
Abstract
Beckwith-Wiedemann syndrome (BWS) is a representative imprinting disorder. Gain of methylation at imprinting control region 1 (ICR1-GOM), leading to the biallelic expression of IGF2 and silencing of H19, is one of the causative alterations in BWS. Twenty percent of BWS patients with ICR1-GOM have genetic defects in ICR1. Evidence of methylation anticipation in familial BWS patients with ICR1 genetic defects has been reported. However, the precise methylation pattern and extent of anticipation in these patients remain elusive. In addition, although age-related IGF2-DMR0 hypomethylation has been reported in the normal population, the period of its occurrence is unknown. In this study, we analyzed 10 sites (IGF2-DMR0, IGF2-DMR2, CTCF binding sites 1–7, and the H19 promoter) within the IGF2/H19 domain in familial BWS patients harboring a pathogenic variant in ICR1. We found that sites near the variant had relatively higher methylation in the first affected generation and observed methylation anticipation through maternal transmission in the next generation. The extent of anticipation was greater at sites far from the variant than nearby sites. The extended and severe GOM might be due to the insufficient erasure/demethylation of pre-acquired ICR1-GOM in primordial germ cells or during the preimplantation stage. In the normal population, age-related IGF2-DMR0 hypomethylation occurred; it became established by young adulthood and continued to old age. Further studies are needed to clarify (1) the precise mechanism of anticipation in patients with familial BWS and (2) the mechanism and biological significance of constitutive hypomethylation of IGF2-DMR0 and/or other imprinted differentially methylated regions.
Similar content being viewed by others

Introduction
Beckwith-Wiedemann syndrome (BWS, OMIM #130650) is a genomic imprinting disorder with various clinical features, including overgrowth, macroglossia, abdominal wall defects, and predisposition to embryonal tumors [1, 2]. The estimated prevalence in the worldwide population ranges from 1 in 13,700 to 1 in 10,340 [3, 4]. Its incidence may increase due to increased use of assisted reproductive technologies [5,6,7]. The disease locus chromosome 11p15.5 contains two imprinting domains, IGF2/H19 and CDKN1C/KCNQ1OT1, which are regulated by imprinting control region 1 (ICR1) and ICR2, respectively [1, 2]. ICR1 is methylated on the paternal chromosome and regulates the expression of two important imprinted genes: paternally expressed IGF2 and maternally expressed non-coding H19. ICR2 is methylated on the maternal chromosome and regulates two other important imprinted genes: paternally expressed non-coding KCNQ1OT1 and maternally expressed CDKN1C.
There are several molecular causes of BWS [8]: 50–60% of cases are caused by loss of methylation (LOM) at ICR2; 5–10% are caused by gain of methylation (GOM) at ICR1; 20–25% are caused by paternal uniparental disomy of 11p; 5% are caused by loss-of-function variants of CDKN1C; and 1–2% are caused by paternal duplication of 11p15. ICR1-GOM results in the biallelic expression of IGF2, i.e., loss of imprinting (LOI) of IGF2 and silencing of H19. Approximately 20% of ICR1-GOM patients carry genetic defects in ICR1: 12% are pathogenic variants or small deletions in the OCT4/SOX2 binding sites; and 8% are microdeletions in ICR1 [9,10,11,12,13,14,15].
IGF2 expression is regulated by the binding of CTCF, an insulator protein, to the CTCF binding sites (CTSs) in ICR1. The binding of CTCF to its seven target sites (CTS1–7) on the unmethylated maternal allele promotes H19 expression from the maternal allele (Fig. 1a). On the other hand, the germline-derived DNA methylation on the paternal allele of ICR1 prevents CTCF binding, which leads to an interaction between the IGF2 promoter and enhancers, resulting in the expression of paternal IGF2 [1]. Further, CTCF function is modulated by neighboring binding factors, such as cohesin, OCT4, and SOX2 [16]. Also, three binding sites (OCT0, OCT1, and OCT2) for OCT4/SOX2 were identified in and around ICR1 (Fig. 1a) [12]. Several pathogenic variants and small deletions of OCT1 on the maternal allele lead to ICR1-GOM [12,13,14,15]. The methylation pattern and methylation anticipation of ICR1-GOM in individuals with genetic defects in ICR1 have been reported [15, 17]. However, these studies have not analyzed all differentially methylated sites (DMSs) within the IGF2/H19 domain as shown in Fig. 1a, namely IGF2-DMR0, IGF2-DMR2, CTS1–7, and the H19 promoter. Also, no patients with familial BWS showing methylation anticipation have been reported since these studies were conducted. Therefore, the precise methylation pattern and the extent of methylation anticipation remain elusive.

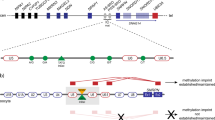
A map of the IGF2/H19 imprinting domain and methylation data of BWS family members. a Schematic representation of the 10 analyzed DMSs within the IGF2/H19 imprinting domain (not to scale). The pathogenic variant of OCT1 was found in ICR1. The transcriptional directions of both IGF2 and H19 are from centromere to telomere. cen centromere, tel telomere, M maternal allele, P paternal allele, open lollipop: unmethylated DMS, and black lollipop: methylated DMS. b Methylation analysis of 10 DMSs in the BWS family. CTS1–5 were highly methylated in affected members. Methylation of CTS1–5 was slightly higher in the third generation than in the second generation, and that of other DMSs (IGF2-DMR0, IGF2-DMR2, CTS6–7, and H19 promoter) was apparently higher in the third generation than in the second generation. The methylation indexes represent the mean methylation of all CpG sites analyzed in each DMS (see Supplementary Table S3). The number of analyzed CpG sites within each DMS is shown in Supplementary Table S1. c Methylation of 10 DMSs in isolated ICR1-GOM patients and control children. There was no significant difference in Δme values between the isolated ICR1–GOM patients and normal children (p = 0.2864, Mann–Whitney U test), indicating that GOM occurred evenly at all DMSs within the IGF2/H19 domain in the isolated ICR1-GOM patients (Supplementary Tables S2 and S4). The vertical bar at each DMS of normal children indicates the standard deviation. Patient IDs beginning with B- and b21- indicate the patients obtained before and after 2009, respectively
Among the DMSs within the IGF2/H19 domain, IGF2-DMR0 is a relatively well-analyzed DMS, but its biological function remains unknown. IGF2-DMR0 is methylated on the paternal allele, and its methylation takes place after implantation under the regulation of ICR1 in cis [18]. IGF2-DMR0 is hypermethylated and hypomethylated in BWS and Silver-Russell syndrome, respectively, and these methylation patterns are similar to the ICR1 methylation statuses in the two disorders [18]. Hypomethylation of IGF2-DMR0 has been observed in tumors from cancers such as Wilms tumor and colorectal cancer [18,19,20,21]. It has also been reported that IGF2-DMR0 hypomethylation is associated with age, suggesting that this epigenetic modification is acquired [22]. However, the period during which it is acquired remains unknown.
Here, we describe DNA methylation anticipation in a family with BWS in which the affected members had a previously reported single nucleotide variant (GRCh37:11:g.2023019A>G) in OCT1 in ICR1 [12, 17]. In addition, we demonstrate that IGF2-DMR0 hypomethylation that is acquired during young adulthood continues into old age in the normal population.
Materials and methods
Patients and controls
A Japanese family comprising three members affected with BWS and three unaffected members (BWS family) was examined in this study. The clinical features of the affected individuals fulfilled three diagnostic criteria for BWS previously described by Elliott et al., DeBaun et al., and Weksberg et al. [23,24,25]. Their Beckwith-Wiedemann spectrum (BWSp) scores exceeded 4, which is a score diagnosed as classical BWS [2] (Fig. 2). In addition, 11 patients with BWS caused by ICR1-GOM without any genetic defects (isolated ICR1-GOM), who fulfilled at least one of the three BWS criteria, were analyzed. The following groups were included as controls: normal children (n = 24, 12 girls and 12 boys, median age = 4.0 [1.8, 6.0] years), adults in their twenties (n = 24, 12 women and 12 men, median age = 24.0 [23.0, 25.0] years), and adults in their forties (n = 24, 12 women and 12 men, median age = 43.0 [41.0, 45.0] years). Genomic DNA was extracted from the peripheral blood lymphocytes of all subjects using a FlexiGene DNA Kit (QIAGEN, Hilden, Germany).

The pedigree of the BWS family. The pathogenic variant was transmitted from the grandmother, who was a carrier of the variant (indicated by a black dot), to her two daughters and to the proband. The clinical features of the aunt and mother were those recorded during their childhood. BWSp scores are also shown. The sequence information of the pathogenic variant is shown in Supplementary Fig. S2. Diagonal: deceased individual, diamond shape: gender not specified, SB stillborn, P proband, y years, m months, Rt right, wt wild type
This study was approved by the Ethics Committee for Human Genome and Gene Analyses of the Faculty of Medicine, Saga University. Written informed consent was obtained from all subjects.
Screening of the ICR1 variant
We screened genetic defects of ICR1 in the BWS family as previously described [26]. In brief, long-range PCR encompassing the entire ICR1 region, which included the seven CTSs and two OCT4/SOX2 binding sites (OCT1 and OCT2), was performed to examine nucleotide insertions and deletions. The PCR products, amplified by several primer sets, were subjected to Sanger sequencing to identify genetic variants. All of the primers used in this study are specified in Supplementary Table S1.
DNA methylation analysis
Genomic DNA (500 ng) was bisulfite-converted using an EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s protocol. We investigated the methylation status of 10 DMSs which were located inside (CTS1–6) and outside (IGF2-DMR0, IGF2-DMR2, CTS7, and H19 promoter) ICR1 (Fig. 1a). The number of CpG sites included in each DMS is presented in Supplementary Table S1. After bisulfite conversion, the relevant sites were amplified by PCR. The PCR products were subjected to pyrosequencing using the Pyromark Q24 instrument (QIAGEN) to quantitatively analyze DNA methylation. All primer sets were examined using varying mixtures of the unmethylated control and the fully methylated control DNA: 0, 25, 50, 75, and 100% methylated DNA, as previously described [21]. Furthermore, we confirmed that the standard deviation (SD) for each CpG site within each DMS in the control children (n = 24) was less than 5% (Supplementary Tables S2 and S5. Only median values are shown in the tables.). These stringent conditions guaranteed the quantitative capability of the methylation analysis.
Statistical analysis
Differences in methylation were analyzed using the non-parametric Mann–Whitney U test or Steel-Dwass test. P values less than 0.05 were considered statistically significant.
Results
Molecular diagnosis of the BWS family
First, we analyzed the microsatellite markers at 11p15.5-p15.4, and the methylation status of ICR1 (only at CTS3 and CTS6) and ICR2, of a proband (III-2) and her parents (II-3, II-4) in the BWS family. We could not find any abnormalities in the microsatellite markers and ICR2 methylation status, but we did find GOM at both CTS3 and CTS6 in the proband and her mother (II-4) (Supplementary Fig. S1, Fig. 1b, Supplementary Table S3). An aunt (II-2) of the proband also showed GOM at CTS3 and CTS6 (Fig. 1b, Supplementary Table S3). The proband (III-2), mother (II-4), and aunt (II-2) exhibited BWS features that fulfilled the clinical criteria, and their BWSp scores were high enough to diagnose these cases as classical BWS (Fig. 2). Therefore, a pathogenic variant of ICR1 was suspected to be the cause of the GOM. Sequence analysis of ICR1 revealed that a pathogenic variant of OCT1 (GRCh37:11:g.2023019A>G), which was the same variant previously reported [12, 17], was found in all affected members, but not in unaffected members, with the exception of the maternal grandmother (I-2) (Fig. 2, Supplementary Fig. S2). We concluded that the pathogenic variant of ICR1 was the cause of the GOM in the BWS family in this study.
Anticipated and extended methylation of ICR1
To clarify whether the methylation anticipation occurred through maternal transmission of the pathogenic variant of OCT1, we analyzed all 10 DMSs in the IGF2/H19 domain within the family and compared the methylation statuses between the affected members of the second (mother and aunt) and third (proband) generations (Fig. 1b, Supplementary Table S3). CTS1–5 were highly methylated in the affected members (mother, aunt, and proband). The average methylation of CTS1–5 in the third generation (proband) was slightly higher, by approximately 2%, than that in the second generation (mother and aunt; Fig. 1b, Supplementary Table S3). In addition, the methylation levels of other DMSs, i.e., IGF2-DMR0, IGF2-DMR2, CTS6–7, and H19 promoter, were also apparently higher in the third generation than in the second generation. Furthermore, the average methylation of all DMSs was also higher in the third generation than in the second generation (Supplementary Table S3). These results clearly indicate that methylation anticipation occurred through maternal transmission and suggest that the extent of anticipation was larger at DMSs further from the pathogenic variant of OCT1 than CTS1–5, which were close to the variant. These results also suggest that CTSs close to the pathogenic variant were more prone to GOM.
We analyzed the methylation statuses of all DMSs in isolated ICR1-GOM patients, whose ICR1 sequences did not harbor any genetic defects, to clarify whether the methylation pattern was different between these patients and those with ICR1-GOM due to the pathogenic variant of OCT1. To do this, we calculated the values of Δ methylation (Δme), which is the difference between the maximum and minimum methylation values among all DMSs. To exclude the effect of anticipation, we used the Δme values of the mother and aunt, who were the first affected generation carrying the variant. The average Δme value of the mother and aunt was higher than that of the isolated ICR1-GOM patients and normal children (the mother and aunt vs. the isolated ICR1-GOM patients: p = 0.0379; the mother and aunt vs. normal children: p = 0.0386; Mann–Whitney U test) (Supplementary Tables S2, S3, and S4). However, there was no significant difference in the Δme values between the isolated ICR1-GOM patients and normal children (p = 0.2864, Mann–Whitney U test). The curves representing the methylation patterns of the mother and aunt were convex shaped (Fig. 1b), whereas those of the isolated ICR1-GOM patients and normal children were not (Fig. 1c). These results confirmed that the pathogenic variant of OCT1 affected the methylation of nearby CTSs, i.e., CTS1–5, and that the anticipation occurred mainly at DMSs far from the variant. In addition, the fact that there was no significant difference in Δme values between the isolated ICR1-GOM patients and normal children indicates that GOM occurred evenly at all DMSs within the IGF2/H19 domain in the isolated ICR1-GOM patients.
All affected members fulfilled the three diagnostic criteria for BWS [23,24,25]. The BWSp scores were: 5 for the aunt; 6 for the mother; and 6 for the proband (Fig. 2). Since the BWSp scores of all affected members did not differ significantly, we surmised that there was no anticipation of clinical features.
Age-related decline of methylation at IGF2-DMR0 in the normal population
Among the unaffected family members, the methylation levels of IGF2-DMR0 of the grandmother and father (I-2 and II-3, respectively) were lower than that of the proband’s sister (III-1; Fig. 1b, Supplementary Table S3). Since hypomethylation of IGF2-DMR0 in the peripheral blood of elderly people (in their sixties) has been previously reported [22], we hypothesized that an age-related decline of IGF2-DMR0 methylation occurs in the normal population. To examine this, we analyzed the IGF2-DMR0 methylation level in normal children and adults in their twenties and forties (Supplementary Table S5). All analyzed CpG sites showed significantly lower methylation levels in the adults in their twenties and forties than in the children (Fig. 3, Supplementary Table S6), and no difference was observed between the adults in their twenties and forties. Similar results were found when the methylation levels were analyzed by sex, although the difference in methylation at specific CpGs varied between females and males (Supplementary Fig. S3). Also, in each cohort, there was no significant difference in methylation levels between females and males (Supplementary Fig. S4). These results indicate that an age-related decline of IGF2-DMR0 methylation does occur, becomes established by young adulthood, and persists until old age.

Methylation analysis of IGF2-DMR0 in the normal population. All CpG sites showed a significantly lower methylation level in adults in their twenties and forties than in children. The methylation indexes represent the median of each CpG site (Supplementary Tables S5 and S6). The methylation levels of adults in their twenties and forties were lower than that of children (*p < 0.01, Steel-Dwass test, Supplementary Table S6), whereas the methylation levels between adults in their twenties and forties did not differ (Steel-Dwass test, Supplementary Table S6)
Discussion
In this study, we analyzed the methylation levels of 10 DMSs within the IGF2/H19 domain in isolated ICR1-GOM BWS patients, normal controls, and members of a BWS family, in which affected members harbored a pathogenic variant of OCT1. Our first major finding is that the extent of methylation anticipation through maternal transmission was greater at DMSs far from the pathogenic variant than at those close to the variant. This is the second report of methylation anticipation in familial BWS patients with a pathogenic variant of OCT1, the first one being by Berland et al. [17] in which the same pathogenic variant was observed. Our second major finding is that, in the normal population, an age-related decline of methylation occurred at IGF2-DMR0 which became established by young adulthood and continued to old age.
Evidence of DNA methylation anticipation through maternal transmission of the pathogenic variant of OCT1, has also been reported by Berland et al. [17], and methylation patterns of ICR1-GOM due to pathogenic variants have been reported by Abi Habib et al. [15]. However, Berland et al. analyzed only three DMSs (CTS1, CTS6, and H19 promoter), and Abi Habib et al. did not analyze CTS5 or IGF2-DMR2 and did not describe methylation anticipation. We, on the other hand, analyzed all 10 DMSs within the IGF2/H19 domain, in detail, with highly stringent conditioned bisulfite pyrosequencing to clarify the precise methylation pattern. The curves representing the methylation patterns of the affected members were convex-shaped at CTS1–5. This methylation pattern was similar to the “moustache” pattern reported by Abi Habib et al. [15]. Our results indicate methylation anticipation of the entire IGF2/H19 domain and its greater effect on DMSs situated far from the pathogenic variant. In addition, our results suggest a predominant GOM susceptibility of DMSs close to the pathogenic variant and the occurrence of uniform GOM at all DMSs in the isolated ICR1-GOM patients.
In mouse P19 cells, Sox-Oct motifs were reported to maintain the unmethylated state of ICR1, and base substitution mutations of the motifs induced ICR1-GOM [27, 28]. Bisulfite sequencing revealed that mutations in Oct motifs affected the methylation status of the nearest site, CTS2, leading to partial GOM [27]. In mouse knock-in mutations of Oct motifs, mature oocytes showed partial aberrant methylation of the mutant ICR1, but the aberrant methylation disappeared in blastocysts, probably due to global demethylation during the pre-implantation stage [29]. After implantation, somatic tissues, such as liver and muscle, again showed GOM on the maternal mutant ICR1. Bisulfite sequencing revealed that partial GOM occurred at CTS2 (the closest DMS to the mutations) in the liver, similar to the P19 cell experiments. However, ICR1 methylation was sufficiently erased in primordial germ cells (PGCs) irrespective of the mutations [29]. These results suggest that ICR1-GOM due to the OCT1 variant in humans occurs mainly after the implantation stage due to insufficient protection from de novo methylation. With respect to the mechanism of anticipation, the ICR1-GOM acquired in the previous generation might be erased insufficiently in PGCs or during the preimplantation stage. The remaining aberrant methylation might then function as “seeds of aberrant methylation”, triggering an extended and severe GOM in the entire IGF2/H19 domain, especially on DMSs other than CTS1–5. These mechanisms likely occur, although the ICR1 genomic structure and the number of CTSs are different between mouse and human.
With respect to clinical features, we could not identify any anticipation between the two generations. A previous report has suggested clinical anticipation; however, the clinical information of the previous generation was insufficient [17]. In our study, we obtained detailed clinical features of the mother and aunt and found they were consistent with the diagnostic criteria for BWS. The BWSp scores of all affected members did not differ substantially. Therefore, there was no clinical anticipation in this BWS family. However, it should be noted that both the mother and aunt developed unilateral Wilms tumor, but the proband did not. There is a possibility that the proband will develop Wilms tumor in the future because ICR1-GOM is associated with a high risk of developing Wilms tumor [30, 31]. If the proband develops Wilms tumor, her BWSp score will increase. In such a case, clinical anticipation may be suggested.
The second finding of this study is the age-related decline of methylation at IGF2-DMR0. Ito et al. also reported IGF2-DMR0 hypomethylation in the peripheral blood of controls whose ages ranged from 60 to 80 years [22]. We found that IGF2-DMR0 hypomethylation was established by age 20–30, indicating the occurrence of constitutive IGF2-DMR0 hypomethylation at a relatively young age. Further studies are required to investigate whether other differentially methylated regions (DMRs) show constitutive hypomethylation and to determine the biological significance of constitutive hypomethylation.
In conclusion, we reported the extent of DNA methylation anticipation due to the ICR1 pathogenic variant in familial BWS patients and the establishment of age-related IGF2-DMR0 hypomethylation by young adulthood in the normal population. Further studies are needed to clarify the precise mechanism of methylation anticipation, the existence of clinical anticipation in familial BWS patients, and the mechanism and biological significance of constitutive hypomethylation of IGF2-DMR0 and/or other imprinted DMRs.
References
Soejima H, Higashimoto K. Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders. J Hum Genet. 2013;58:402–9.
Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14:229–49.
Thorburn MJ, Wright ES, Miller CG, Smith-Read EH. Exomphalos-macroglossia-gigantism syndrome in Jamaican infants. Am J Dis Child. 1970;119:316–21.
Mussa A, Russo S, De Crescenzo A, Chiesa N, Molinatto C, Selicorni A, et al. Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am J Med Genet A. 2013;161A:2481–6.
Uyar A, Seli E. The impact of assisted reproductive technologies on genomic imprinting and imprinting disorders. Curr Opin Obstet Gynecol. 2014;26:210–21.
Lazaraviciute G, Kauser M, Bhattacharya S, Haggarty P, Bhattacharya S. A systematic review and meta-analysis of DNA methylation levels and imprinting disorders in children conceived by IVF/ICSI compared with children conceived spontaneously. Hum Reprod Update. 2014;20:840–52.
Mussa A, Molinatto C, Cerrato F, Palumbo O, Carella M, Baldassarre G, et al. Assisted reproductive techniques and risk of Beckwith-Wiedemann syndrome. Pediatrics. 2017;140:e20164311.
Eggermann T, Algar E, Lapunzina P, Mackay D, Maher ER, Mannens M, et al. Clinical utility gene card for: Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2014. https://www.nature.com/articles/ejhg2013132
Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC, Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet. 2004;36:958–60.
Prawitt D, Enklaar T, Gartner-Rupprecht B, Spangenberg C, Oswald M, Lausch E, et al. Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms’ tumor. Proc Natl Acad Sci USA. 2005;102:4085–90.
Sparago A, Russo S, Cerrato F, Ferraiuolo S, Castorina P, Selicorni A, et al. Mechanisms causing imprinting defects in familial Beckwith-Wiedemann syndrome with Wilms’ tumour. Hum Mol Genet. 2007;16:254–64.
Demars J, Shmela ME, Rossignol S, Okabe J, Netchine I, Azzi S, et al. Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum Mol Genet. 2010;19:803–14.
Poole RL, Leith DJ, Docherty LE, Shmela ME, Gicquel C, Splitt M, et al. Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur J Hum Genet. 2012;20:240–3.
Higashimoto K, Jozaki K, Kosho T, Matsubara K, Fuke T, Yamada D, et al. A novel de novo point mutation of the OCT-binding site in the IGF2/H19-imprinting control region in a Beckwith-Wiedemann syndrome patient. Clin Genet. 2013;86:539–44.
Abi Habib W, Azzi S, Brioude F, Steunou V, Thibaud N, Das Neves C, et al. Extensive investigation of the IGF2/H19 imprinting control region reveals novel OCT4/SOX2 binding site defects associated with specific methylation patterns in Beckwith-Wiedemann syndrome. Hum Mol Genet. 2014;23:5763–73.
Weth O, Renkawitz R. CTCF function is modulated by neighboring DNA binding factors. Biochem Cell Biol. 2011;89:459–68.
Berland S, Appelbäck M, Bruland O, Beygo J, Buiting K, Mackay DJ, et al. Evidence for anticipation in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2013;21:1344–8.
Murrell A, Ito Y, Verde G, Huddleston J, Woodfine K, Silengo MC, et al. Distinct methylation changes at the IGF2-H19 locus in congenital growth disorders and cancer. PLoS ONE. 2008;3:e1849.
Sullivan MJ, Taniguchi T, Jhee A, Kerr N, Reeve AE. Relaxation of IGF2 imprinting in Wilms tumours associated with specific changes in IGF2 methylation. Oncogene. 1999;18:7527–34.
Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62:6442–6.
Hidaka H, Higashimoto K, Aoki S, Mishima H, Hayashida C, Maeda T, et al. Comprehensive methylation analysis of imprinting-associated differentially methylated regions in colorectal cancer. Clin Epigenetics. 2018;10:150.
Ito Y, Koessler T, Ibrahim AE, Rai S, Vowler SL, Abu-Amero S, et al. Somatically acquired hypomethylation of IGF2 in breast and colorectal cancer. Hum Mol Genet. 2008;17:2633–43.
Elliott M, Bayly R, Cole T, Temple IK, Maher ER. Clinical features and natural history of Beckwith-Wiedemann syndrome: presentation of 74 new cases. Clin Genet. 1994;46:168–74.
DeBaun MR, Tucker MA. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pedia. 1998;132(3 Pt 1):398–400.
Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8–14.
Higashimoto K, Nakabayashi K, Yatsuki H, Yoshinaga H, Jozaki K, Okada J, et al. Aberrant methylation of H19-DMR acquired after implantation was dissimilar in soma versus placenta of patients with Beckwith-Wiedemann syndrome. Am J Med Genet A. 2012;158A:1670–5.
Hori N, Nakano H, Takeuchi T, Kato H, Hamaguchi S, Oshimura M, et al. A dyad oct-binding sequence functions as a maintenance sequence for the unmethylated state within the H19/Igf2-imprinted control region. J Biol Chem. 2002;277:27960–7.
Hori N, Yamane M, Kouno K, Sato K. Induction of DNA demethylation depending on two sets of Sox2 and adjacent Oct3/4 binding sites (Sox-Oct motifs) within the mouse H19/insulin-like growth factor 2 (Igf2) imprinted control region. J Biol Chem. 2012;287:44006–16.
Zimmerman DL, Boddy CS, Schoenherr CS. Oct4/Sox2 binding sites contribute to maintaining hypomethylation of the maternal igf2/h19 imprinting control region. PLoS ONE. 2013;8:e81962.
Mussa A, Molinatto C, Baldassarre G, Riberi E, Russo S, Larizza L, et al. Cancer risk in Beckwith-Wiedemann syndrome: a systematic review and meta-analysis outlining a novel (epi)genotype specific histotype targeted screening protocol. J Pedia. 2016;176:e1.
Maas SM, Vansenne F, Kadouch DJ, Ibrahim A, Bliek J, Hopman S, et al. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A. 2016;170:2248–60.
Acknowledgements
We thank the Analytical Research Center for Experimental Sciences, Saga University, for their experimental support. This study was supported by the following: grants from the Grant-in-Aid for Scientific Research (C) program of the Japan Society for the Promotion of Science [16K09970, awarded to KH; 17K08687, awarded to HS], grants for Practical Research Projects for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development (AMED) [17ek0109280h0001, 17ek0109234h0001, and 17ek0109205h0001, awarded to HS], a grant for Child Health and Development research from the National Center for Child Health and Development [26–13, awarded to HS], a grant for Research on Intractable Diseases from the Ministry of Health, Labor, and Welfare [H29-nanchitou(nan)-ippan-025, awarded to HS], a grant from the Joint Research Program of the Institute for Molecular and Cellular Regulation at Gunma University [16029, awarded to KH], and a grant from the 2018 Liaoning Provincial Natural Science Key Project of China [no. 20180530064, awarded to FS].
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sun, F., Higashimoto, K., Awaji, A. et al. The extent of DNA methylation anticipation due to a genetic defect in ICR1 in Beckwith-Wiedemann syndrome. J Hum Genet 64, 937–943 (2019). https://doi.org/10.1038/s10038-019-0634-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0634-0
