
Abstract
We recently reported that myeloid cell-expressed sirtuin 6 (Sirt6) plays a crucial role in M1 macrophage polarization and chemotaxis. Given the prominent role of macrophages during wound repair and macrophage heterogeneity, we hypothesized that a Sirt6 deficiency in myeloid cells would delay skin wound closure by affecting the phenotypes of macrophages in wounds. To address this question, a full-thickness excisional lesion was made in the dorsal skin of myeloid cell-specific Sirt6 knockout (KO) and wild-type mice. Wound closure was delayed in the KO mice, which exhibited less collagen deposition, suppressed angiogenesis, and reduced expression of wound healing-related genes compared to the wild-type mice. Using immunohistochemical, flow cytometric, and gene-expression analyses of macrophage subpopulations from wound tissue, we identified increased infiltration of M1 macrophages with a concomitant decrease in M2 macrophage numbers in the KO mice compared to the wild-type mice. Consistent with the in vivo wound closure defects observed in the KO mice, keratinocytes and fibroblasts treated with KO macrophage-derived conditioned medium migrated slower than those treated with wild-type macrophage-derived conditioned medium. An analysis of downstream signaling pathways indicated that impaired Akt signaling underlies the decreased M2 phenotypic switching in KO mice. These results suggest that a macrophage phenotypic switch induced by Sirt6 deficiency contributes to impaired wound healing in mice.
Similar content being viewed by others


Introduction
Skin wound healing is a highly ordered process comprising several overlapping stages: (i) an inflammatory stage that cleans out debris and bacteria, (ii) a proliferative stage that refills the dermal wound space, and (iii) a long-term remodeling stage that involves the resolution of inflammation and reorganization of connective tissue into a scar1. Thus, the recruitment of inflammatory cells into the wound site is an initial event in the tissue repair process. Neutrophils form the first line of defense against infection and are a source of proinflammatory cytokines2,3. Macrophages also regulate wound healing by producing various growth factors such as transforming growth factor-β, basic fibroblast growth factor, and platelet-derived growth factor4,5,6. In response to these growth factors, epithelial cells proliferate and migrate to cover the wound, endothelial cells participate in angiogenesis, and fibroblasts contribute to the process of dermal healing7. In support of this view, suppressed recruitment of macrophages into wound sites impairs wound healing8,9. However, due to the release of proinflammatory and cytotoxic mediators, uncontrolled macrophage activity may also be detrimental to tissue repair1. Macrophages have been subdivided into two subpopulations based on their distinct gene-expression profiles: classically activated M1 macrophages and alternatively activated M2 macrophages10. Interestingly, not only does the number of macrophages infiltrating the wound site change but macrophage phenotypes also shift in the various stages of wound repair. In the inflammatory stage, M1 macrophages initiate an acute inflammatory response, whereas during the proliferative stage, M2 macrophages promote angiogenesis and granulation tissue formation11. These findings suggest that proper macrophage polarization is critical to effective wound healing. However, the mechanism that regulates macrophage polarization is unclear.
Sirtuins are a class of NAD-dependent histone deacetylases that consists of seven enzymes (Sirt1 to Sirt7) that differ in their cellular localization. Among the seven sirtuin family members, Sirt6 is localized in the nucleus and is involved in transcriptional silencing, genome stability, and longevity12. As a histone deacetylase, Sirt6 deacetylates histone H3 lysine 9 (H3K9)13 and histone H3 lysine 56 (H3K56)14 and represses the transcriptional activities of several transcription factors. As a nonhistone protein deacetylase, Sirt6 deacetylates forkhead box protein O115,16, C-terminal binding protein interacting protein17, GCN518, pyruvate kinase M219, and GATA binding protein 320. Since Sirt6 is a critical determinant of phenotypic switching and the migratory responses of macrophages21, we hypothesized that myeloid cell-expressed Sirt6 could play a role in the wound healing process. To test this hypothesis, we constructed myeloid cell-specific Sirt6 knockout (mS6KO) mice and studied the effects of Sirt6 deficiency on cell migration in vitro and inflammation and wound healing in vivo.
Materials and methods
Animals
Sirt6flox/flox mice (B6;129-Sirt6tm1Ygu/J) and LysM-Cre mice (B6.129P2-lyz2tm1(cre)Ifo/J) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Sirt6flox/flox and homozygous LysM-Cre mice were crossed to obtain mS6KO mice. To avoid potential variations due to sex and/or genetic background, female mice from the F2 generation [Sirt6flox/flox; LysM-Cre+ (mS6KO) and Sirt6flox/flox; LysM-Cre− (WT)] were used for all experiments. The mice had free access to food and water and were maintained in a room with controlled humidity (50%) and temperature (22 °C) on a 12-h light/dark cycle. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 2011). The study protocol was approved by the Institutional Animal Care and Use Committee of Chonbuk National University (Permit No. CBNU 2015-083).
Wound modeling
Twelve-week-old WT and KO mice were intraperitoneally anaesthetized with ketamine (100 mg/kg) and xylazine (5 mg/kg), shaved, and cleaned. An immediate-bonding adhesive (Grace Bio-Labs, Bend, OR, USA) was used to fix a splint to the skin. Full-thickness wounds were made in the doughnut region of the splint using 6-mm-diameter punches (Acuderm, Fort Lauderdale, FL, USA). Tegaderm film (3 M Health Care, St. Paul, MN, USA) was placed over the wounds to stabilize the wound site. The percentage of the initial wound that remained open was quantitated at different time points (days 0, 3, 5, 7, and 14). After the mice were sacrificed, the skin wounds were collected for histopathological analysis and RNA isolation.
Histology
Tissue was removed and immediately placed in fixative (10% formalin solution in 0.1 M phosphate-buffered saline (PBS)). Histological sections (5 μm) were cut from formalin-fixed, paraffin-embedded tissue blocks. To compare histopathology between lesions, we harvested skin near the center of the wound. The tissue sections were stained with hematoxylin-eosin (H&E) under standard conditions. Immunohistochemical staining was performed using a DAKO Envision system (DAKO, Carpinteria, CA, USA), which uses dextran polymers conjugated with horseradish peroxidase to avoid contamination with endogenous biotin. After deparaffinization, the tissue sections were treated using a microwave antigen-retrieval procedure with 0.01 M sodium citrate buffer. After blocking endogenous peroxidase activity, the sections were incubated with Protein Block Serum-Free (DAKO) to block nonspecific staining. The sections were then incubated with antibodies against F4/80 and von Willebrand factor (vWF; Millipore, Beverly, MA, USA). Peroxidase activity was detected with 3-amino-9-ethyl carbazole. The number of F4/80-positive macrophages was counted in five microscopic fields (magnification, 100×) for each sample in the fields with the highest numbers of F4/80-positive macrophages. The results are expressed as the average number of F4/80-positive macrophages per field. Masson’s trichrome staining was performed with a commercial kit from Abcam (ab150686, Cambridge, UK). Double-staining immunofluorescence analysis was performed to determine the types of macrophages in the skin wounds. Frozen sections were incubated with a combination of anti-F4/80 and anti-iNOS antibodies (Santa Cruz Biochemicals, Dallas, TX, USA) or anti-F4/80 and anti-MRC1 antibodies (also known as CD206; Abcam) at 4 °C overnight. Myofibroblasts were identified by staining with antibodies to vimentin (Santa Cruz Biochemicals) and α-smooth muscle actin (α-SMA; Abcam). After incubation with the corresponding fluorochrome-conjugated secondary antibodies, the sections were mounted and visualized using a LSM510 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
ELISA analysis
Tissue levels of TNF-α, IL-1β, IL-6, IL-4, IL-13, and IL-10 were measured using specific ELISA kits (all from eBioscience, San Diego, CA, USA).
Flow cytometric analysis
Wound cells were isolated by an enzymatic digestion with collagenase V. For flow cytometric analysis, Fc receptors were blocked with mouse SeroBlock FcR (CD16/CD32, eBioscience). The cells were stained with a PerCP- or FITC-conjugated anti-F4/80, a FITC- or PE-conjugated anti-Ly6g, an APC-conjugated anti-CD11b, a PE-conjugated anti-Ly6c, or a FITC-conjugated anti-MHCII antibody for 30 min at 4 °C. After washing with FACS buffer (2% fetal bovine serum (FBS) in PBS) three times, the cells were analyzed using an Accuri flow cytometer (BD Biosciences, San Jose, CA, USA).
Cell culture
The human keratinocyte cell line HaCaT was kindly donated by Dr. Jeong HS (Chonnam National University Medical School, Gwangju, Korea). The cells were grown in Dulbecco’s modified Eagle’s medium (Lonza, Walkersville, MD, USA) supplemented with 10% (v/v) FBS and antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin) at 37 °C in a humidified atmosphere with 5% CO2. A mouse dermal fibroblast cell line (MDFB) was obtained from iXCells Biotechnologies (San Diego, CA, USA) and grown in fibroblast growth medium (iXCells Biotechnologies). Adenoviruses expressing Sirt6 (AdSirt6) and β-galactosidase (AdLacZ) were prepared as described previously22.
Preparation of conditioned medium and the wound scratch assay
Bone marrow was isolated from the femurs and tibias of WT and KO mice and cultured in α-MEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS. Floating cells were defined as bone marrow macrophages (BMMs). To prepare conditioned medium (CM), BMMs (2 × 106) were grown in α-MEM supplemented with 10% FBS. Confluent cells were treated with TNF-α (10 ng/ml), IL-1β (10 ng/ml), and IL-6 (10 ng/ml) for 3 h; washed 3 times; and cultured for a further 24 h; then, the supernatants were collected and used. Cell migration was assessed by determining the ability of the cells to move into a cell-free area in a two-dimensional scratch assay. Briefly, HaCaT cells (2 × 106 cells) or MDFB cells (2 × 106 cells) were grown in a 12-well plate. When cell confluence reached 90% or higher, fresh medium containing 10 μg/ml mitomycin C was added for 2 h. The cells in the center of the well were scratched with a 100-μl sterile pipette tip to create a cell-free area. The medium was changed to WT or KO BMM-derived CM. The scratched area was photographed using a microscopy system (Carl Zeiss) soon after scratching and 12 and 36 h later. The scratch area was measured using iSolution DT 36 software (Carl Zeiss).
M2 polarization
BMMs grown in α-MEM were stimulated with IL-4 (10 ng/ml, Invitrogen) and macrophage colony-stimulating factor (10 ng/ml, Thermo Fisher Scientific, Waltham, MA, USA) for 6 h. To exogenously express Akt in BMMs, cells were transduced with adenoviruses expressing a constitutively active form of Akt (S473D/T308D, AdAkt). The adenoviruses were a kind gift from Dr. Ahn J.Y. (Sungkyunkwan University, Suwon, Korea)23.
Western blotting
Cells were homogenized in Mammalian Protein Extraction Reagent (Thermo Fisher Scientific). The homogenates (20 μg of total protein) were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The blots were probed with primary antibodies against Sirt6 (Abcam), p-Akt, Akt, p-FoxO1, p-STAT6 (Cell Signaling Technology, Beverly, MA, USA), HSP90, α-tubulin, GAPDH (Bioworld, Irving, TX, USA), Arg1 (Santa Cruz Biochemicals), and Ym1 (STEMCELL Technologies, Vancouver, Canada). Immunoreactive bands were detected with a Las-4000 imager (GE Healthcare Life Science, Pittsburgh, PA, USA).
RNA isolation and real-time RT-PCR
Total RNA was extracted from tissue using Trizol reagent (Invitrogen). RNA was precipitated with isopropanol and dissolved in diethyl pyrocarbonate-treated distilled water. First-strand cDNA was generated with oligo dT-adaptor primers by reverse transcription (TaKaRa). Specific primers were designed using qPrimerDepot (http://mouseprimerdepot.nci.nih.gov, Table S1). The real-time reverse transcription polymerase chain reaction (RT-PCR) reaction systems had a final volume of 10 μl and contained 10 ng of reverse-transcribed total RNA, 200 nM forward and reverse primers, and a PCR master mix. RT-PCR was performed in 384-well plates using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Reverse transcription and PCR were performed using a One-Step RT-PCR kit (Invitrogen). The PCR products were separated by electrophoresis on 2% agarose gels, followed by staining with ethidium bromide.
Statistical analyses
Data are expressed as the mean ± standard error of the mean. GraphPad Prism software was used to perform the statistical analyses (GraphPad Prism version 5.2, San Diego, CA, USA). Significant differences between groups were determined using an unpaired Student’s t test. A p value less than 0.05 was considered significant.
Results
Wound healing is impaired in mS6KO mice
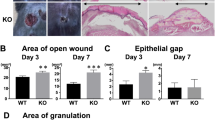
To understand the role of myeloid cell-expressed Sirt6 in excisional wound healing, we generated mS6KO mice by breeding Sirt6flox/flox mice with LysM-Cre mice. By Western blot analysis, the successful deletion of Sirt6 was confirmed in BMMs from mS6KO mice (Fig. 1a). Excision wounds were made in the dorsal skin of female mS6KO mice and their WT littermates. We observed slower wound healing in the mS6KO mice than in the WT mice, suggesting that the myeloid cell-specific Sirt6 deficiency delayed wound healing (Fig. 1b, c). Histological comparison of the wounds further confirmed delayed wound closure in the mS6KO mice (Fig. 1d). Trichrome staining of tissue harvested on day 14 after wounding showed reduced collagen content in the granulation tissue of the mS6KO mice (Fig. 2a). Similarly, the intensity of vWF-positive immunostaining was decreased in KO tissue, indicating that angiogenesis was also suppressed in the mS6KO mice (Fig. 2a). The mRNA levels of extracellular matrix genes (Col1a1, Col3a1, Timp1, Pdgfra, and Tgfb1) and the angiogenesis gene Vegfa were significantly suppressed in the skin tissue of the mS6KO mice (Fig. 2b). Consistent with these results, the number of myofibroblasts (vimentin+α-SMA+ cells) was significantly decreased in the skin wounds of the mS6KO mice (Fig. 2c). Collectively, these data suggest that myeloid cell-specific Sirt6 deficiency delays wound healing through the repression of collagen deposition, epithelial regrowth, and angiogenesis.

a The process used to generate myeloid cell-specific Sirt6 knockout (mS6KO) mice is shown. b Excisional wounds were made by 6-mm-diameter punches. The wound sites were photographed at the indicated time. A day 0 picture was taken immediately after injury. Representative results from five individual mice from each group are shown. c Wound photographs were analyzed at the indicated times to determine wound closure in WT and KO mice. Values represent the mean ± SEM (n = 8). *p < 0.05 and **p < 0.01 vs. WT. d Histological sections of mouse skin wounds were examined by H&E staining. Bars = 250 μm

a Representative photomicrographs of wounds stained with Masson’s trichrome or immunostained with an anti-vWF antibody at 0 and 14 days after injury are shown (original magnification, 200×). Bars = 50 μm. b The mRNA levels of extracellular matrix-related genes in wounds were determined by real-time RT-PCR (n = 8). c Skin wounds of WT and KO mice were double stained for vimentin and α-SMA expression to identify myofibroblasts. The vimentin and α-SMA-positive cells were counted in sections of the wounds harvested on day 14 (n = 6). Values represent the mean ± SEM. *p < 0.05 and **p < 0.01 vs. WT
Myeloid cell-specific Sirt6 deficiency decreases M2 macrophage infiltration into skin wounds
We examined the infiltration of macrophages, which are critical inflammatory cells for wound healing, into wound sites by counting F4/80-positive cells. The accumulation of F4/80-immunopositive cells was significantly increased in mS6KO mice compared to WT mice (Fig. 3a, b). Real-time RT-PCR for F4/80 mRNA (Adgre1) expression and flow cytometric analysis for wound-associated macrophages (F480hiLy6gloCD11bhi) corroborated the increased accumulation of macrophages in mS6KO mice compared to WT mice (Figs. 3c, d, and S1A).

a Immunohistochemistry was used to identify infiltrating macrophages (F4/80+ cells) at 0, 3, and 7 days after injury. Bars = 50 μm. b F4/80-positive macrophages were counted in wound sections. c The mRNA level of F4/80 (Adgre1) in the wound site was determined by real-time RT-PCR (n = 8). d, e Single-cell suspensions were prepared by enzymatically digesting and gently dissociating skin wounds. After excluding dead cells, the cells were analyzed by flow cytometry to identify all macrophages (F4/80hiLy6gloCD11bhi) and M2 macrophages (F4/80hiLy6cloMHCIIhi) (n = 5–6). Values represent the mean ± SEM. *p < 0.05 and **p < 0.01 vs. WT
Next, we immunostained skin wounds with an M1-specific anti-iNOS antibody and M2-specific anti-MRC1 antibody. Myeloid cell-specific Sirt6 deficiency increased the number of F4/80+iNOS+ macrophages and decreased the number of F4/80+MRC1+ macrophages in the skin wounds of mS6KO mice compared to those of WT mice (Fig. 4a, b). Flow cytometric analysis also showed a decrease in the accumulation of M2 macrophages (F480hiLy6cloMHCIIhi) in the skin wounds of mS6KO mice (Figs. 3e and S1B). Consistently, mRNA expression levels for M1 marker genes (Ccl2, Tnfa, Il6, Il1b, Nos2, and Il1b) and M1 cytokines (TNF-α, IL-1β, and IL-6) were increased, while the levels of M2 marker genes (Clec7a, Arg1, Chil3, Mrc1, and Mgl1) and M2 cytokines (IL-4, IL-13, and IL-10) were decreased in mS6KO tissue (Fig. 5a–d). These results indicate a decrease in the accumulation of M2-type macrophages in the skin wounds of mS6KO mice.

The skin wounds of WT and KO mice were double stained for F4/80 and iNOS to identify M1 macrophages (a) or F4/80 and MRC1 to identify M2 macrophages (b). Digitally merged signals are shown in the right panels. Bars = 50 μm. Representative results from three independent experiments are shown

a, b The mRNA levels of M1 and M2 marker genes in the wound site were determined by real-time RT-PCR (n = 8). c, d The levels of M1 and M2 cytokines in the tissue were measured using ELISA (n = 6). Values represent the mean ± SEM. *p < 0.05 and **p < 0.01 vs. WT
CM from mS6KO BMMs decreases scratch wound closure
To examine whether secreted factors from M1 macrophages suppress cell migration during wound closure, we conducted scratch wound assays (Fig. S2). We chose epidermal keratinocytes (HaCaT cells) and dermal fibroblasts (MDFB cells) for the cell migration study. CM from KO BMMs significantly decreased the rate of HaCaT cell migration compared to CM from WT BMMs (Fig. 6a). Complete gap closure was seen with treatment with CM from WT BMMs by 36 h, as opposed to treatment with CM from KO BMMs resulting in only 58% closure at the same time. In experiments with MDFB cells, the overall results were similar: the fibroblasts treated with CM from WT BMMs migrated more quickly to fill the scratch area than those treated with CM from KO BMMs (Fig. 6b). Next, we assessed whether exogenous Sirt6 expression in KO BMMs could rescue the migratory defect seen in HaCaT and MDFB cells. Ectopic overexpression of Sirt6 in KO BMMs rescued cells from the migratory defect (Fig. 6a, b).

Bone marrow macrophages (BMMs, 2 × 106) isolated from WT or KO mice were treated with TNF-α (10 ng/ml), IL-1β (10 ng/ml), and IL-6 (10 ng/ml) for 3 h, and then conditioned medium (CM) was harvested. Scratch wounds were made in monolayers of HaCaT (a) and MDFB cells (b). For rescue experiments, KO BMMs were transduced with AdSirt6. Representative images show the progression of wound closure at the indicated times. The scratch area is indicated by a dotted line. The rate of wound closure was calculated by measuring the scratch area under the same magnification at different times. Values represent the mean ± SEM (n = 8) **p < 0.01 vs. WT; #p < 0.05; and ##p < 0.01 vs. KO
Myeloid cell-specific Sirt6 deficiency suppresses M2 polarization through downregulation of the PI3K-Akt pathway
To explain the decreased number of M2 macrophages in skin wounds in mS6KO mice, we compared M2 polarization in vitro. BMMs isolated from mS6KO mice expressed markedly lower levels of M2 marker genes (Arg1, Il10, Clec7a, Mrc1, and Chil3) in IL-4-stimulated M2-polarizing conditions than BMMs from WT mice (Fig. 7a). The classic IL-4 signaling pathway involves the activation of phosphoinositide 3-kinase (PI3K)-Akt through the recruitment of IRS-1/224. We observed increased levels of p-Akt and the downstream molecule p-FoxO1 after IL-4 stimulation in BMMs from WT mice, whereas this increase was not observed in BMMs from mS6KO mice (Figs. 7b and S3A). In addition to activating Akt, IL-4 treatment also leads to the activation of JAK2-STAT625. The phosphorylation levels of STAT6 were similar in BMMs from mS6KO and WT mice (Fig. 7b), indicating that the change in M2 marker expression was mediated through the downregulation of components of the PI3K-Akt pathway and not through the JAK2-STAT6 pathway. To further investigate the regulation of the PI3K-Akt signaling pathway by Sirt6 under M2-polarizing conditions, we transduced BMMs with either Sirt6 (AdSirt6) or a constitutively active form of Akt (AdAkt). Treatment with IL-4 led to a moderate increase in M2 marker protein expression in the BMMs from mS6KO mice expressing Sirt6 or Akt (Figs. 7c and S3B). Accordingly, the levels of M2 marker genes were increased in the Sirt6- or Akt-overexpressing KO cells (Fig. 7d). The role of Akt signaling in the enhancement of M2 polarization was further tested by blocking Akt signals with MK2206 or Akti. When WT BMMs were pretreated with these Akt inhibitors prior to stimulation with IL-4, Akt phosphorylation and M2 marker gene expression were markedly suppressed (Figs. 7e, S3C, and 7F). Finally, consistent with these in vitro results, in vivo results showed that Akt phosphorylation was increased in skin wounds in WT mice but was significantly impaired in skin wounds in KO mice (Fig. S4).

a BMMs were treated with 10 ng/ml IL-4 and 10 ng/ml MCSF for 6 h, and the expression patterns of M2 marker genes were compared by RT-PCR. b BMMs were treated with 10 ng/ml IL-4 for the indicated periods, and then whole-cell extracts were generated and analyzed by Western blotting. c, d BMMs from mS6KO mice were transduced with an adenovirus expressing β-galactosidase (AdLacZ), Sirt6 (AdSirt6), or CA-Akt (AdAkt) at a multiplicity of infection (MOI) of 100 and treated with IL-4 for the indicated times. Protein and mRNA levels were analyzed. e, f BMMs from WT mice were treated with 10 ng/ml IL-4 and 10 ng/ml MCSF for 6 h with or without Akt inhibitors (8 μM MK2206 and 10 μM Akti), and then the protein levels in the whole cell extract and mRNA levels of M2 marker genes were determined. Values represent the mean ± SEM (n = 3). *p < 0.05 and **p < 0.01 vs. AdLacZ or Veh; #p < 0.05 vs. IL-4. Veh Vehicle, MK MK2206, i Akti
Discussion
In this study, we demonstrated that myeloid cell-specific Sirt6 deficiency leads to delayed wound closure compared to WT control. This aberrant wound closure pattern was associated with the augmented infiltration of macrophages that failed to phenotypically switch from M1 to M2 macrophages.
Previously, Thandavarayan et al.26 observed that Sirt6 knockdown impairs diabetic wound closure with concomitantly increased levels of oxidative stress, inflammatory cytokines, and NF-κB activation in skin wounds. Similarly, the sirtuin activator resveratrol accelerates wound repair by increasing keratinocyte proliferation, while the sirtuin inhibitor sirtinol retards wound closure27. More recently, Hu et al.28 showed that Sirt6 KO mice display delayed and incomplete healing of the cornea after wounding. These reports suggest a beneficial role for sirtuin (specifically Sirt6) activation in wound healing. Based on this background, we aimed to understand how a myeloid cell-specific Sirt6 deficiency would affect macrophage infiltration and wound closure using mS6KO mice. Our results showed that the number of macrophages in excisional wounds was markedly increased in mS6KO mice. The significantly increased expression of a macrophage chemoattractant, CCL2, in mS6KO mice is possibly responsible for the increased infiltration of macrophages seen in the wounds of this strain compared to those of WT mice. Remarkably, we clearly demonstrated heterogeneity in the macrophage populations recruited to the wounds of mS6KO mice. The expression levels of established M1-specific marker genes were upregulated in skin wounds in mS6KO mice, while the levels of M2 marker genes were downregulated. These results imply that the wound environment in mS6KO mice favors the proinflammatory M1 status.
Ample evidence points to a phenotypic switch from an M1 to an M2 macrophage in the process of wound repair29. In the early inflammatory phase, M1 macrophages are the predominant cells in the tissue and are involved in the clearance of pathogens and dead cells and in the modulation of the adaptive immune system. In the later proliferative phase, M2 macrophages contribute to the resolution of inflammation and tissue remodeling. While STAT6 has been shown to be required for IL-4 to exert its effects24, the level of p-STAT6 was shown to be unaffected by Sirt6 deficiency, indicating that IL-4 impacts other signaling pathways. We observed enhanced levels of p-Akt in BMMs after exposure to IL-4, a finding consistent with that of a previous study showing that the activation of the PI3K-Akt pathway results in stronger polarization of macrophages toward the M2 phenotype25. Moreover, the transduction of KO BMMs with CA-Akt selectively restored the expression of M2 marker genes. These results indicate that the activation of the PI3K-Akt pathway is required for the expression of M2 markers; however, future studies are needed to determine how Sirt6 deficiency affects the PI3K-Akt pathway.
Using in vivo and in vitro approaches, we demonstrated that myeloid cell-specific Sirt6 deficiency utilizes the following mechanisms to delay wound closure. First, more proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 were produced in mS6KO mice. It is well known that M1-type macrophages are a source of proinflammatory cytokines. These cytokines alter the normal functioning of epithelial cells and dermal fibroblasts and ultimately delay the rates of re-epithelialization (epithelium closure) and wound closure (dermis closure)29. Second, we observed a lower number of vWF-positive cells in mS6KO mice than in WT mice. M1 macrophages are described as having an antiangiogenic profile, which is considered detrimental to recovery after an inflammatory event30,31. Third, the delayed wound closure observed in mS6KO mice was accompanied by decreases in the migration of keratinocytes and dermal fibroblasts, suggesting that myeloid cell-specific Sirt6 deficiency might impair epithelial and dermal closure by suppressing cell migration. Although the underlying mechanisms by which myeloid cell-specific Sirt6 deficiency suppresses keratinocyte and dermal fibroblast migration remain elusive, these findings suggest, for the first time, that myeloid cell-specific Sirt6 deficiency has a deleterious role in excisional wound healing. Therefore, myeloid cell-specific Sirt6 activation might be a therapeutic strategy for accelerating wound healing.
References
Eming, S. A., Krieg, T. & Davidson, J. M. Inflammation in wound repair: molecular and cellular mechanisms. J. Invest. Dermatol. 127, 514–525 (2007).
Kanno, E. et al. Wound healing in skin promoted by inoculation with Pseudomonas aeruginosa PAO1: The critical role of tumor necrosis factor-α secreted from infiltrating neutrophils. Wound Repair Regen. 19, 608–621 (2011).
Kim, M. H. et al. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J. Invest. Dermatol. 128, 1812–1820 (2008).
Al-Mulla, F., Leibovich, S. J., Francis, I. M. & Bitar, M. S. Impaired TGF-β signaling and a defect in resolution of inflammation contribute to delayed wound healing in a female rat model of type 2 diabetes. Mol. Biosyst. 7, 3006–3020 (2011).
Numata, Y. et al. The accelerating effect of histamine on the cutaneous wound-healing process through the action of basic fibroblast growth factor. J. Invest. Dermatol. 126, 1403–1409 (2006).
Plasari, G. et al. Nuclear factor I-C links platelet-derived growth factor and transforming growth factor β1 signaling to skin wound healing progression. Mol. Cell Biol. 29, 6006–6017 (2009).
Barrientos, S., Stojadinovic, O., Golinko, M. S., Brem, H. & Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 16, 585–601 (2008).
Goren, I. et al. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am. J. Pathol. 175, 132–147 (2009).
Mirza, R., DiPietro, L. A. & Koh, T. J. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am. J. Pathol. 175, 2454–2462 (2009).
Jablonski, K. A. et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE 10, e0145342 (2015).
Lucas, T. et al. Differential roles of macrophages in diverse phases of skin repair. J. Immunol. 184, 3964–3977 (2010).
Kugel, S. & Mostoslavsky, R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem. Sci. 39, 72–81 (2014).
Kawahara, T. L. et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell 136, 62–74 (2009).
Michishita, E. et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8, 2664–2666 (2009).
Song, M. Y., Wang, J., Ka, S. O., Bae, E. J. & Park, B. H. Insulin secretion impairment in Sirt6 knockout pancreatic β cells is mediated by suppression of the FoxO1-Pdx1-Glut2 pathway. Sci. Rep. 6, 30321 (2016).
Woo, S. J. et al. Myeloid sirtuin 6 deficiency accelerates experimental rheumatoid arthritis by enhancing macrophage activation and infiltration into synovium. EBioMedicine 38, 228–237 (2018).
Kaidi, A., Weinert, B. T., Choudhary, C. & Jackson, S. P. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329, 1348–1353 (2010).
Dominy, J. E. Jr. et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 48, 900–913 (2012).
Bhardwaj, A., Bhardwaj, A. & Das, S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl Acad. Sci. USA 113, E538–E547 (2016).
Jang, H. Y., Gu, S., Lee, S. M. & Park, B. H. Overexpression of sirtuin 6 suppresses allergic airway inflammation through deacetylation of GATA3. J. Allergy Clin. Immunol. 138, 1452–1455 (2016).
Lee, Y. et al. Myeloid sirtuin 6 deficiency causes insulin resistance in high-fat diet-fed mice by eliciting macrophage polarization toward an M1 phenotype. Diabetes 66, 2659–2668 (2017).
Lee, H. S. et al. Overexpression of sirtuin 6 suppresses inflammatory responses and bone destruction in mice with collagen-induced arthritis. Arthritis Rheum. 65, 1776–1785 (2013).
Ko, H. R. et al. Akt1-Inhibitor of DNA binding2 is essential for growth cone formation and axon growth and promotes central nervous system axon regeneration. Elife 5, e20799 (2016).
Barrett, J. P., Minogue, A. M., Falvey, A. & Lynch, M. A. Involvement of IGF-1 and Akt in M1/M2 activation state in bone marrow-derived macrophages. Exp. Cell Res. 335, 258–268 (2015).
Gordon, S. & Martinez, F. O. Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604 (2010).
Thandavarayan, R. A. et al. Sirtuin-6 deficiency exacerbates diabetes-induced impairment of wound healing. Exp. Dermatol 24, 773–778 (2015).
Spallotta, F. et al. A nitric oxide-dependent cross-talk between class I and III histone deacetylases accelerates skin repair. J. Biol. Chem. 288, 11004–11012 (2013).
Hu, X. et al. Sirt6 deficiency impairs corneal epithelial wound healing. Aging 10, 1932–1946 (2018).
Mahdavian Delavary, B., van der Veer, W. M., van Egmond, M., Niessen, F. B. & Beelen, R. H. Macrophages in skin injury and repair. Immunobiology 216, 753–762 (2011).
Chambers, S. E., O’Neill, C. L., O’Doherty, T. M., Medina, R. J. & Stitt, A. W. The role of immune-related myeloid cells in angiogenesis. Immunobiology 218, 1370–1375 (2013).
Mosser, D. M. & Edwards, J. P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008).
Acknowledgements
This research was supported by grants from the Medical Research Center Program (2017R1A5A2015061) and Basic Science Research Program (2017R1A2B2005730, 2018R1D1A1B07050772, and 2019R1A2C2002281) through the National Research Foundation of Korea (NRF), which is funded by the Korean government, and by a fund of the Biomedical Research Institute, Chonbuk National University Hospital (to S.K.Y.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koo, JH., Jang, HY., Lee, Y. et al. Myeloid cell-specific sirtuin 6 deficiency delays wound healing in mice by modulating inflammation and macrophage phenotypes. Exp Mol Med 51, 1–10 (2019). https://doi.org/10.1038/s12276-019-0248-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-019-0248-9
