
Abstract
The stress hormone cortisol modulates fuel metabolism, cardiovascular homoeostasis, mood, inflammation and cognition. The CORtisol NETwork (CORNET) consortium previously identified a single locus associated with morning plasma cortisol. Identifying additional genetic variants that explain more of the variance in cortisol could provide new insights into cortisol biology and provide statistical power to test the causative role of cortisol in common diseases. The CORNET consortium extended its genome-wide association meta-analysis for morning plasma cortisol from 12,597 to 25,314 subjects and from ~2.2 M to ~7 M SNPs, in 17 population-based cohorts of European ancestries. We confirmed the genetic association with SERPINA6/SERPINA1. This locus contains genes encoding corticosteroid binding globulin (CBG) and α1-antitrypsin. Expression quantitative trait loci (eQTL) analyses undertaken in the STARNET cohort of 600 individuals showed that specific genetic variants within the SERPINA6/SERPINA1 locus influence expression of SERPINA6 rather than SERPINA1 in the liver. Moreover, trans-eQTL analysis demonstrated effects on adipose tissue gene expression, suggesting that variations in CBG levels have an effect on delivery of cortisol to peripheral tissues. Two-sample Mendelian randomisation analyses provided evidence that each genetically-determined standard deviation (SD) increase in morning plasma cortisol was associated with increased odds of chronic ischaemic heart disease (0.32, 95% CI 0.06–0.59) and myocardial infarction (0.21, 95% CI 0.00–0.43) in UK Biobank and similarly in CARDIoGRAMplusC4D. These findings reveal a causative pathway for CBG in determining cortisol action in peripheral tissues and thereby contributing to the aetiology of cardiovascular disease.
Similar content being viewed by others

Introduction
Cortisol plays a vital role in adaptation to environmental stress, modulating fuel metabolism, cardiovascular homoeostasis, mood, memory and inflammation [1]. Cortisol levels vary throughout the day under the control of the hypothalamic–pituitary–adrenal (HPA) axis. Patients with tumours causing excess cortisol develop Cushing’s syndrome, characterised by a host of features including obesity, hypertension, diabetes mellitus, depression, cognitive impairment and osteoporosis with an excess mortality due primarily to cardiovascular disease [2]. Similarly, higher plasma cortisol in the population associates with hypertension, hyperglycaemia, cardiovascular disease, type 2 diabetes, cognitive dysfunction and depression, while lower cortisol associates with immunological abnormalities and post-traumatic stress disorder [3,4,5,6,7,8,9,10].
We established the CORtisol NETwork (CORNET) consortium with the initial aim of identifying genetic determinants of inter-individual variation in HPA axis function. A genome-wide association meta-analysis (GWAMA), investigating ~2.2 M SNPs in 12,597 individuals from 11 European cohorts, identified a single locus on chromosome 14 associated with morning plasma cortisol at genome-wide significance [11]. The locus spans SERPINA6 and SERPINA1 and influences function of corticosteroid-binding globulin (CBG, the product of SERPINA6), a protein that binds cortisol in the blood. However, it was unclear if the effect is mediated directly through SERPINA6 or indirectly through the product of SERPINA1, α1-antitrypsin, which is involved in regulating cleavage and inactivation of CBG [12]. Using these genetic variants as a proxy for morning plasma cortisol levels in Mendelian randomisation analyses we provided evidence to suggest that cortisol is a causal risk factor for coronary heart disease, but the odds ratio was not statistically significant (OR: 1.06, 95% CI: 0.98–1.15) [13]. An independent study has confirmed recently that common variants in SERPINA6 are associated with plasma cortisol and with coronary artery disease [14].
The genetic variants in the SERPINA6/A1 locus explain only ~0.5% of the variance in morning plasma cortisol. Moreover, attempts to identify genetic variants associated with an alternative phenotype of salivary cortisol have not been successful [15]. Identifying additional genetic variants that explain more of the variance in cortisol could provide new insights into cortisol biology and statistical power to test the causative role of cortisol in the aetiology of other common diseases. We aimed: to identify additional specific loci influencing cortisol; to refine where in the SERPINA6/A1 locus there is an influence on cortisol; to establish whether SERPINA6/A1 variation influences tissue-specific expression of CBG and α1-antitrypsin; and to confirm whether high cortisol is causal in ischaemic heart disease and test if it is causal in other common diseases. To achieve these aims, we undertook an extended GWAMA analysis, with more subjects and more SNPs than the original CORNET GWAMA [11], and used the results to provide instruments for expression quantitative trait loci (eQTL) and Mendelian randomisation analyses.
Methods
Genome-wide association meta-analysis study
We performed a meta-analysis of genome-wide association studies of morning plasma cortisol in 25,314 subjects from 17 European population-based cohorts: CROATIA-Vis (n = 886), CROATIA-Korcula (n = 897), CROATIA-Split (n = 493), ORCADES (n = 1974), Rotterdam Study (n = 2870), NFBC1966 (n = 1324), Helsinki Birth Cohort Study 1934–44 (n = 399), ALSPAC (n = 1487), PREVEND (n = 1151), PIVUS (n = 919), Raine Study (n = 860), ET2DS (n = 847), MrOS-Sweden (n = 969), KORA (n = 1651), TwinsUK (n = 5654), SHIP (n = 910) and VIKING (n = 2073). Characteristics of the study populations are presented in Table S1 and details of each cohort are provided in Supplementary Material. All individuals were of European ancestries. Exclusion criteria were current glucocorticoid use, pregnant or breast-feeding women, and twins (exclusion of one of each twin pair). Cortisol was measured by immunoassay in blood samples collected from study participants between 0700 and 1100 h in all cohorts except for in TwinsUK which measured cortisol using liquid chromatography-mass spectrometry. All participants provided written informed consent and studies were approved by local Research Ethics Committees and/or Institutional Review Boards.
Each study performed linear regression on z-scores of log-transformed morning plasma cortisol (additive genetic effects), adjusted for sex, age and cohort-specific genetic ancestry. Additional models also adjusted for smoking and body mass index. Imputation of the gene-chip results used the 1000 Genomes European population reference panel. Details of the genotyping, imputation and cohort-specific adjustment for genetic ancestry are provided in Table S2 and Supplementary Material.
Quality control was carried out on the imputed genome-wide data for all 17 studies prior to meta-analysis; this excluded all SNPs with a minor allele frequency (MAF) <0.5%, call rate <95%, and poor imputation quality (MACH R2_HAT <0.30, IMPUTE PROPER_INFO <0.60, BEAGLE INFO <0.30, as appropriate). Furthermore, only SNPs with estimates from at least four studies were included, resulting in a final number of 8,452,427 SNPs. Quantile–quantile (QQ) plots and genomic control (lambda) were used to assess evidence for population structure that was not accounted for in association analyses. Sex chromosomes were not analysed. Quality control at the study-level and meta-level was performed using EasyQC software [16].
The results from all cohorts were combined into a fixed-effects meta-analysis using Stouffer’s method with weighting of Z-scores proportional to the square-root of the number of individuals in each sample, using METAL software [17]. Manhattan and QQ plots visualised the results using EasyStrata software [18].
SNP-based heritability
Linkage disequilibrium (LD) score regression exploits the relationship between SNP-phenotype association strengths and LD patterns [19]. Assuming true causal effects, the SNPs which are in higher LD with nearby SNPs are expected to have more inflated test statistics, because they are more likely to tag causal variants with stronger effects. The SNP heritability was estimated using LD score regression v1.9.0 [19]. Since imputation quality can confound LD score regression results, we restricted the analysis to a list of well-imputed SNPs, as recommended by the software authors. After applying default quality control settings, the final SNP number was 1,028,327. The genome-wide summary statistics were partitioned into functional categories using the method described by Finucane et al. [20]. More details of the partitioned heritability method can be found in the Supplementary Material.
Genetic correlations
Genetic correlations between morning plasma cortisol and selected diseases and traits from UK Biobank were estimated using bivariate LD score regression. This technique examines the correlation structure of genetic effects of SNPs across the genome. The data processing pipeline devised by Bulik-Sullivan et al. [21] was followed using LD Hub v1.9.0 software [22].
Gene- and pathway-based association analysis
Gene- and pathway- based associations, which assign SNPs to genes and biological pathways respectively, were performed using MAGMA and FUMA software [23, 24]. SNPs were assigned to 18,062 genes using the National Centre for Biotechnology Information build 37.3. The gene boundary was defined as the start and stop site of each gene. The European panel of the 1000 Genomes data (phase 1, release 3) was used as a reference panel to account for LD between the SNPs. A Bonferroni correction was used to control for 18,062 tests (α = 0.05/18 062; P < 2.768 × 10−6).
eQTL analysis in the Stockholm Tartu Atherosclerosis Reverse Networks Engineering Task (STARNET) study
eQTL analyses were undertaken in the STARNET study which is composed of Caucasian individuals of Eastern European origin (30% female), with a confirmed diagnosis of coronary artery disease. Of these individuals 27% had diabetes, 77% had hypertension and 37% had suffered a myocardial infarction before the age of 60 years. Genotyping and RNA sequencing of seven vascular and metabolic tissue samples from 600 patients undergoing coronary artery bypass surgery was performed [25]. The tissue sets available in STARNET include; whole blood, atherosclerotic-lesion free internal mammary artery, atherosclerotic aortic root, subcutaneous fat, visceral abdominal fat, skeletal muscle, and liver. Whole blood samples were taken pre-operatively and the remaining tissue biopsies were obtained during surgery.
For the analysis, we included 580 SNPs within SERPINA6/SERPINA1 locus on chromosome 14 identified from the CORNET plasma cortisol GWAMA, defined as within 100 Kb of SERPINA6 (±of the transcription start/end point), with no missing data, and MAF >5%. All transcripts were annotated using the Human Genome Reference Consortium Human Build 37 (GRCh37).
Transcriptome-wide eQTL associations between selected SNPs and all genes expressed in the STARNET tissues were tested using the Kruskal–Wallis test, a non-parametric ANOVA used to determine whether sample groups originate from the same distribution that has been used previously in genetic association studies [26]. Kruskal–Wallis test statistics and p values were computed using the kruX algorithm [27], using available pre-processed and normalised genotype and expression data matrices [25] as input. Multiple testing correction was performed for all SNP associations with every STARNET gene in each tissue separately by calculating q values [28] using the qvalue package in Python. eQTL associations with SERPINA6 were visualised using the GWAS visualisation tool LocusZoom [29]. The allelic effect of individual SNPs on SERPINA6 was visualised by constructing boxplots for each allele.
The global tissue-specific effect on gene expression for SNPs associated with plasma cortisol at a genome-wide level of significance was depicted using Q–Q plots showing the observed transcriptome-wide SNP-gene associations against the expected uniform distribution. Deviation from the uniform distribution was tested using the Kolmogorov–Smirnov test statistic and p value for each SNP.
Bayes factor colocalisation analysis was performed in R using the package Coloc [30]. eQTL analysis was repeated using linear regression with the R package MatrixEQTL [31] to obtain beta values required for colocalisation. linear regression and Kruskal–Wallis p values were consistent (Fig. S7). For visualisation of the colocalisation event, LD with the lead SNP was calculated using the package LDlinkR [32].
Two-sample Mendelian randomisation
The instrument for morning plasma cortisol consisted of independent (r2 < 0.3) genetic variants that reached a genome-wide level of significance (P < 5 × 10−8). To detect independent top SNPs we used the clumping function as implemented in PLINK. The European samples from the 1000 Genomes Project were used to estimate LD between SNPs. Amongst those SNPs within 1000 kb and r2 < 0.3, only the SNP with the lowest P value was retained.
Two-sample Mendelian randomisation [33] was used to estimate the causal effect of morning plasma cortisol on hypothetically cortisol-related diseases and traits (chronic ischaemic heart disease, myocardial infarction, diabetes mellitus, body mass index and osteoporosis) available in UK Biobank and publicly available GWAS consortia in the MR Base platform [34].
Genetic instruments for various traits/conditions were also constructed to estimate the causal effect these had on morning plasma cortisol (bidirectional Mendelian randomisation). Details of the selected disease and traits, the population, consortia details and genetic instruments are provided in Table S3. The SNP-cortisol estimate was divided by the SNP-outcome estimate (Wald ratio method [35]) and then combined using inverse variance weighting. Additional analyses described in the Supplementary Material were performed to investigate the robustness of this causal estimate and any potential pleiotropic effects.
Results
Genome-wide association meta-analysis
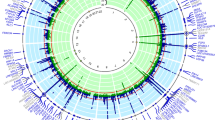
The GWAMA of morning plasma cortisol levels in 25,314 identified a single locus on chromosome 14 reaching a genome-wide level of significance (p < 5 × 10−8) (Fig. 1). This is the same locus as the CORNET consortium previously identified [11] and includes the SERPINA6 gene, encoding corticosteroid binding globulin (CBG), and the SERPINA1 gene, encoding α1-antitrypsin, an inhibitor of neutrophil elastase which cleaves and inactivates CBG. In an additive genetic model, the top SNP rs9989237 reported a per minor allele effect of 0.11 cortisol z-score (p = 2.2 × 10−19). The effect allele frequency was 0.22 and this variation explained 0.13% of the morning plasma cortisol variance. Within this locus we identified four blocks of SNPs in low LD (r2 < 0.3) visualised in Figs. 1, S1.

a Manhattan plot of −log10 P values of the SNP-based association analysis of morning plasma cortisol (n = 25,314). The locus on chr14 spans SERPINA6 and SERPINA1 genes; no other loci reached genome-wide significance. b, c Zoomed in Manhattan plot (LocusZoom plot) of −log10 P values of the SNP-based association analysis of morning plasma cortisol (n = 25,314). These show two (of the four) LD blocks (r2 > 0.3) in this locus
SNP-based heritability
Using LD score regression, common SNPs across the genome were found to explain 4.2% (s.e. 1.9%) of the phenotypic variation of morning plasma cortisol. There was no evidence of enrichment in a particular cell type, and particularly not in the adrenal/pancreas or liver cell type groups of SNPs, when partitioning the heritability into functional cell types (all P values > 0.5, Table S4).
Genetic correlations
LD score regression was used to test whether genetic variants associated with morning plasma cortisol also contribute to health-related traits. Estimated genetic correlations are presented in Fig. S2. There was evidence of a positive genetic correlation between morning plasma cortisol and acute myocardial infarction (rg = 0.50, 95% CI 0.04–0.97) and a negative genetic correlation with BMI (rg = −0.32, 95% CI −0.54 to −0.10).
Gene-based and pathway-based association analyses
Gene-based association analysis identified three genes, SERPINA6, SERPINA1 and SERPINA10, all located on chromosome 14, that attained genome-wide significance following correction for multiple comparisons (Fig. S3). Pathway-based association analysis identified the top pathway as metabolism of lipids and lipoproteins (P = 1.6 × 10−5) (Table S5).
Tissue-specific eQTL analyses
The statistical effects of 580 SNPs in the SERPINA6 region on gene expression were obtained using individual-level genotype and RNA-sequencing data from seven vascular and metabolic tissues from the STARNET study [25]. Following multiple testing correction (q ≤ 0.05), 32 cis-eQTLs for SERPINA6 were identified in the liver, the only tissue where SERPINA6 is highly expressed (Fig. 2a). Of these 32 cis-eQTLs, 21 were also at genome-wide significance in CORNET (p ≤ 5 × 10−8), and therefore also associated with variation for plasma cortisol (Table S6).

Tissue-specific association of cortisol-related SNPs with gene expression in STARNET. a LocusZoom plot showing genomic loci of given SNPs against measure of significance (−log10 (p value)) for an eQTL analysis in liver for all SNPs within 100 Kb of SERPINA6. Squares represent the 21 significant cis-eQTLs (q ≤ 0.05) that are also at genome-wide significance in CORNET (p ≤ 5 × 10−8). b Genotypic effect of representative SNP for LD block 2 (rs2736898) on SERPINA6 gene expression in liver. c Global tissue-specific effects on gene expression for rs2736898 represented as Q–Q plots for genes in liver, subcutaneous fat and visceral abdominal fat describing observed p values vs. those expected by chance. Deviation from expected uniform distribution described by Kolmogorov–Smirnov test p value (Ks-test)
The global effect on tissue-specific gene expression of representative SNPs from each LD block was assessed using the distribution of transcriptome-wide eQTL p values (Figs. S4, S5). LD block 2, represented by the SNP rs2736898, with the alternate allele C, exerted a negative effect on SERPINA6 expression in liver (q = 0.00015) (Fig. 2b) and showed the strongest tissue-specific effects, particularly in visceral abdominal fat, subcutaneous fat and liver (Fig. 2c). In all cases the allele associated with higher plasma cortisol in the GWAMA was the allele associated with higher SERPINA6 expression in STARNET (Fig. S6).
To determine if the signal identified for SERPINA6 cis-eQTLs in liver and SNPs associated with plasma cortisol are driven by the same causal variant, Bayes factor colocalisation analysis was performed while accounting for allelic heterogeneity [30] (Fig. 3). The probability of both traits sharing a causal variant was low (40.6%) when examining all SNPs within 100 Kb of SERPINA6. However, when examining each LD block individually, the block represented by rs2736898 returns a 99.2% probability of shared causal variant in this region (Table S7).

Scatterplot showing colocalisation of joint signal from CORNET GWAMA and SERPINA6 cis-eQTLs from STARNET-liver. Includes all SNPs within 100 Kb of SERPINA6 that were present in both datasets (n = 535). Colour bar indicates degree of LD with rs2736898. Formal colocalisation analysis with Coloc indicates 99.2% probability of the presence of a shared causal variant within LD block 2 mediating GWAMA and SERPINA6 cis-eQTL signal
Two-sample Mendelian randomisation
The clumping procedure identified four SNPs (rs9989237, rs2736898, rs11620763, rs7146221) as markers representing genome-wide significant signals in this region. Two-sample Mendelian randomisation analyses provided evidence that each genetically-determined standard deviation (SD) increase in morning plasma cortisol was associated with an increased risk of chronic ischaemic heart disease (0.32, 95% CI 0.06–0.59) and myocardial infarction (0.21, 95% CI 0.00–0.43) in UK Biobank (Fig. 4a). Similar estimates were observed for these disease outcomes in non-UK Biobank cohorts (CARDIoGRAM plus C4D, Fig. 4b).

Causal estimates of a 1 SD increase in morning plasma cortisol on relevant disease and trait outcomes. Estimates are from two-sample Mendelian randomisation analyses using inverse variance weighting to combine estimates from each genetic variant. a Outcomes from UK Biobank (chronic ischaemic heart disease, cases = 8755, controls = 328,444; myocardial infarction, cases = 7790, controls = 328,893; diabetes mellitus, cases = 16,183, controls = 320,290; body mass index, sample size = 336,107; osteoporosis, cases = 5266, controls = 331,893); (b) Equivalent outcomes from non-UK Biobank sources—CARDIoGRAMplusC4D (coronary heart disease, cases = 60,801, controls = 123,504; myocardial infarction, cases = 43,676, controls = 128,199), DIAGRAM (type 2 diabetes, cases = 26,488, controls = 83,964), GIANT (BMI, sample size = 339,224) and GEFOS (lumber spine mineral density, sample size = 28,498)
The bidirectional Mendelian Randomisation analyses, estimating the genetically predicted effect of various traits or disease on plasma cortisol, did not support any causal associations (Fig. 5).

Bidirectional causal estimates of the effect of the disease or trait on morning plasma cortisol. Estimates are from two-sample Mendelian randomisation analyses using inverse variance weighting to combine estimates from each genetic variant
Discussion
These results confirm that genetic variation in the SERPINA6/A1 locus on chromosome 14 is associated with morning plasma cortisol. Despite doubling the sample size and trebling the SNP density from the previous genome-wide association study [11] no new genetic loci were identified. The improved imputation of genetic markers used in this analysis allowed identification of new SNPs within the SERPINA6/A1 locus and strengthened the instrument used in two-sample Mendelian randomisation analyses. This additional information coupled with tissue-specific gene expression data suggests that genetic variation within this locus influences expression of SERPINA6 rather than SERPINA1 in the liver. Furthermore, it appears that the eQTL and GWAMA signals colocalise within the block of SNPs in LD, represented by rs2736898, suggesting this block is primarily responsible for driving CBG-mediated variation for cortisol. Moreover, it demonstrates effects on adipose tissue gene expression, suggesting that resulting variations in CBG levels in turn influence the delivery of cortisol to peripheral tissues.
Given previous evidence of heritability of plasma cortisol it is surprising that the increase in sample size and SNP coverage from our earlier GWAMA did not identify any new loci associated with morning plasma cortisol. Twin studies have estimated the heritability of plasma cortisol ranging from 14 to 45% [36,37,38]. However, SNP heritability estimates of plasma cortisol are considerably lower at 6% [15], and were confirmed in this sample at 4%. Nevertheless, despite poor prediction of plasma cortisol by common autosomal SNPs, our interrogation of the SERPINA6/SERPINA1 locus demonstrates important insights.
To test the functional significance of SERPINA6/A1 variants we used eQTL analyses in the STARNET cohort. A hypothesis-free approach that investigated the effect of all 580 SNPs within the SERPINA6/SERPINA1 locus on expression of all STARNET genes in each of the seven tissue types identified cis-eQTLs for SERPINA6 expression in the liver, the only tissue where SERPINA6 is highly expressed. This effect was refined to one of the LD blocks, represented by rs2736898. Importantly, genetic variation in SERPINA6/SERPINA1 was also associated in trans-eQTL analyses with gene expression in visceral abdominal fat and subcutaneous fat as well as liver. These results suggest that not only does cortisol-associated genetic variation influence CBG expression in the liver, but it also influences cortisol signalling in peripheral tissues, an effect that is likely to be mediated by CBG.
CBG binds ~90% of cortisol in plasma but it is usually thought that only the free cortisol can access tissues and have biological effects. Patients with mutations in SERPINA6 [39] and animals with deletion of CBG, however, exhibit features consistent with cortisol deficiency despite biochemical changes in the CBG-bound rather than free cortisol pool. Our data support the hitherto speculative evidence that CBG is involved actively in delivery of cortisol to peripheral tissues. We and others had proposed that this is mediated by altered cleavage of CBG by neutrophil elastase in tissues [11, 12], and evidence from immunoassays suggested that variants in SERPINA6/SERPINA1 might mediate their effect through altered inhibition of neutrophil elastase by α1-antitrypsin. However, the more detailed analyses facilitated by the expanded GWAMA presented here do not support this interpretation since we did not identify eQTLs for α1-antitrypsin expression. Moreover, the reliability of immunoassays to determine CBG cleavage has since been called into question [40].
Epidemiological analysis of cortisol has demonstrated associations with a large number of diseases and traits [2,3,4,5,6,7,8,9], but the direction of causality, if any, has not been established. The improved imputation of genetic markers used in this analysis allowed identification of new SNPs within the SERPINA6/A1 locus and strengthened the instrument used in the two-sample Mendelian randomisation analyses. We tested whether genetically-elevated cortisol is causally associated with cortisol-related outcomes selected from the features of Cushing’s syndrome—a rare condition caused by tumours secreting ACTH or cortisol—that are highly prevalent in the general population. Consistent with previous reports based on a less refined genetic instrument [13, 14] we found evidence that cortisol causally increases the risk of heart disease. However, we did not find conclusive evidence that elevated cortisol causes type 2 diabetes, osteoporosis or obesity; given the relatively small variance in cortisol accounted for by the genetic instrument, it is possible that causal associations with additional diseases and traits would be revealed by analysis of larger sample sizes. We also tested whether common diseases and traits underlie elevated plasma cortisol, using bidirectional Mendelian randomisation analyses. The strongest evidence suggested that higher BMI reduces cortisol levels, consistent with prior epidemiological and experimental evidence that obesity enhances clearance of cortisol from the circulation [1].
It is arguable that morning plasma cortisol or salivary cortisol are poor surrogates for overall cortisol exposure given their diurnal and ultradian fluctuations and variation with acute stress. A more robust assessment of cortisol phenotype, perhaps from hair cortisol [41] or ambulatory sampling of interstitial fluid over 24 h, may reveal stronger epidemiological and genetic associations.
In summary, this large GWAMA has revealed the limited magnitude and range of genetic effects on plasma cortisol, but has identified a pathway from variation in the SERPINA6/SERPINA1 locus through variation in liver SERPINA6 expression to variation in CBG-mediated gene transcription including in adipose tissue that is causally associated with cardiovascular disease.
References
Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007;157:545–59.
Ragnarsson O, Olsson DS, Papakokkinou E, Chantzichristos D, Dahlqvist P, Segerstedt E, et al. Overall and disease-specific mortality in patients with cushing disease: a Swedish Nationwide Study. J Clin Endocrinol Metab. 2019;104:2375–84.
Phillips DIW, Barker DJP, Fall CHD, Seckl JR, Whorwood CB, Wood PJ, et al. Elevated plasma cortisol concentrations: a link between low birth weight and the insulin resistance syndrome? J Clin Endocrinol Metab. 1998;83:757–60.
Filipovsky J, Ducimetiere P, Eschwege E, Richard JL, Rosselin G, Claude JR. The relationship of blood pressure with glucose, insulin, heart rate, free fatty acids and plasma cortisol levels according to degree of obesity in middle-aged men. J Hypertens. 1996;14:229–35.
Fraser Robert, Ingram Mary C, Anderson Niall H, Morrison Caroline, Davies Eleanor, Connell John MC. Cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension. 1999;33:1364–8.
Reynolds RM, Walker BR, Syddall HE, Andrew R, Wood PJ, Whorwood CB, et al. Altered control of cortisol secretion in adult men with low birth weight and cardiovascular risk factors. J Clin Endocrinol Metab. 2001;86:245–50.
Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NPV, et al. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1:69–73.
Holsboer F. Stress, hypercortisolism and corticosteroid receptors in depression: implicatons for therapy. J Affect Disord. 2001;62:77–91.
Seckl JR, Meaney MJ. Glucocorticoid “Programming” and PTSD risk. Ann NY Acad Sci. 2006;1071:351–78.
Ball TM. Cortisol circadian rhythms and stress responses in infants at risk of allergic disease. NIM. 2006;13:294–300.
Bolton JL, Hayward C, Direk N, Lewis JG, Hammond GL, Hill LA, et al. Genome wide association identifies common variants at the SERPINA6/SERPINA1 locus influencing plasma cortisol and corticosteroid binding globulin. PLOS Genet. 2014;10:e1004474.
Hammond GL, Smith CL, Paterson NAM, Sibbald WJ. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J Clin Endocrinol Metab. 1990;71:34–9.
Crawford AA, Soderberg S, Kirschbaum C, Murphy L, Eliasson M, Ebrahim S, et al. Morning plasma cortisol as a cardiovascular risk factor: findings from prospective cohort and Mendelian randomization studies. Eur J Endocrinol. 2019;181:429–38.
Pott J, Bae YJ, Horn K, Teren A, Kühnapfel A, Kirsten H, et al. Genetic association study of eight steroid hormones and implications for sexual dimorphism of coronary artery disease. J Clin Endocrinol Metab. 2019;104:5008–23.
Neumann A, Direk N, Crawford AA, Mirza S, Adams H, Bolton J, et al. The low single nucleotide polymorphism heritability of plasma and saliva cortisol levels. Psychoneuroendocrinology. 2017;85:88–95.
Winkler TW, Day FR, Croteau-Chonka DC, Wood AR, Locke AE, Mägi R, et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc. 2014;9:1192–212.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Winkler TW, Kutalik Z, Gorski M, Lottaz C, Kronenberg F, Heid IM. EasyStrata: evaluation and visualization of stratified genome-wide association meta-analysis data. Bioinformatics. 2015;31:259–61.
Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5.
Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–35.
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41.
Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2017;33:272–9.
Leeuw CA, de, Mooij JM, Heskes T, Posthuma D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLOS Comput Biol. 2015;11:e1004219.
Watanabe K, Taskesen E, Bochoven Avan, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1–11.
Franzén O, Ermel R, Cohain A, Akers NK, Narzo AD, Talukdar HA, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016;353:827–30.
Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, et al. Mapping the Genetic Architecture of Gene Expression in Human Liver. PLOS Biol. 2008;6:e107.
Qi J, Asl HF, Björkegren J, Michoel T. kruX: matrix-based non-parametric eQTL discovery. BMC Bioinform. 2014;15:11.
Storey JD, Tibshirani R. Statistical significance for genomewide studies. PNAS. 2003;100:9440–5.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7.
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genet. 2014;10:e1004383.
Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–8.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–7.
Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, EPIC- InterAct Consortium. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30:543–52.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. ELife. 2018;7:e34408.
Halperin M. Fitting of straight lines and prediction when both variables are subject to error. J Am Stat Assoc. 1961;56:657–69.
Inglis GC, Ingram MC, Holloway CD, Swan L, Birnie D, Hillis WS, et al. Familial pattern of corticosteroids and their metabolism in adult human subjects—the Scottish Adult Twin Study. J Clin Endocrinol Metab. 1999;84:4132–7.
Meikle AW, Stringham JD, Woodward MG, Bishop DT. Heritability of variation of plasma cortisol levels. Metab Clin Exp. 1988;37:514–7.
Froehlich JC, Zink RW, Li T-K, Christian JC. Analysis of heritability of hormonal responses to alcohol in twins: beta-endorphin as a potential biomarker of genetic risk for alcoholism. Alcohol Clin Exp Res. 2000;24:265–77.
Meyer EJ, Nenke MA, Rankin W, Lewis JG, Torpy DJ. Corticosteroid-binding globulin: a review of basic and clinical advances. Horm Metab Res. 2016;48:359–71.
Hill LA, Vassiliadi DA, Dimopoulou I, Anderson AJ, Boyle LD, Kilgour AHM, et al. Neutrophil elastase-cleaved corticosteroid-binding globulin is absent in human plasma. J Endocrinol. 2019;240:27–39.
Wester VL, Rossum EFCvan. Clinical applications of cortisol measurements in hair. Eur J Endocrinol. 2015;173:M1–0.
Acknowledgements
CORNET consortium: CORNET has been supported by grants from the Chief Scientist Office of the Scottish Government, the British Heart Foundation and Wellcome Trust.
ALSPAC: Ethical approval for the study was obtained from the ALSPAC Ethics and Law Committee and the Local Research Ethics Committees. Consent for biological samples has been collected in accordance with the Human Tissue Act (2004). Informed consent for the use of data collected via questionnaires and clinics was obtained from participants following the recommendations of the ALSPAC Ethics and Law Committee at the time. We are extremely grateful to all the families who took part in this study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses.
Croatia Vis, Korcula, Split: The CROATIA_Vis,CROATIA_Korcula and CROATIA_Split studies were funded by grants from the Medical Research Council (UK), European Commission Framework 6 project EUROSPAN (Contract No. LSHG-CT-2006-018947) and Republic of Croatia Ministry of Science, Education and Sports research grants. (108-1080315-0302). We would like to acknowledge the staff of several institutions in Croatia that supported the field work, including but not limited to The University of Split and Zagreb Medical Schools, Institute for Anthropological Research in Zagreb and Croatian Institute for Public Health. CH is supported by an MRC University Unit Programme Grant MC_UU_00007/10 (QTL in Health and Disease.
ET2DS: The sponsor for the ET2DS was the University of Edinburgh. The study was funded by the Medical Research Council (UK) (ProjectGrant G0500877), the Chief Scientist Office of the Scottish Executive (Programme Support Grant CZQ/1/38), Pfizer plc. and DiabetesUK (Clinical Research Fellowship 10/0003985). The funders had no other role in the design, analysis or writing of this paper.
HBCS: We thank all study participants as well as everybody involved in the Helsinki Birth Cohort Study. Helsinki Birth Cohort Study has been supported by grants from the Academy of Finland, the Finnish Diabetes Research Society, Folkhälsan Research Foundation, Novo Nordisk Foundation, Finska Läkaresällskapet, Juho Vainio Foundation, Signe and Ane Gyllenberg Foundation, University of Helsinki, Ministry of Education, Ahokas Foundation, Emil Aaltonen Foundation.
KORA: The KORA study was initiated and financed by the Helmholtz Zentrum München—German Research Centre for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Centre of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. The KORA-Study Group consists of A. Peters (speaker), J. Heinrich, R. Holle, R. Leidl, C. Meisinger, K. Strauch and their co-workers, who are responsible for the design and conduct of the KORA studies. We gratefully acknowledge the contribution of all members of field staff conducting the KORA study. Finally, we are grateful to all study participants of KORA for their invaluable contributions to this study. All KORA participants have given written informed consent and the study was approved by the Ethics Committee of the Bavarian Medical Association.
MrOS Sweden: MrOS in Sweden is supported by the Swedish Research Council, the Swedish Foundation for Strategic Research, the ALF/LUA research grant in Gothenburg, the Lundberg Foundation, the Knut and Alice Wallenberg Foundation, the Torsten Soderberg Foundation, and the Novo Nordisk Foundation.
North Finland Birth Cohort (1966): NFBC1966 Acknowledgements: We thank all cohort members and researchers who participated in the 31 years study. We also wish to acknowledge the work of the NFBC project centre. Funding: NFBC1966 received financial support from University of Oulu Grant no. 65354, Oulu University Hospital Grant no. 2/97, 8/97, Ministry of Health and Social Affairs Grant no. 23/251/97, 160/97, 190/97, National Institute for Health and Welfare, Helsinki Grant no. 54121, Regional Institute of Occupational Health, Oulu, Finland Grant no. 50621, 54231.
ORCADES: The Orkney Complex Disease Study (ORCADES) was supported by the Chief Scientist Office of the Scottish Government (CZB/4/276, CZB/4/710), a Royal Society URF to JFW, the MRC Human Genetics Unit quinquennial programme “QTL in Health and Disease”, Arthritis Research UK and the European Union framework programme 6 EUROSPAN project (contract no. LSHG-CT-2006-018947). DNA extractions were performed at the Edinburgh Clinical Research Facility, University of Edinburgh. We would like to acknowledge the invaluable contributions of the research nurses in Orkney, the administrative team in Edinburgh and the people of Orkney.
PIVUS: Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) was supported by Wellcome Trust Grants (WT098017, WT064890, WT090532), Uppsala University, Uppsala University Hospital, the Swedish Research Council and the Swedish Heart-Lung Foundation.
PREVEND: The PREVEND study programme has been made possible by an initial grant of the Dutch Kidney Foundation. Its continuation has been made possible by grants from the University Medical Centre Groningen, the Dutch Kidney Foundation and the Dutch Heart Foundation.
The Raine Study: The Raine Study was supported by the National Health and Medical Research Council of Australia [grant numbers 572613, 403981 and 1059711] and the Canadian Institutes of Health Research [grant number MOP-82893]. The authors are grateful to the Raine Study participants and their families, and to the Raine Study team for cohort coordination and data collection. The authors gratefully acknowledge the NHMRC for their long term funding to the study over the last 30 years and also the following institutes for providing funding for Core Management of the Raine Study: The University of Western Australia (UWA), Curtin University, Women and Infants Research Foundation, Telethon Kids Institute, Edith Cowan University, Murdoch University, The University of Notre Dame Australia and The Raine Medical Research Foundation. This work was supported by resources provided by the Pawsey Supercomputing Centre with funding from the Australian Government and Government of Western Australia.
Rotterdam: The Rotterdam Study is supported by the Erasmus MC University Medical Centre and Erasmus University Rotterdam; The Netherlands Organisation for Scientific Research (NWO); The Netherlands Organisation for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly (RIDE); The Netherlands Genomics Initiative (NGI); the Ministry of Education, Culture and Science; the Ministry of Health, Welfare and Sports; the European Commission (DG XII); and the Municipality of Rotterdam. The contribution of inhabitants, general practitioners and pharmacists of the Ommoord district to the Rotterdam Study is gratefully acknowledged. AN and HT were supported by a grant of the Dutch Ministry of Education, Culture, and Science and the Netherlands Organisation for Scientific Research (NWO grant No. 024.001.003, Consortium on Individual Development). The work of HT is further supported by a NWO-VICI grant (NWO-ZonMW: 016.VICI.170.200). We would like to thank Anis Abuseiris, Karol Estrada, Tobias A. Knoch and Rob de Graaf as well as their institutions, the Biophysical Genomics, Erasmus MC Rotterdam, The Netherlands, and especially the national German MediGRID and Services@MediGRID part of the German D-Grid, both funded by the German Bundesministerium fuer Forschung und Technology under grants #01 AK 803 A-H and # 01 IG 07015 G for access to their grid resources.
Scientific computing at Mount Sinai: This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. Research reported in this paper was supported by the Office of Research Infrastructure of the National Institutes of Health under award numbers S10OD018522 and S10OD026880.
SHIP: SHIP is part of the Community Medicine Research Net of the University Medicine Greifswald, which is supported by the German Federal State of Mecklenburg-West Pomerania.
TwinsUK: TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London.
VIKING: The Viking Health Study—Shetland (VIKING) was supported by the MRC Human Genetics Unit quinquennial programme grant “QTL in Health and Disease”. DNA extractions and genotyping were performed at the Edinburgh Clinical Research Facility, University of Edinburgh. We would like to acknowledge the invaluable contributions of the research nurses in Shetland, the administrative team in Edinburgh and the people of Shetland.
The CORNET consortium
Rotterdam Study: AN, HT; Vis (Croatia): CH, HC; Korcula (Croatia): CH, HC; Split (Croatia): CH, IR; ORCADES: CH, JFW; Helsinki Birth Cohort (1934–44) Study: JL, JE; North Finland Birth Cohort (1966) Study: VK, MRJ; ALSPAC: NT, GDS; PREVEND: PvdH, SJLB; PIVUS: APM, AM; Edinburgh type 2 Diabetes Study: SM, JP; MrOS Sweden: MN, CO, Dan Mellström47; The Raine Study: CAW, CEP; TwinsUK: MM, TS; KORA: EA, CG; SHIP: MP, NF, HV; VIKING: DC, JFW; ORCADES: CLKB, PKJ, JFW.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Crawford, A.A., Bankier, S., Altmaier, E. et al. Variation in the SERPINA6/SERPINA1 locus alters morning plasma cortisol, hepatic corticosteroid binding globulin expression, gene expression in peripheral tissues, and risk of cardiovascular disease. J Hum Genet 66, 625–636 (2021). https://doi.org/10.1038/s10038-020-00895-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-020-00895-6
This article is cited by
-
Maternal plasma cortisol’s effect on offspring birth weight: a Mendelian Randomisation study
BMC Pregnancy and Childbirth (2024)
-
Links between the genetic determinants of morning plasma cortisol and body shape: a two-sample Mendelian randomisation study
Scientific Reports (2024)
-
Insights into Causal Cardiovascular Risk Factors from Mendelian Randomization
Current Cardiology Reports (2023)
-
Sex steroid hormones and risk of breast cancer: a two-sample Mendelian randomization study
Breast Cancer Research (2022)
