
Abstract
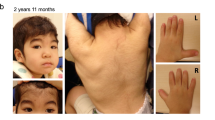
The RNA-binding motif protein 10, RBM10, is an RNA splicing regulator essential for development. Loss-of-function RBM10 variants are associated with TARP syndrome, a severe X-linked recessive condition in males. We report a 3-year-old male with a mild phenotype, consisting of cleft palate, hypotonia, developmental delay, and minor dysmorphisms, associated with a missense RBM10 variant, c.943T>C, p.Ser315Pro, affecting the RRM2 RNA-binding domain. His clinical features were similar to a previously reported case associated with a missense variant. The p.Ser315Pro mutant protein was expressed normally in the nucleus, but its expression level and protein stability were slightly reduced. Nuclear magnetic resonance spectroscopy showed that the structure and the RNA-binding ability of the RRM2 domain with the p.Ser315Pro were unaffected. However, it affects the alternative splicing regulations of downstream genes, NUMB and TNRC6A, and its splicing alteration patterns were variable depending on target transcripts. In summary, a novel germline missense RBM10 p.Ser315Pro variant that causes functional changes in the expression of its downstream genes results in a non-lethal phenotype associated with developmental delays. The functional alteration effects depend on the residues affected by missense variants. Our findings are expected to bring broader insights into the RBM10-associated genotype-phenotype relationships by delineating the molecular mechanism of RBM10 functions.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others


Data availability
Data are available from corresponding authors upon reasonable request.
References
Inoue A. RBM10: Structure, functions, and associated diseases. Gene. 2021;783:145463.
Wang K, Bacon ML, Tessier JJ, Rintala-Maki ND, Tang V, Sutherland LC. RBM10 modulates apoptosis and influences TNF-alpha gene expression. J Cell Death. 2012;5:1–19.
Hernandez J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcarcel J. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016;13:466–72.
Guan G, Li R, Tang W, Liu T, Su Z, Wang Y, et al. Expression of RNA-binding motif 10 is associated with advanced tumor stage and malignant behaviors of lung adenocarcinoma cancer cells. Tumour Biol. 2017;39:1010428317691740.
Mohan N, Kumar V, Kandala DT, Kartha CC, Laishram RS. A splicing-independent function of RBM10 controls specific 3’ UTR processing to regulate cardiac hypertrophy. Cell Rep. 2018;24:3539–53.
Wang Y, Gogol-Doring A, Hu H, Frohler S, Ma Y, Jens M, et al. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol Med. 2013;5:1431–42.
Rodor J, FitzPatrick DR, Eyras E, Caceres JF. The RNA-binding landscape of RBM10 and its role in alternative splicing regulation in models of mouse early development. RNA Biol. 2017;14:45–57.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008;456:470–6.
Yang X, Coulombe-Huntington J, Kang S, Sheynkman GM, Hao T, Richardson A, et al. Widespread expansion of protein interaction capabilities by alternative splicing. Cell 2016;164:805–17.
Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, Center NIHIS. et al. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet. 2010;86:743–8.
Imagawa E, Konuma T, Cork EE, Diaz GA, Oishi K. A novel missense variant in RBM10 can cause a mild form of TARP syndrome with developmental delay and dysmorphic features. Clin Genet. 2020;98:606–12.
Kumps C, D’Haenens E, Vergult S, Leus J, van Coster R, Jansen A, et al. Phenotypic spectrum of the RBM10-mediated intellectual disability and congenital malformation syndrome beyond classic TARP syndrome features. Clin Genet. 2021;99:449–56.
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18:696–704.
Collins KM, Kainov YA, Christodolou E, Ray D, Morris Q, Hughes T, et al. An RRM-ZnF RNA recognition module targets RBM10 to exonic sequences to promote exon exclusion. Nucleic Acids Res. 2017;45:6761–74.
Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell. 2013;52:720–33.
Gripp KW, Hopkins E, Johnston JJ, Krause C, Dobyns WB, Biesecker LG. Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. Am J Med Genet Part A. 2011;155A:2516–20.
Johnston JJ, Sapp JC, Curry C, Horton M, Leon E, Cusmano-Ozog K, et al. Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. Am J Med Genet Part A. 2014;164A:120–8.
Powis Z, Hart A, Cherny S, Petrik I, Palmaer E, Tang S, et al. Clinical diagnostic exome evaluation for an infant with a lethal disorder: genetic diagnosis of TARP syndrome and expansion of the phenotype in a patient with a newly reported RBM10 alteration. BMC Med Genet. 2017;18:60.
Hojland AT, Lolas I, Okkels H, Lautrup CK, Diness BR, Petersen MB, et al. First reported adult patient with TARP syndrome: a case report. Am J Med Genet Part A. 2018;176:2915–18.
Kaeppler KE, Stetson RC, Lanpher BC, Collura CA. Infant male with TARP syndrome: review of clinical features, prognosis, and commonalities with previously reported patients. Am J Med Genet Part A. 2018;176:2911–14.
Niceta M, Barresi S, Pantaleoni F, Capolino R, Dentici ML, Ciolfi A, et al. TARP syndrome: long-term survival, anatomic patterns of congenital heart defects, differential diagnosis and pathogenetic considerations. Eur J Med Genet. 2019;62:103534.
Manotas H, Payan-Gomez C, Roa MF, Pineros JG. TARP syndrome associated with renal malformation and optic nerve atrophy. BMJ Case Rep. 2021;14:e240601.
Daicheng H, Shiwen X, Jingxuan Z, Junbo H, Bo W. A frameshift RBM10 variant associated with TARP syndrome. Front Genet. 2022;13:922048.
Poulton C, Baynam G, Pugh K, Mason M, Kiraly-Borri C, Gration D, et al. Further evidence for distinct traits associated with RBM10 missense variants. Clin Genet. 2022;102:161–63.
Potter AB, O’Brien TD, Kulkarni A, McCabe S, Matthews K, Kovak K, et al. Missense variant in RBM10 associated with mild and non-lethal form of TARP syndrome. Clin Genet. 2023. In press.
Zhao J, Sun Y, Huang Y, Song F, Huang Z, Bao Y, et al. Functional analysis reveals that RBM10 mutations contribute to lung adenocarcinoma pathogenesis by deregulating splicing. Sci Rep. 2017;7:40488.
Wang LY, Xiao SJ, Kunimoto H, Tokunaga K, Kojima H, Kimura M, et al. Sequestration of RBM10 in nuclear bodies: targeting sequences and biological significance. Int J Mol Sci. 2021;22:10526.
Goto Y, Kimura H. Inactive X chromosome-specific histone H3 modifications and CpG hypomethylation flank a chromatin boundary between an X-inactivated and an escape gene. Nucleic Acids Res. 2009;37:7416–28.
Martin HC, Gardner EJ, Samocha KE, Kaplanis J, Akawi N, Sifrim A, et al. The contribution of X-linked coding variation to severe developmental disorders. Nat Commun. 2021;12:627.
Acknowledgements
We would like to thank the patient and his family for contributing to this study and the personnel of Microscopy Core Facility at the Icahn School of Medicine at Mount Sinai.
Author information
Authors and Affiliations
Contributions
Conceptualization: EI, KO. Investigation: EI, LM, TK. Supervision: KO. Patient recruitment and consent acquisition: VKM, CN. Writing - original draft: EI, TK, KO. Writing - review and editing: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
It was a non-interventional single case report describing clinical data that was collected as part of routine clinical care. No research or non-clinical testing or evaluations were done on the patient or patient-derived samples. IRB approval was not required according to the institution’s policy (Central Michigan University School of Medicine), while written informed consent was obtained from the patient’s parents for the publication as stated above. The original study regarding the functional assays of the p.Pro322Leu variant was approved by the Institutional Review Board of Icahn School of Medicine at Mount Sinai (GCO# 05-1150, HS# 10-00584).
Informed consent
Written informed consent was obtained from the patient’s parents for the publication of the patient’s clinical information and photographs.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Imagawa, E., Moreta, L., Misra, V.K. et al. Functional insight into a neurodevelopmental disorder caused by missense variants in an RNA-binding protein, RBM10. J Hum Genet 68, 643–648 (2023). https://doi.org/10.1038/s10038-023-01162-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-023-01162-0
