirus, Coxsackievirus, Rotavirus, Dengue virus (DENV), WNV, and murine hepatitis virus10,11 (Table 3). MDA5 recognizes long dsRNA, AU-rich motifs, and RNase L cleavage products10,11,60. MDA5 activation is similar to that of RIG-I; however, the MDA5 RD binds to the RNA backbone and not to the 5′end. This difference allows the HEL2 loop of MDA5 insert to the major groove of viral RNA, which is not limited to the RNA end16. This interaction triggers the two CARDs to form a tetrameric structure that transduces a signal to the adapter molecule MAVS, which is shared with the RIG-I mediated pathway16,61.
Several regulators and PTMs also regulate MDA5 activation and inactivation (Fig. 1). MDA5 activation involves dephosphorylation of S88 on the MDA5 CARD by PP1α/γ36. MDA5 also undergoes SUMOylation by PIAS2β, which promotes interferon signaling62, and TRIM38 acts as a SUMO E3 ligase to mediate downstream signaling via SUMOylation of K43/K865 of MDA563. K63-linked ubiquitination at the CARDs is also a critical mechanism underlying MDA5 activation. In addition, TRIM65 mediates K63-linked ubiquitination of K743 in the CTD of MDA5 to induce MDA5 oligomerization and activation64, and DEXD/H box helicase DDX60 also acts as a positive regulator by binding to MDA565. In contrast, right open reading kinase 3 (RIOK) phosphorylates the RD of MDA5 at S828 to inhibit filament formation66. Notably, the E3 ubiquitin ligase RNF125 negatively regulates MDA5 via its ligase function, whereas RNF123 performs the same role independently of its ligase function31,43. USP3 and USP21 inhibit MDA5 function via deubiquitination29,30, and proteins such as dihydroxyacetone kinase (DAK), Atg5-Atg-12, NLRC5, and TRIM13 interact with and inhibit MDA5 (Table 2)63,67,68,69.
MDA5 is also subject to several types of activation that resemble those that activate RIG-I. Indeed, PP1α/γ, USP3, RNF123, and TRIM40 target RIG-I and MDA5 simultaneously, likely because the domain structures of these two proteins are similar. Upon activation, these two proteins transmit signals through their CARDs to MAVS and share subsequent pathways involved in cytokine secretion. Hence, these two proteins are commonly involved in virus recognition and play a complementary role in the initiation of innate immunity. Regardless, further studies on the molecular mechanisms that regulate MDA5 activation and inactivation are necessary.
Different viral proteins have been reported to inhibit MDA5. The V proteins of paramyxoviruses limit the induction of IFN-β by interfering with MDA5 but not RIG-I70. In addition, the V protein of Parainfluenza virus (PIV) 5, Mumps virus, MV, Menangle virus, Hendra virus, Nipah virus, Maquera virus, SeV, and Salem virus binds to MDA570. In particular, structural studies have demonstrated that the V protein of PIV5 recognizes a structural motif within MDA5, thereby disrupting its ATP-hydrolysis function as well as filament formation71. The helicase C domain of MDA5 is sufficient for association with V proteins from PIV2, PIV5, MV, Mumps virus, Hendra virus, and Nipah virus. In addition, human herpesvirus tegument protein US11 and the HRSV N protein antagonize innate immune responses initiated by MDA545,50. The 2A protease of Coxsackievirus B (CVB) 3 and Poliovirus mediates degradation of MDA5 in a proteasome- and caspase-dependent manner, whereas EV 71 2A cleaves MDA5 to inactivate it72.
LGP2 and viral evasion
LGP2 is an RLR that lacks N-terminal CARDs, and thus LGP2 cannot transmit signals to MAVS; however, it can bind to viral RNA and modulate the activities of RIG-I and MDA511. Overall, the exact role of LGP2 in innate immunity is still unclear, though based on previous studies, LGP2 is a negative regulator of RLRs73,74 and acts synergistically with MDA563,64. Although recent evidence shows that LGP2 strengthens MDA5-mediated innate immune responses against HCV infection75, other studies suggest that LGP2 acts as a negative regulator by interacting with TRAF family proteins and interfering with their ubiquitin ligase activity76.
Because LGP2 lacks CARDs and signaling activity, few studies have examined how it is regulated. Nonetheless, research suggests that pumilio protein 1 (PUM1) regulates expression of innate immune genes by acting as a biphasic negative regulator of LGP277. In addition, PACT amplifies innate immune responses when expressed together with both LGP2 and MDA578.
Although the role of LGP2 is not clear, interactions between viruses and LGP2 have been reported. For example, the Paramyxovirus V protein interacts with LGP2 and interferes with its ability to coactivate MDA579. Foot and mouth disease virus (FMDV) 2B also directly interacts with LGP2, and expression of LGP is decreased by the C-terminal region of 2B80. Furthermore, FMDV leader protease (Lpro) directly interacts with and cleaves LGP280; this event is thought to affect the function of LGP2 that regulates MDA5, which is responsible for FMDV genome recognition. Further research will be required to address the functions or molecular mechanisms of LGP2 and how its activity is affected by viral proteins.
Host viral DNA sensors and viral evasion mechanisms
Upon DNA virus invasion, viral DNA is released into the host cell cytoplasm, and viral protein synthesis begins. Because the DNA of eukaryotic cells is located in the nucleus or mitochondrion, the presence of viral DNA in the cytoplasm acts as a PAMP, which is detected by several intracellular sensor molecules81.
Based on recent reports, cytoplasmic viral DNA is recognized by cyclic GMP-AMP synthase (cGAS), interferon gamma inducible protein 16 (IFI16), interferon-inducible protein (AIM2), DDX41, and RNA PolIII, among others. Similar to detection of viral RNA, immediate detection of viral DNA triggers host innate immune responses and enhances expression of antiviral-related cytokines74,81,82. cGAS and IFI16 transmit signals to the endoplasmic reticulum (ER) adapter protein STING, whereas AIM2 and IFI16 mediate activation of the inflammasome82,83. Ultimately, these reactions activate type I IFN signaling and antiviral responses, similar to those observed in RLR signaling. The activities of DNA-sensing factors are also modulated by a number of positive and negative regulators. However, DNA viruses have evolved numerous and elaborate strategies to counteract viral DNA sensing by host sensor molecules. Below, we summarize the regulation of major sensor factors and viral evasion mechanisms.
cGAS and viral evasion
cGAS is a well-known cytosolic DNA sensor essential for early innate immune responses to DNA viruses. In the cytoplasm, cGAS detects self and nonself DNA and induces production of type I IFNs and proinflammatory cytokines. Ligands for cGAS are present in the genomes of HSV-1, Kaposi’s sarcoma-associated herpesvirus (KSHV), Vaccinia virus, murine gammaherpesvirus 68 (MHV68), Adenovirus, and Hepatitis B virus (HBV)18,74,82. cGAS binds to the sugar-phosphate backbone of dsDNA without sequence specificity. This interaction is mediated by positively charged DNA-binding sites of cGAS and induces a conformational change in cGAS, opening activation sites for cGAMP synthesis18,82. After recognition of viral DNA, cGAS generates 2′,3′-cGAMP, along with ATP and GTP, all of which play roles as second messengers to activate STING84,85, which undergoes conformational changes and is translocated from the ER to ER-Golgi intermediate compartments86. TBK1 is also activated, resulting in phosphorylation of transcription factors that potentiate cytokine-mediated antiviral responses82.
cGAS is also controlled by PTMs, such as phosphorylation, ubiquitination, SUMOylation, acetylation, and glutamylation5 (Fig. 2). Polyubiquitination of cGAS is mediated by E3 ligase, TRIM14, RINCK/TRIM41, and TRIM5687,88,89,90, whereas ER-resident RNF185 catalyzes K27-linked ubiquitination in response to HSV-1 infection. Importantly, RNF185-mediated ubiquitination at K173 and K384, two major ubiquitination sites, activates cGAS, thus generating more cGAMP88. RINCK/TRIM41 and TRIM56 mediate cGAS monoubiquitination and promote innate immune responses to DNA viruses; cGAS K335 is also monoubiquitinated by TRIM56 and E1 and UbcH5 E2 enzymes89,90.

Schematic presentation of positive and negative regulators of cGAS through PTMs or non-PTMs and immune invasion viral proteins interacting with cGAS. cGAS induces signaling through the adapter protein STING, resulting in dimerization of STING and activation of the transcription factors NF-κB and IRF3/IRF7 via cytosolic kinases IKK and TBK1, respectively. Activated transcription factors NF-κB, IRF7, and IRF3 translocate to the nucleus and induce transcription of type I IFN and pro-inflammatory genes
K48-linked ubiquitination of cGAS is a recognition signal that triggers selective autophagic degradation, whereby TRIM14 recruits USP14 to cleave K48-linked ubiquitination and stabilize cGAS87. SUMOylation also plays an important role in regulating cGAS. TRIM38 mediates SUMOylation of cGAS to inhibit its degradation, whereas sentrin-specific protease (SENP) 2 induces SUMOylation and ultimately degradation of cGAS91,92. SENP7 targets cGAS for deSUMOylation, thereby stabilizing it and protecting it from degradation92. cGAS is also regulated by glutamylation, phosphorylation, and acetylation. Tubulin tyrosine ligase-like (TTLLs) glutamylases 4 and 6 target E302 for monoglutamylation and E272 for polyglutamylation, respectively, whereas cytosolic carboxypeptidases (CCP) 5 and 6 antagonize TTLLs93. Phosphorylation of cGAS at S305 and S291, as catalyzed by protein kinase B (PKB/Akt), strongly suppresses cGAS94. In addition, acetylation inhibits cGAS-mediated production of interferon95. There are also other mechanisms of cGAS regulation that do not rely on PTMs. As positive regulators, manganese and the Ras-GAP SH3 domain-binding protein (G3BP1) target cGAS to promote its DNA-binding activity96,97; PI(4,5)P2 localizes cGAS to the plasma membrane95. The CCHC-type zinc-finger (ZF) protein ZCCHC3 acts with cGAS as a cosensor to enable recognition of dsDNA98. In contrast, inflammasome activation triggers caspase-mediated cleavage of cGAS. Oligoadenylate-synthetase-family (OASL) protein downregulates cGAS enzyme activity, and becline 1 targets cGAS to suppress cGAMP synthesis99,100. Recent studies have also reported glutamylation and monoubiquitination as novel PTMs of cGAS, and two novel carboxypeptidases and glutamylases regulate cGAS via differential glutamylation93. TRIM56 monoubiquitinates cGAS and affects antiviral signaling by promoting a marked increase in cGAS dimerization and DNA-binding activity, which eventually increases cGAMP production90.
To antagonize host innate immune activation, Zika virus (ZIKV) NS1 stabilizes caspase 1 and protects it from proteasomal degradation101. K11-linked ubiquitin chains of caspase 1 at K134 are cleaved by USP8, which is recruited by NS1. Thus, ZIKV triggers degradation of cGAS by caspase 1, thereby blocking antiviral innate immunity101. HSV-1 also evades cGAS-mediated innate immune responses through two viral proteins: the UL37 tegument protein deamidates cGAS, a process that determines species-specific inactivation of HSV-1102; and VP22 of HSV-1 interacts with cGAS to inhibit its enzymatic activity103.
To evade cGAS-mediated innate immune responses, UL31 of human cytomegalovirus (HCMV) interacts with cGAS to inhibit cGAMP synthesis; this is achieved by preventing cGAS from binding to DNA, whereas pp65 of HCMV inhibits cGAS activity104,105.
ORF52 and LANA of KSHV also inhibit cGAS activity. LANA of KSHV interacts with cGAS directly to inhibit cGAS-mediated pathways, whereas ORF52 of KSHV prevents cGAS from sensing DNA by inhibiting its enzymatic activity106. In addition, cGAS detects mitochondrial DNA released during DENV infection; however, the NS2B of DENV mediates lysosomal degradation of cGAS107.
The mechanisms by which virus proteins interfere with cGAS involve inhibition of DNA-binding activity and enzymatic activity or degradation. In particular, HSV-1 and HCMV express multiple proteins that interfere with cGAS-triggered innate immunity102,103,104,105. It is thought that many viruses have evolved diverse mechanisms that hinder cGAS function because cGAS is an intracellular sensor that is critical for detecting viral DNA. cGAS is inhibited not only by DNA viruses but also by RNA viruses such as ZIKV and DENV101,107. Moreover, DNA as a byproduct of RNA viral infection is recognized by cGAS, and viruses possess a mechanism to avoid this type of recognition. Taken together, these findings suggest that viral pathogenesis involving host immunity is more complex and sophisticated than previously thought, indicating that more research is needed in this area.
IFI16 and viral evasion
IFI16 is a nuclear protein located predominantly in the nucleus; however, it shuttles between the nucleus and cytoplasm to sense viral DNA derived from Herpesvirus, human immunodeficiency virus (HIV), and bacteria such as listeria monocytogenes74,81. IFI16 contains an N-terminal pyrin domain (PYD) and two C-terminal HIN domains: it recognizes viral DNA via the HIN domain and then interacts with cGAS to promote cGAMP production and plays a vital role in cGAMP-mediated signaling, which activates TBK1 within the STING complex5,108. IFI16 is also able to detect viral DNA in the nucleus, activating ASC, an adapter molecule for the inflammasome and leading to production of IL-1β and IL-18109,110.
As IFI16 recognizes the Herpesvirus genome, it has been the subject of intense study. Upon Herpesvirus infection, IFI16 is acetylated by P300 in the nucleus and activates STING after its translocation to the cytoplasm111. cGAS also stabilizes IFI16 to promote innate immune signaling during HSV infection, though cGAS generates less cGAMP112. BRCA1 forms a complex with IFI16 in the nucleus that is strengthened upon viral infection; this triggers translocation of IFI16 to the cytoplasm and inflammasome activation113. In contrast, DNA viruses produce specific proteins that enable escape from IFI16-mediated immune responses. The HSV-1 viral E3 ubiquitin ligase ICP0 suppresses IFI16 by mediating its proteasomal degradation114. In addition, it has been reported that KSHV lytic protein(s) potentially degrade IFI16 to maintain latency115. Finally, HCMV possesses proteins that interfere with IFI16; Vps4 and TGN46 induce trafficking of IFI16 to multivesicular bodies, whereas pUL83 interacts with the PYRIN domain, which interferes with DNA sensing or inhibits expression of interferon-inducible genes116,117,118.
To date, few studies have been conducted on viral proteins that interfere with the recognition and signaling mechanisms of IFI16. However, as mentioned above, recent papers suggest the existence of a relationship between IFI16 and cGAS-cGAMP signaling112,119. A study in keratinocytes showed that IFI16 is required for STING activation by cGAMP and that IFI16 interacts with STING to promote its phosphorylation112, and there is another report that IFI16 interacts with the cGAS–STING pathway in macrophages119. These findings indicate that IFI16 is more closely related to DNA virus recognition and viral defense mechanisms than once thought. For this reason, it is expected that new virus proteins that interfere with the recognition and signaling mechanisms of IFI16 will be reported in the near future.
AIM2 and viral evasion
AIM2, a member of the PYRIN protein family, consists of two domains: the PYRIN domain at the N-terminus and HIN200 domain at the C-terminus81. The HIN200 domain is responsible for DNA binding, whereas the PYRIN domain interacts with the PYRIN domain of ASC to activate caspase-174,81. AIM2 has affinity for viral DNA derived from murine cytomegalovirus (MCMV) and Vaccinia virus; activation of AIM2 leads to secretion of IL-1β of IL-18 and mediates inflammation in response to viral infection18,81. As regulatory molecules, nuclear factor E2-related factor-2 (Nrf2) and pyruvate kinase isozyme M2 (PKM2) act as positive regulators of AIM2 inflammasome activation120,121. In contrast, high-mobility group box 1 (HMGB1) and DNA complexes induce autophagy to reduce activation of the AIM2 inflammasome122. TRIM11 mediates autopolyubiquitination and negatively regulates the AIM2 inflammasome by recruiting p62 and triggering selective autophagy123.
In contrast, pUL83 of HCMV binds to AIM2 and disrupts AIM2-mediated inflammasome activation. Upon HCMV infection, pUL83 interacts with AIM2 in macrophages, thereby inhibiting activated inflammasome components124. Recent reports show that VP22 of HSV-1 negatively regulates AIM2 inflammasome formation and IL-1β secretion: VP22 interacts with the HIN200 domain, but not with the PYRIN domain, to inhibit oligomerization of AIM2. Consequently, VP22-mediated inactivation of the inflammasome promotes virus replication in vivo125.
Taken together, these studies indicate that the AIM2 inflammasome serves as a key element in innate immunity against DNA viruses and that several viral proteins specifically inhibit AIM2 activation. Because the AIM2 inflammasome is also involved in the sensing of other DNA viruses, it is expected that further research will identify even more viral proteins that interact with AIM2. Furthermore, inflammasome activation not only constitutes a barrier to DNA viral infection but also is injurious to the host. In this respect, further studies are needed to determine the mechanisms that modulate the activity of the AIM2 inflammasome to enhance our understanding of host responses to DNA viral infection.
Conclusions
Intracellular sensing of viral RNA or DNA by PRRs is indispensable for host cells to mount an antiviral innate immune response to inhibit replication and spread of invading viruses and prime an effective adaptive immune response18,73. This review summarizes our current knowledge of the key intracellular sensors and how they are modulated by various molecules to mediate IFN responses and the maintenance of immune homeostasis. Moreover, we discuss viral proteins that interact with these host cytosolic sensing molecules and facilitate evasion of host defenses.
Over the past decade, our understanding of intracellular sensor-mediated antiviral responses has expanded, and we know much more about the molecular mechanisms by which they are regulated via host and viral factors. This knowledge not only allows us to understand viral pathogenesis but also reveals how intracellular sensors are activated and regulated. Extensive knowledge of these mechanisms will allow for research and development of novel anti-inflammatory agents, immunostimulatory agents, new vaccines, and antiviral agents that target cellular regulators or specific viral proteins. Regardless, further work is needed to identify other cytosolic sensors (such as novel sensors of nucleic acids), other positive or negative regulatory molecules and related pathways, and novel escape mechanisms utilized by new viruses or variants.
References
Goubau, D., Deddouche, S. & Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 38, 855–869 (2013).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
Mogensen, T. H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol Rev. 22, 240–273 (2009). Table of Contents.
Chiang, C. & Gack, M. U. Post-translational control of intracellular pathogen sensing pathways. Trends Immunol. 38, 39–52 (2017).
Chan, Y. K. & Gack, M. U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol 14, 360–373 (2016).
Hur, S. Double-stranded RNA sensors and modulators in innate immunity. Annu Rev. Immunol. 39, 349–375 (2019).
Loo, Y. M. & Gale, M. Jr. Immune signaling by RIG-I-like receptors. Immunity 34, 680–692 (2011).
Chen, N. et al. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 69, 297–304 (2017).
Said, E. A., Tremblay, N., Al-Balushi, M. S., Al-Jabri, A. A. & Lamarre, D. Viruses seen by our cells: the role of viral RNA sensors. J. Immunol. Res. 2018, 9480497 (2018).
Chow, K. T., Gale, M. Jr. & Loo, Y. M. RIG-I and other RNA sensors in antiviral immunity. Annu. Rev. Immunol. 36, 667–694 (2018).
Chazal, M. et al. RIG-I recognizes the 5' region of dengue and zika virus genomes. Cell Rep. 24, 320–328 (2018).
Hagmann, C. A. et al. RIG-I detects triphosphorylated RNA of Listeria monocytogenes during infection in non-immune cells. PLoS One 8, e62872 (2013).
Luo, D. et al. Structural insights into RNA recognition by RIG-I. Cell 147, 409–422 (2011).
Leung, D. W. & Amarasinghe, G. K. Structural insights into RNA recognition and activation of RIG-I-like receptors. Curr. Opin. Struc. Biol. 22, 297–303 (2012).
Luo, D. Toward a crystal-clear view of the viral RNA sensing and response by RIG-I-like receptors. RNA Biol. 11, 25–32 (2014).
Takahasi, K. et al. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 29, 428–440 (2008).
Ori, D., Murase, M. & Kawai, T. Cytosolic nucleic acid sensors and innate immune regulation. Int. Rev. Immunol. 36, 74–88 (2017).
Gao, D. et al. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS One 4, e5760 (2009).
Oshiumi, H., Matsumoto, M., Hatakeyama, S. & Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 284, 807–817 (2009).
Oshiumi, H., Miyashita, M., Matsumoto, M. & Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 9, e1003533 (2013).
Gack, M. U. et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 (2007).
Yan, J., Li, Q., Mao, A. P., Hu, M. M. & Shu, H. B. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell Biol. 6, 154–163 (2014).
Inn, K. S. et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 41, 354–365 (2011).
Wang, P. et al. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat. Immunol. 11, 912–919 (2010).
Pauli, E. K. et al. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal 7, ra3 (2014).
Kuniyoshi, K. et al. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc. Natl Acad. Sci. USA 111, 5646–5651 (2014).
Friedman, C. S. et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9, 930–936 (2008).
Cui, J. et al. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 24, 400–416 (2014).
Fan, Y. et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 211, 313–328 (2014).
Arimoto, K. et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl Acad. Sci. USA 104, 7500–7505 (2007).
Wang, W. et al. RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. Proc. Natl Acad. Sci. USA 113, 9581–9586 (2016).
Chen, W. et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152, 467–478 (2013).
Wang, L. et al. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J. Virol. 87, 4507–4515 (2013).
Maharaj, N. P., Wies, E., Stoll, A. & Gack, M. U. Conventional protein kinase C-alpha (PKC-alpha) and PKC-beta negatively regulate RIG-I antiviral signal transduction. J. Virol. 86, 1358–1371 (2012).
Wies, E. et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38, 437–449 (2013).
Sun, Z., Ren, H., Liu, Y., Teeling, J. L. & Gu, J. Phosphorylation of RIG-I by casein kinase II inhibits its antiviral response. J. Virol. 85, 1036–1047 (2011).
Choi, S. J. et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 35, 429–442 (2016).
Liu, H. M. et al. Regulation of retinoic acid inducible gene-I (RIG-I) activation by the histone deacetylase 6. EBioMedicine 9, 195–206 (2016).
Su, Z. Z., Sarkar, D., Emdad, L., Barral, P. M. & Fisher, P. B. Central role of interferon regulatory factor-1 (IRF-1) in controlling retinoic acid inducible gene-I (RIG-I) expression. J. Cell Physiol. 213, 502–510 (2007).
Hayakawa, S. et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 12, 37–44 (2011).
Liu, H. M. et al. The mitochondrial targeting chaperone 14-3-3epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 11, 528–537 (2012).
Wang, S. et al. RNF123 has an E3 ligase-independent function in RIG-I-like receptor-mediated antiviral signaling. EMBO Rep. 17, 1155–1168 (2016).
Rajput, A. et al. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 34, 340–351 (2011).
Xing, J., Wang, S., Lin, R., Mossman, K. L. & Zheng, C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 86, 3528–3540 (2012).
Fang, P. et al. Porcine deltacoronavirus accessory protein NS6 antagonizes interferon beta production by interfering with the binding of RIG-I/MDA5 to double-stranded RNA. J. Virol. 92, e00712–18 (2018).
Barral, P. M., Sarkar, D., Fisher, P. B. & Racaniello, V. R. RIG-I is cleaved during picornavirus infection. Virology 391, 171–176 (2009).
Santiago, F. W. et al. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J. Virol. 88, 4572–4585 (2014).
Xu, C., Sun, L., Liu, W. & Duan, Z. Latent membrane protein 1 of Epstein-Barr virus promotes RIG-I degradation mediated by proteasome pathway. Front Immunol. 9, 1446 (2018).
Lifland, A. W. et al. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 86, 8245–8258 (2012).
Jureka, A.S., Kleinpeter, A.B., Cornilescu, G., Cornilescu, C.C. & Petit, C.M. Structural basis for a novel interaction between the NS1 protein derived from the 1918 influenza virus and RIG-I. Structure 23, 2001–2010 (2015).
Gack, M. U. et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449 (2009).
Hu, Y. et al. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 91, e02143–16 (2017).
Ban, J. et al. Human respiratory syncytial virus NS 1 targets TRIM25 to suppress RIG-I ubiquitination and subsequent RIG-I-mediated antiviral signaling. Viruses 10, E716 (2018).
Sanchez-Aparicio, M. T., Feinman, L. J., Garcia-Sastre, A. & Shaw, M. L. Paramyxovirus V proteins interact with the RIG-I/TRIM25 regulatory complex and inhibit RIG-I signaling. J. Virol. 92, e01960–17 (2018).
Chiang, C. et al. The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 to suppress RIG-I-mediated innate immune signaling. J. Virol. 92, e01737–17 (2018).
Gupta, S. et al. Herpesvirus deconjugases inhibit the IFN response by promoting TRIM25 autoubiquitination and functional inactivation of the RIG-I signalosome. PLoS Pathog. 14, e1006852 (2018).
Zhang, H. L. et al. West nile virus NS1 antagonizes interferon beta production by targeting RIG-I and MDA5. J. Virol. 91, e02396–16 (2017).
Mesman, A. W. et al. Measles virus suppresses RIG-I-like receptor activation in dendritic cells via DC-SIGN-mediated inhibition of PP1 phosphatases. Cell Host Microbe 16, 31–42 (2014).
Luecke, S. & Paludan, S. R. Molecular requirements for sensing of intracellular microbial nucleic acids by the innate immune system. Cytokine 98, 4–14 (2017).
Wu, B. et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152, 276–289 (2013).
Fu, J., Xiong, Y., Xu, Y., Cheng, G. & Tang, H. MDA5 is SUMOylated by PIAS2beta in the upregulation of type I interferon signaling. Mol. Immunol. 48, 415–422 (2011).
Narayan, K. et al. TRIM13 is a negative regulator of MDA5-mediated type I interferon production. J. Virol. 88, 10748–10757 (2014).
Lang, X. et al. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 214, 459–473 (2017).
Miyashita, M., Oshiumi, H., Matsumoto, M. & Seya, T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell Biol. 31, 3802–3819 (2011).
Takashima, K., Oshiumi, H., Takaki, H., Matsumoto, M. & Seya, T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 11, 192–200 (2015).
Diao, F. et al. Negative regulation of MDA5- but not RIG-I-mediated innate antiviral signaling by the dihydroxyacetone kinase. Proc. Natl Acad. Sci. USA 104, 11706–11711 (2007).
Jounai, N. et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl Acad. Sci. USA 104, 14050–14055 (2007).
Cui, J. et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 141, 483–496 (2010).
Childs, K. et al. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359, 190–200 (2007).
Parisien, J. P. et al. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J. Virol. 83, 7252–7260 (2009).
Feng, Q. et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 88, 3369–3378 (2014).
Radoshevich, L. & Dussurget, O. Cytosolic innate immune sensing and signaling upon infection. Front Microbiol. 7, 313 (2016).
McFadden, M. J., Gokhale, N. S. & Horner, S. M. Protect this house: cytosolic sensing of viruses. Curr. Opin. Virol. 22, 36–43 (2017).
Hei, L. & Zhong, J. Laboratory of genetics and physiology 2 (LGP2) plays an essential role in hepatitis C virus infection-induced interferon responses. Hepatology 65, 1478–1491 (2017).
Parisien, J. P. et al. RNA sensor LGP2 inhibits TRAF ubiquitin ligase to negatively regulate innate immune signaling. EMBO Rep. 19, e45176 (2018).
Liu, Y., Qu, L., Liu, Y., Roizman, B. & Zhou, G. G. PUM1 is a biphasic negative regulator of innate immunity genes by suppressing LGP2. Proc. Natl Acad. Sci. USA 114, E6902–E6911 (2017).
Miyamoto, M. & Komuro, A. PACT is required for MDA5-mediated immunoresponses triggered by Cardiovirus infection via interaction with LGP2. Biochem. Biophys. Res. Commun. 494, 227–233 (2017).
Rodriguez, K. R. & Horvath, C. M. Paramyxovirus V protein interaction with the antiviral sensor LGP2 disrupts MDA5 signaling enhancement but is not relevant to LGP2-mediated RLR signaling inhibition. J. Virol. 88, 8180–8188 (2014).
Zhu, Z. et al. Foot-and-mouth disease virus infection inhibits LGP2 protein expression to exaggerate inflammatory response and promote viral replication. Cell Death Dis. 8, e2747 (2017).
Ma, Z., Ni, G. & Damania, B. Innate sensing of DNA virus genomes. Annu Rev. Virol. 5, 341–362 (2018).
Ni, G., Ma, Z. & Damania, B. cGAS and STING: at the intersection of DNA and RNA virus-sensing networks. PLoS Pathog. 14, e1007148 (2018).
Chen, Q., Sun, L. & Chen, Z. J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 (2016).
Ablasser, A. et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013).
Li, X. D. et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013).
Cai, X., Chiu, Y. H. & Chen, Z. J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296 (2014).
Chen, M. et al. TRIM14 Inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 64, 105–119 (2016).
Wang, Q. et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 13, e1006264 (2017).
Liu, Z. S. et al. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 8, 35 (2018).
Seo, G. J. et al. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 9, 613 (2018).
Hu, M. M. et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA Virus. Immunity 45, 555–569 (2016).
Cui, Y. et al. SENP7 potentiates cGAS activation by relieving SUMO-mediated inhibition of cytosolic DNA sensing. PLoS Pathog. 13, e1006156 (2017).
Xia, P. et al. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol. 17, 369–378 (2016).
Seo, G. J. et al. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. 13, 440–449 (2015).
Dai, J. et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell 176, 1447–1460 e1414 (2019).
Wang, C. et al. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity 48, 675–687 e677 (2018).
Liu, Z. S. et al. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol. 20, 18–28 (2019).
Lian, H. et al. ZCCHC3 is a co-sensor of cGAS for dsDNA recognition in innate immune response. Nat. Commun. 9, 3349 (2018).
Liang, Q. et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15, 228–238 (2014).
Ghosh, A. et al. Oligoadenylate-synthetase-family protein OASL inhibits activity of the DNA sensor cGAS during DNA virus infection to limit interferon production. Immunity 50, 51–63 e55 (2019).
Zheng, Y. et al. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 37, e99347 (2018).
Zhang, J. et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 24, 234–248 e235 (2018).
Huang, J. et al. Herpes simplex virus 1 tegument protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity. J. Virol. 92, e00841–18 (2018).
Biolatti, M. et al. Human cytomegalovirus tegument proteinpp65 (pUL83) dampens type I interferon production by inactivating the DNA sensor cGAS without affecting STING. J. Virol. 92, e01774–17 (2018)i.
Huang, Z. F. et al. Human cytomegalovirus protein UL31 inhibits DNA sensing of cGAS to mediate immune evasion. Cell Host Microbe 24, 69–80 e64 (2018).
Zhang, G. et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl Acad. Sci. USA 113, E1034–1043 (2016).
Aguirre, S. et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2, 17037 (2017).
Unterholzner, L. et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 (2010).
Duan, X. et al. Differential roles for the interferon-inducible IFI16 and AIM2 innate immune sensors for cytosolic DNA in cellular senescence of human fibroblasts. Mol. Cancer Res. 9, 589–602 (2011).
Kerur, N. et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 (2011).
Li, T., Diner, B. A., Chen, J. & Cristea, I. M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl Acad. Sci. USA 109, 10558–10563 (2012).
Almine, J. F. et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 8, 14392 (2017).
Dutta, D. et al. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-beta responses. PLoS Pathog. 11, e1005030 (2015).
Cuchet-Lourenco, D., Anderson, G., Sloan, E., Orr, A. & Everett, R. D. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 87, 13422–13432 (2013).
Roy, A. et al. Nuclear innate immune DNA sensor IFI16 is degraded during lytic reactivation of Kaposi's Sarcoma-associated herpesvirus (KSHV): role of IFI16 in maintenance of KSHV latency. J. Virol. 90, 8822–8841 (2016).
Cristea, I. M. et al. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 84, 7803–7814 (2010).
Li, T., Chen, J. & Cristea, I. M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14, 591–599 (2013).
Dell'Oste, V. et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J. Virol. 88, 6970–6982 (2014).
Jonsson, K. L. et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 8, 14391 (2017).
Zhao, C., Gillette, D. D., Li, X., Zhang, Z. & Wen, H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J. Biol. Chem. 289, 17020–17029 (2014).
Xie, M. et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 7, 13280 (2016).
Liu, L. et al. HMGB1-DNA complex-induced autophagy limits AIM2 inflammasome activation through RAGE. Biochem. Biophys. Res Commun. 450, 851–856 (2014).
Liu, T. et al. TRIM11 suppresses AIM2 inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep. 16, 1988–2002 (2016).
Huang, Y. et al. Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome. Virol. J. 14, 34 (2017).
Maruzuru, Y. et al. Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe 23, 254–265 e257 (2018).
Shi, Y. et al. Ube2D3 and Ube2N are essential for RIG-I-mediated MAVS aggregation in antiviral innate immunity. Nat. Commun. 8, 15138 (2017).
Liu, W. et al. Cyclophilin A-regulated ubiquitination is critical for RIG-I-mediated antiviral immune responses. Elife 6, e24425 (2017).
Tan, P. et al. Assembly of the WHIP-TRIM14-PPP6C mitochondrial complex promotes RIG-I-mediated antiviral signaling. Mol. Cell 68, 293–307.e5 (2017).
Lee, N. R., Choi, J. Y., Yoon, I. H., Lee, J. K. & Inn, K. S. Positive regulatory role of c-Src-mediated TRIM25 tyrosine phosphorylation on RIG-I ubiquitination and RIG-I-mediated antiviral signaling pathway. Cell Immunol. 332, 94–100 (2018).
Hu, M. M., Liao, C. Y., Yang, Q., Xie, X. Q. & Shu, H. B. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J. Exp. Med 214, 973–989 (2017).
Zhao, K. et al. Cytoplasmic STAT4 promotes antiviral type I IFN production by blocking CHIP-mediated degradation of RIG-I. J. Immunol. 196, 1209–1217 (2016).
Li, H. et al. USP14 promotes K63-linked RIG-I deubiquitination and suppresses antiviral immune responses. Eur. J. Immunol. 49, 42–53 (2019).
Zhou, P. et al. MLL5 suppresses antiviral innate immune response by facilitating STUB1-mediated RIG-I degradation. Nat. Commun. 9, 1243 (2018).
Zhao, C. et al. The E3 ubiquitin ligase TRIM40 attenuates antiviral immune responses by targeting MDA5 and RIG-I. Cell Rep. 21, 1613–1623 (2017).
Willemsen, J. et al. Phosphorylation-dependent feedback inhibition of RIG-I by DAPK1 identified by Kinome-wide siRNA Screening. Mol. Cell 65, 403–415 e408 (2017).
Kim, S. S., Sze, L. & Lam, K. P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance IFN-beta response. J. Biol. Chem. 294, 6430–6438 (2019).
Kok, K. H. et al. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9, 299–309 (2011).
Lian, H. et al. The zinc-finger protein ZCCHC3 binds RNA and facilitates viral RNA sensing and activation of the RIG-I-like receptors. Immunity 49, 438–448 e435 (2018).
Nunez, R. D. et al. The RNA helicase DDX6 associates with RIG-I to augment induction of antiviral signaling. Int J. Mol. Sci. 19, E1877 (2018).
Mahony, R., Broadbent, L., Maier-Moore, J. S., Power, U. F. & Jefferies, C. A. The RNA binding protein La/SS-B promotes RIG-I-mediated type I and type III IFN responses following Sendai viral infection. Sci. Rep. 7, 14537 (2017).
Song, Y. et al. E3 ligase FBXW7 is critical for RIG-I stabilization during antiviral responses. Nat. Commun. 8, 14654 (2017).
Soonthornvacharin, S. et al. Systems-based analysis of RIG-I-dependent signalling identifies KHSRP as an inhibitor of RIG-I receptor activation. Nat. Microbiol 2, 17022 (2017).
Sun, X. et al. MCPIP1 attenuates the innate immune response to influenza A virus by suppressing RIG-I expression in lung epithelial cells. J. Med Virol. 90, 204–211 (2018).
Nguyen, N. T., Now, H., Kim, W. J., Kim, N. & Yoo, J. Y. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci. Rep. 6, 23377 (2016).
Li, M. T. et al. Negative regulation of RIG-I-mediated innate antiviral signaling by SEC14L1. J. Virol. 87, 10037–10046 (2013).
Lin, R. et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J. Biol. Chem. 281, 2095–2103 (2006).
Du, Y. et al. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 37, 351–366 (2018).
Arimoto, K., Konishi, H. & Shimotohno, K. UbcH8 regulates ubiquitin and ISG15 conjugation to RIG-I. Mol. Immunol. 45, 1078–1084 (2008).
Meng, J. et al. ARRDC4 regulates enterovirus 71-induced innate immune response by promoting K63 polyubiquitination of MDA5 through TRIM65. Cell Death Dis. 8, e2866 (2017).
Lin, J. P., Fan, Y. K. & Liu, H. M. The 14-3-3eta chaperone protein promotes antiviral innate immunity via facilitating MDA5 oligomerization and intracellular redistribution. PLoS Pathog. 15, e1007582 (2019).
Zhu, Q. et al. DHX29 functions as an RNA co-sensor for MDA5-mediated EMCV-specific antiviral immunity. PLoS Pathog. 14, e1006886 (2018).
MacDuff, D. A. et al. HOIL1 is essential for the induction of type I and III interferons by MDA5 and regulates persistent murine noroviral infection. J. Virol. 92, e01368–18 (2018).
Li, L. F. et al. Interferon-inducible oligoadenylate synthetase-like protein acts as an antiviral effector against classical swine fever virus via the MDA5-mediated type I interferon-signaling pathway. J. Virol. 91, e01514–16 (2017).
Diao, F. et al. Negative regulation of MDA5- but not RIG-I-mediated innate antiviral signaling by the dihydroxyacetone kinase. Proc. Natl Acad. Sci. USA 104, 11706–11711 (2007).
Kitai, Y. et al. Negative regulation of melanoma differentiation-associated gene 5 (MDA5)-dependent antiviral innate immune responses by Arf-like protein 5B. J. Biol. Chem. 290, 1269–1280 (2015).
Siu, K. L. et al. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J. Virol. 88, 4866–4876 (2014).
Edwards, M. R. et al. Differential regulation of interferon responses by ebola and marburg virus VP35 proteins. Cell Rep. 14, 1632–1640 (2016).
Gupta, S. et al. Herpesvirus deconjugases inhibit the IFN response by promoting TRIM25 autoubiquitination and functional inactivation of the RIG-I signalosome. PLoS Pathog. 14, e1006852 (2018).
Gori-Savellini, G., Valentini, M. & Cusi, M. G. Toscana virus NSs protein inhibits the induction of type I interferon by interacting with RIG-I. J. Virol. 87, 6660–6667 (2013).
Wang, D. et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 85, 3758–3766 (2011).
Li, D. et al. Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-beta signaling pathway. Sci. Rep. 6, 21888 (2016).
Zhu, Z. et al. Foot-and-mouth disease virus viroporin 2B antagonizes RIG-I-Mediated Antiviral Effects by Inhibition of Its Protein Expression. J. Virol. 90, 11106–11121 (2016).
Hou, Z. et al. Hepatitis B virus inhibits intrinsic RIG-I and RIG-G immune signaling via inducing miR146a. Sci. Rep. 6, 26150 (2016).
Oshiumi, H., Miyashita, M., Matsumoto, M. & Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 9, e1003533 (2013).
Zhao, J. et al. A viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 20, 770–784 (2016).
Manokaran, G. et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221 (2015).
Chan, Y. K. & Gack, M. U. A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 17, 523–530 (2016).
Rui, Y. et al. Disruption of MDA5-mediated innate immune responses by the 3C proteins of coxsackievirus A16, coxsackievirus A6, and enterovirus D68. J. Virol. 91, e00546–17 (2017).
Li, L. et al. Encephalomyocarditis virus 2C protein antagonizes interferon-beta signaling pathway through interaction with MDA5. Antivir. Res 161, 70–84 (2019).
Kuo, R. L. et al. Role of enteroviral RNA-dependent RNA polymerase in regulation of MDA5-mediated interferon-beta activation. J. Virol. 93, e00132–19 (2019).
Lahaye, X. et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 39, 1132–1142 (2013).
Dell'Oste, V. et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegaloviral infection and is entrapped in the egressing virions during the late stage. J. Virol. 88, 6970–6982 (2014).
Acknowledgements
This work was supported by the National Research Foundation (Grant No. 2018M3A9H4079660, 2018M3A9H4078703, 2019R1A2C2008283), Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, HC., Chathuranga, K. & Lee, JS. Intracellular sensing of viral genomes and viral evasion. Exp Mol Med 51, 1–13 (2019). https://doi.org/10.1038/s12276-019-0299-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-019-0299-y
This article is cited by
-
Epigenetic regulation as a therapeutic target in the malaria parasite Plasmodium falciparum
Malaria Journal (2024)
-
Generic model to unravel the deeper insights of viral infections: an empirical application of evolutionary graph coloring in computational network biology
BMC Bioinformatics (2024)
-
BATF and BATF3 deficiency alters CD8+ effector/exhausted T cells balance in skin transplantation
Molecular Medicine (2024)
-
Targeting hypoxia-inducible factors: therapeutic opportunities and challenges
Nature Reviews Drug Discovery (2024)
-
Integration-free induced pluripotent stem cells from three endangered Southeast Asian non-human primate species
Scientific Reports (2024)
