
Abstract
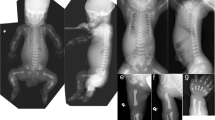
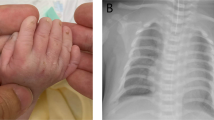
A null mutation in a patient can facilitate phenotype assignment and uncovers the function of that specific gene. We present five sibs of a consanguineous Pakistani family afflicted with a new syndrome with an unusual combination of skeletal anomalies including cranial asymmetry, fused sagittal sutures deviating from the medial axis, mandibular prognathia, maxillary hypoplasia, misaligned and crowded teeth, delayed bone age, multiple dislocations, hypoplastic and malpositioned patellae, humeral intracondylar fissures, scapular dyskinesis, long limbs, lumbar lordosis, protruding chest, prominent clavicles, short 5th digital rays, and ventral transverse digital creases plus features of cutis laxa. We mapped the disease gene locus to a 3.62-Mb region at 17q25.3 and identified a homozygous deletion of maximal 7.3 kb deduced to totally inactivate MYADML2 and downstream gene PYCR1, biallelic variants in which cause autosomal recessive cutis laxa (ARCL). All five affected sibs had the most common features of ARCL but not many of the less common ones. We attributed the anomalies not typical for ARCL to MYADML2 deficit, because no other genetic defect possibly a candidate to underlie the skeletal phenotype was found. MYADML2 is a gene of unknown function, has not been studied, and has not been associated with disease. Our findings present a possible phenotype for MYADML2 deficit that includes impaired bone patterning and maturation, definitely show that the gene is not essential for survival, and provide a start point for future studies on the function of MYADML2 protein. Detection of new patients is needed to confirm and delineate MYADML2-deficiency phenotype.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

Data availability
Additional data that support the findings of this study are available from the corresponding authors upon request.
References
Dougherty KM, Brandriss MC, Valle D. Cloning human pyrroline-5-carboxylate reductase cDNA by complementation in Saccharomyces cerevisiae. J Biol Chem. 1992;15:871–5.
Guernsey DL, Jiang H, Evans SC, Ferguson M, Matsuoka M, Nightingale M, et al. Mutation in pyrroline-5-carboxylate reductase 1 gene in families with cutis laxa type 2. Am J Hum Genet. 2009;85:120–9.
Reversade B, Escande-Beillard N, Dimopoulou A, Fischer B, Chng SC, Li Y, et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet. 2009;41:1016–21.
Kretz R, Bozorgmehr B, Kariminejad MH, Rohrbach M, Hausser I, Baumer A, et al. Defect in proline synthesis: pyrroline-5-carboxylate reductase 1 deficiency leads to a complex clinical phenotype with collagen and elastin abnormalities. J Inherit Metab Dis. 2011;34:731–9.
Lin DS, Chang JH, Liu HL, Wei CH, Yeung CY, Ho CS, et al. Compound heterozygous mutations in PYCR1 further expand the phenotypic spectrum of De Barsy syndrome. Am J Med Genet A. 2011;155A:3095–9.
Lin DS, Yeung CY, Liu HL, Ho CS, Shu CH, Chuang CK, et al. A novel mutation in PYCR1 causes an autosomal recessive cutis laxa with premature aging features in a family. Am J Med Genet A. 2011;155A:1285–9.
Dimopoulou A, Fischer B, Gardeitchik T, Schroter P, Kayserili H, Schlack C, et al. Genotype-phenotype spectrum of PYCR1-related autosomal recessive cutis laxa. Mol Genet Metab. 2013;110:352–61.
Scherrer DZ, Baptista MB, Matos AH, Maurer-Morelli CV, Steiner CE. Mutations in PYCR1 gene in three families with autosomal recessive cutis laxa, type 2. Eur J Med Genet. 2013;56:336–9.
Gardeitchik T, Mohamed M, Fischer B, Lammens M, Lefeber D, Lace B, et al. Clinical and biochemical features guiding the diagnostics in neurometabolic cutis laxa. Eur J Hum Genet. 2014;22:888–95.
Alazami AM, Al-Qattan SM, Faqeih E, Alhashem A, Alshammari M, Alzahrani F, et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum Genet. 2016;135:525–40.
Vahidnezhad H, Karamzadeh R, Saeidian AH, Youssefian L, Sotoudeh S, Zeinali S, et al. Molecular dynamics simulation of the consequences of a PYCR1 mutation (p.Ala189Val) in patients with complex connective tissue disorder and severe intellectual disability. J Investig Dermatol. 2017;137:525–8.
Ritelli M, Palit A, Giacopuzzi E, Inamadar AC, Dordoni C, Mujja A, et al. Clinical and molecular characterization of a 13-year-old Indian boy with cutis laxa type 2B: Identification of two novel PYCR1 mutations by amplicon-based semiconductor exome sequencing. J Dermatol Sci. 2017;88:141–3.
Lessel D, Ozel AB, Campbell SE, Saadi A, Arlt MF, McSweeney KM, et al. Analyses of LMNA-negative juvenile progeroid cases confirms biallelic POLR3A mutations in Wiedemann-Rautenstrauch-like syndrome and expands the phenotypic spectrum of PYCR1 mutations. Hum Genet. 2018;137:921–39.
Onoufriadis A, Nanda A, Sheriff A, Tomita K, Gomaa NS, Simpson MA, et al. Consanguinity and double recessive gene pathology: cutis laxa (PYCR1) and nephrotic syndrome (PLCE1). JAMA Dermotol. 2019;155:257–9.
Morava E, Guillard M, Lefeber DJ, Wevers RA. Autosomal recessive cutis laxa syndrome revisited. Eur J Hum Genet. 2009;17:1099–110.
Kariminejad A, Afroozan F, Bozorgmehr B, Ghanadan A, Akbaroghli S, Khorram Khorshid HR, et al. Discriminative features in three autosomal recessive cutis laxa syndromes: cutis laxa IIA, cutis laxa IIB, and geroderma osteoplastica. Int J Mol Sci. 2017;18:E635.
Aranda JF, Reglero-Real N, Kremer L, Marcos-Ramiro B, Ruiz-Saenz A, Calvo M, et al. MYADM regulates Rac1 targeting to ordered membranes required for cell spreading and migration. Mol Biol Cell. 2011;22:1252–62.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–61.
Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46:D649–55.
The Gene Ontology C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47:D330–8.
Gudbjartsson DF, Sulem P, Helgason H, Gylfason A, Gudjonsson SA, et al. Sequence variants from whole genome sequencing a large group of Icelanders. Sci Data. 2015;2:150011.
Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352:474–7.
Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–9.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;9:877–85.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–94.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–6.
Acknowledgements
We thank the family members for their cooperation. This study was supported by the Scientific and Technological Research Council of Turkey (114Z829 to AT) and URF-QAU Pakistan (QAU-URF-2017-18 to SM).
Author information
Authors and Affiliations
Contributions
Conception and design: AT and SM. Analysis and interpretation: EYB, SM, and AT. Clinical data collection: RMKS and SM. Obtained funding: AT and SM.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yıldız Bölükbaşı, E., Shabbir, R.M.K., Malik, S. et al. Homozygous deletion of MYADML2 in cranial asymmetry, reduced bone maturation, multiple dislocations, lumbar lordosis, and prominent clavicles. J Hum Genet 66, 171–179 (2021). https://doi.org/10.1038/s10038-020-0817-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-020-0817-8

{kind=link}