
Abstract
Pediatric hypertension can cause hypertensive emergencies, including hemorrhagic stroke, contributing to rare but serious childhood morbidity and mortality. Renovascular hypertension (RVH) is one of the major causes of secondary hypertension in children. Grange syndrome (MIM#602531) is a rare disease characterized by multiple stenosis or occlusion of the renal, abdominal, coronary, and cerebral arteries, which can cause phenotypes of RVH and fibromuscular dysplasia (MIM#135580). We report the case of a 7-year-old girl with Grange syndrome who showed RVH and multiple seizure episodes. At 1 year of age, she experienced seizures and sequential hemiparesis caused by a left thalamic hemorrhage without cerebral vascular anomalies. Chronic hypertension was observed, and abdominal computed tomography angiography showed characteristic bilateral renal artery stenosis. Whole-exome sequencing revealed a novel homozygous pathogenic variant in the YY1AP1 gene (NM_001198903.1: c.1169del: p.Lys390Argfs*12). Biallelic YY1AP1 mutations are known to cause Grange syndrome. Unlike previously reported patients, our patient presented with intracerebral hemorrhagic stroke without anomalous brain artery or bone fragility. The phenotype in our patient may help better understand this ultra-rare syndrome. Grange syndrome should be considered in patients presenting with childhood-onset hypertension and/or hemorrhagic stroke for early clinical intervention.
Similar content being viewed by others
Introduction
Hypertension in children and adolescents is becoming a more common problem [1]. It is usually asymptomatic and sometimes causes hypertensive emergencies including hemorrhagic stroke, a rare but life-threatening and notable contributor to childhood morbidity and mortality[2, 3]. Renovascular hypertension (RVH) is a vascular disease that renal artery stenosis/occlusion coexists with chronic hypertension, and a major cause of secondary hypertension in childhood. Hypertension may lead to seizures especially during infancy [4,5,6]. The leading cause of RVH in children is fibromuscular dysplasia (FMD) (MIM#135580), which accounts for 35–50% of all pediatric RVH cases [4, 7,8,9], but its pathogenesis is poorly understood [10, 11]. RVH and FMD are clinically heterogeneous and there are no established diagnostic criteria. Generally, they are clinically diagnosed with using imaging studies, which sometimes makes an accurate diagnosis difficult.
Grange syndrome (MIM#602531) is an ultra-rare autosomal-recessive syndrome characterized by brachydactyly, syndactyly, bone fragility, and multi-focal vascular disease accompanied by RVH and FMD [12, 13]. This syndrome was originally described in a large family with four affected siblings, and then followed by additional case reports with similar vascular and skeletal manifestations [13,14,15,16]. Pathogenic truncating variants in the YY1AP1 gene (MIM#607860) were identified in Grange syndrome in 2017 [12]. YY1AP1 encodes yin yang 1 (YY1)-associated protein 1. YY1AP1 and YY1 are components of the INO80 chromatin remodeling complex and act as transcriptional regulators of proliferation and differentiation in smooth muscle cells (SMCs) [12]. To date, only eleven patients in six pedigrees with Grange syndrome have been reported. Therefore, clinical information of Grange syndrome is limited. We report the case of a female with Grange syndrome who showed hemorrhagic strokes and RVH with a novel pathogenic variant in YY1AP1.
Materials and methods
Samples
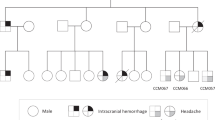
Genomic DNA samples were extracted from the peripheral blood leukocytes of an affected girl, unaffected her parents and sister (Fig. 1a), using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Informed consent was obtained from the parents and this study was approved by the Institutional Review Board of Yokohama City University Faculty of Medicine.

Familial pedigree and pathogenic YY1AP1 variants. a Familial pedigree. b Electropherograms of the YY1AP1 variant (c.1169del: p.Lys390Argfs*12) in the patient (II-1), her parents (I-1 and I-2) and sister (II-2). c Schematic presentation of the YY1AP1 gene and pathogenic variants. Previously reported variants (in black) and the current variant (in red) are shown above the gene. (color figure online)
Genetic analysis
Whole-exome sequencing (WES) of the patient was performed. Targeted enrichment was performed using SureSelect All Exon v6 kit (Agilent Technologies, Santa Clara, CA) and captured libraries were loaded onto a HiSeq 2500 platform (Illumina, San Diego, CA) as described previously [17]. Analyses for the autosomal-dominant (de novo) or autosomal-recessive (homozygous, compound heterozygous) models were conducted. Specifically, analyses for the autosomal-recessive model involved selection of candidate genetic variants in exons and canonical splice sites (±2 bp) with a minor allele frequency of <0.005 in the Exome Aggregation Consortium browser (http://exac.broadinstitute.org/), NHLBI Exome Variant Server (ESP6500) (http://evs.gs.washington.edu/EVS/), or in-house exome data (n = 575). The candidate variants were prioritized based on the biological and clinical relevance of each gene to the phenotype of the patient. The causative variants were validated by Sanger sequencing on an ABI 3500 Genetic analyzer (Applied Biosystems, Foster City, CA) and analyzed with Sequencher software (Gene Codes, Madison, WI).
Results
Clinical course
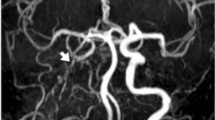
A 7-year-old Brazilian girl was born to healthy parents with a healthy sister (Fig. 1a). Her parents were both originally from a small city named Pariconha, northeast region of Brazil. During pregnancy, oligohydramnios and mild intrauterine growth retardation were detected at 32 gestational weeks. The girl was delivered at 38 weeks’ gestation by caesarean section due to persistent oligohydramnios. Her birth weight and length were 2430 g (−1.5 SD) and 42 cm (−2.7 SD), respectively. She was first admitted to hospital at 15 months of age due to seizures, right-sided hemiparesis, and deteriorated speech and motor function. Brain computed tomography (CT) revealed a left thalamic-caudate hemorrhage, but no cerebrovascular abnormalities such as arteriovenous malformation, aneurysm, or cerebral artery stenosis were observed (Fig. 2a–c). Unfortunately, her accurate blood pressure (BP) during this episode was unknown. Thereafter, she showed drastic recovery upon rehabilitation, and regained speech and motor function. At 3 years of age, hypertension was noted (130/80 mmHg [ >95th percentile]). Despite initiation of amlodipine (calcium channel blocker), her BP remained high at approximately the 95th percentile. She was admitted to the hospital due to the second episode of seizure at 5 years of age. No hemorrhage was observed on intracranial imaging and the cause of the second seizure episode remained unknown. Abdominal CT revealed bilateral renal artery stenosis. Hypertension was well controlled since starting an additional anti-hypertensive drug carvedilol (β-blocker). An antiepileptic drug (oxcarbazepine) and immunosuppressants (prednisolone and azathioprine) were initiated for seizures of unknown causes and suspected inflammatory vasculitis. No further seizure episodes have been reported.

Clinical features of the patient. a, b Axial and coronal views of head CT taken just after stroke at 1 year old, respectively. A high-density area indicated left thalamic caudate hemorrhage without midline displacement with normal ventricles. c Magnetic resonance angiography showed no abnormal findings. d, e Spinal bone X-rays, with frontal and lateral views, which show mild scoliosis. f Photograph of the patient’s face. g Photograph and h X-ray of the right hand. Clinobrachydactyly of the 5th fingers can be noted. i, j Frontal and lateral view of three-dimensional CT angiography of the abdominal and renal arteries, respectively. White arrows demonstrate bilateral stenosis of the proximal renal arteries. The superior mesenteric artery and celiac artery showed no stenosis
The patient was suspected to have an unknown vascular disease and referred for genetic counseling at 7 years of age. A physical examination showed normal height (121 cm [−0.67 SD]), weight (21.65 kg [−0.92 SD]), and occipitofrontal circumference (51 cm [−0.1 SD]) with scapular asymmetry and pectus excavatum. Her BP was 110/60 mmHg (95th percentile) and heart rate was 74 beats/min. Neurological examinations showed mild cognitive impairment, no dysmetria, normal cranial nerves, and normal deep tendon reflexes. Funduscopy was normal. Livedo reticularis, follicular hyperkeratosis, and strabismus were observed, but no characteristic facial features were determined (Fig. 2f). Proportionate limbs and hands with clinodactyly of the fifth fingers were noted (Fig. 2g). She had no history of bone fracture. Her hand X-ray showed middle phalanx shortening of the fifth finger, with an appropriate bone age for her chronological age (Fig. 1h). Spine radiographs showed mild dorso-lumbar scoliosis (Fig. 2d, e). Abdominal three-dimensional CT revealed bilateral renal artery stenosis (Fig. 2i, j). Measurements of the right and left kidneys were 8.2 × 3.7 cm (−0.2 SD) and 7.2 × 3.2 cm (−2.2 SD), respectively, which indicated that her left kidney was relatively small for her age. Electrocardiogram and ultrasonography of her heart and carotid arteries were normal. A laboratory examination was normal, including whole blood cell counts, renal function (creatinine: 0.34 mg/dL and blood urea nitrogen: 27 mg/dL), electrolyte levels (sodium: 139 mEq/L, potassium: 4.5 mEq/L, and chloride: 101 mEq/L), blood clotting tests, and inflammatory tests.
Genetic analysis
The mean depth of the RefSeq coding region was 87.79, with 95.6% of total coding sequences covered by 20 reads or more. WES revealed a novel homozygous 1-bp deletion in YY1AP1 (NM_001198903.1; c.1169del: p.Lys390Argfs*12). The variant was absent from the public databases (ExAC, ESP6500, HGVD) and our in-house control [n = 575]. The web-based in silico software, Mutation Taster (http://www.mutationtaster.org/) predicted this variant to be pathogenic. Sanger sequencing confirmed that this variant was homozygous in the patient and heterozygous in the unaffected carrier parents and the sister (Fig. 1b). This variant resided in the 3.3-Mb homozygous stretch in chromosome 1 (Supplementary Fig. 1).
Discussion
A novel homozygous protein-truncating YY1AP1 variant (NM_001198903.1: c.1169del: p.Lys390Argfs*12) was identified in a girl with a history of hemorrhagic stroke and RVH. The variant is predicted to cause nonsense-mediated mRNA decay and is consistent with the fact that loss-of-function mutation in YY1AP1 causes multiple vascular occlusive disease, Grange syndrome. YY1AP1 is expressed ubiquitously in various human tissues and co-localizes with YY1 in the nucleus [18]. Loss of YY1AP1 leads to cell-cycle arrest and disrupts TGF-β-driven differentiation of vascular SMCs [12]. Although the detailed pedigree information could not be obtained in this family, there is a possibility that her parents have a common ancestor because they originated from the same small city in Brazil and the variant was found in a homozygous stretch in the patient.
Although subarchnoid hemorrhage (SAH) have been observed in two patients with Grange syndrome[14, 16], cerebral hemorrhage has not been previously reported. The exact cause of intracranial hemorrhage of our patient is unknown; however, SAH is generally associated with cerebral aneurysm and intraparenchymal hemorrhage is accompanied by systemic hypertension [18]. The absence of obvious cerebral artery abnormalities suggests that the cause of hemorrhage of the patient would simply have been due to severe hypertension. Prompt diagnosis and precise control of systemic hypertension may prevent the potential risk for stroke and other complications caused by hypertension. The clinical features associated with Grange syndrome in previously reported and current patients are summarized in Table 1. Variable degrees of stenosis or occlusion of multiple arteries throughout the body were seen in affected individuals with Grange syndrome. Specifically, unilateral or bilateral renal arteries stenosis was associated with early-onset uncontrolled hypertension in the majority of patients (90.9%, n = 10/11). They all require multiple anti-hypertensive drugs and four patients underwent renal artery angioplasty (33.3%, 4/12) [13, 15, 16]. Another frequently observed feature is digital anomalies. The majority of patients with Grange syndrome show brachyclinodactyly or syndactyly of the hands and feet (91.7%, 11/12). Our patient also showed mild scoliosis. Skeletal screening is helpful in diagnosing Grange syndrome. A recent report suggested bone fragility as a variable feature and less frequent than previously thought [19]. Our patient has shown no bone fragility, too. Most affected individuals showed cerebral artery stenosis (81.8%, 10/11), and strokes were observed in more than half of them, possibly by hemorrhage or ischemic attacks (54.5%, 6/11). Seizures can occur in patients with Grange syndrome at a higher rate for various reasons with or without cerebral artery stenosis.
Mild learning disabilities and left ventricular hypertrophy (LVH) were also relatively frequently observed in affected individuals; however, these symptoms may be secondary (i.e., LVH could be due to hypertension and mild developmental delay may be caused by silent stroke episodes). Genotype-phenotype correlation is difficult to be determined because of the wide clinical variability in affected individuals even in the same family [13, 19]. Accumulation of patients with Grange syndrome and further genetic studies would clarify the pathogenesis and clinical entity of these diseases, including RVH and FMD. We should note that some symptoms are present after adolescence. One patient died unexpectedly at 18 years of age [13]. Chest or abdominal pain complaints should be considered as urgent issues as they are indicators of the onset of ischemic events. Periodic follow-up examinations are also required in Grange syndrome.
In conclusion, we report the case of a female with Grange syndrome due to a novel biallelic YY1AP1 variant, and clinically showed hemorrhagic stroke and RVH. To the best of our knowledge, YY1AP1 is the only mutated gene known to cause RVH and FMD with a clear Mendelian inheritance pattern [20]. Grange syndrome should be considered in patients with stroke combined with renal artery stenosis.
References
Flynn JT, Kaelber DC, Baker-Smith CM, Blowey D, Carroll AE, Daniels SR et al. Clinical practice guideline for screening and management of high blood pressure in children and adolescents. Pediatrics. 2017;140:e20171904.
Jordan LC, Hillis AE. Hemorrhagic stroke in children. Pedia Neurol. 2007;36:73–80.
Broderick J, Talbot GT, Prenger E, Leach A, Brott T. Stroke in children within a major metropolitan area: the surprising importance of intracerebral hemorrhage. J Child Neurol. 1993;8:250–5.
Tullus K, Brennan E, Hamilton G, Lord R, McLaren CA, Marks SD, et al. Renovascular hypertension in children. Lancet. 2008;371:1453–63.
Hung YM, Weng KP, Lin CC, Huang JS, Chiou YH, Hsieh KS. Brain stem hemorrhage in a 2-year-10-month-old child with renovascular hypertension related to fibromuscular dysplasia. Acta Cardiol Sin. 2015;31:564–7.
Gumer LB, Del Vecchio M, Aronoff S. Strokes in children: a systematic review. Pediatr Emerg Care. 2014;30:660–4.
Gill DG, Mendes de Costa B, Cameron JS, Joseph MC, Ogg CS, Chantler C. Analysis of 100 children with severe and persistent hypertension. Arch Dis Child. 1976;51:951–6.
Bayazit AK, Yalcinkaya F, Cakar N, Duzova A, Bircan Z, Bakkaloglu A, et al. Reno-vascular hypertension in childhood: a nationwide survey. Pediatr Nephrol. 2007;22:1327–33.
Piercy KT, Hundley JC, Stafford JM, Craven TE, Nagaraj SK, Dean RH, et al. Renovascular disease in children and adolescents. J Vasc Surg. 2005;41:973–82.
Perdu J, Boutouyrie P, Bourgain C, Stern N, Laloux B, Bozec E, et al. Inheritance of arterial lesions in renal fibromuscular dysplasia. J Hum Hypertens. 2007;21:393–400.
Ganesh SK, Morissette R, Xu Z, Schoenhoff F, Griswold BF, Yang J, et al. Clinical and biochemical profiles suggest fibromuscular dysplasia is a systemic disease with altered TGF-beta expression and connective tissue features. FASEB J. 2014;28:3313–24.
Guo DC, Duan XY, Regalado ES, Mellor-Crummey L, Kwartler CS, Kim D, et al. Loss-of-function mutations in YY1AP1 lead to grange syndrome and a fibromuscular dysplasia-like vascular disease. Am J Hum Genet. 2017;100:21–30.
Grange DK, Balfour IC, Chen SC, Wood EG. Familial syndrome of progressive arterial occlusive disease consistent with fibromuscular dysplasia, hypertension, congenital cardiac defects, bone fragility, brachysyndactyly, and learning disabilities. Am J Med Genet. 1998;75:469–80.
Weymann S, Yonekawa Y, Khan N, Martin E, Heppner FL, Schinzel A, et al. Severe arterial occlusive disorder and brachysyndactyly in a boy: a further case of Grange syndrome? Am J Med Genet. 2001;99:190–5.
Wallerstein R, Augustyn AM, Wallerstein D, Elton L, Tejeiro B, Johnson V, et al. A new case of Grange syndrome without cardiac findings. Am J Med Genet A. 2006;140:1316–20.
Volonghi I, Frigerio M, Mardighian D, Gasparotti R, Del Zotto E, Giossi A, et al. Grange syndrome: an identifiable cause of stroke in young adults. Am J Med Genet A. 2012;158A:2894–8.
Sekiguchi F, Nasiri J, Sedghi M, Salehi M, Hosseinzadeh M, Okamoto N, et al. A novel homozygous DPH1 mutation causes intellectual disability and unique craniofacial features. J Hum Genet. 2018;63:487–91.
Wang CY, Liang YJ, Lin YS, Shih HM, Jou YS, Yu WC. YY1AP, a novel co-activator of YY1. J Biol Chem. 2004;279:17750–5.
Rath M, Spiegler S, Strom TM, Trenkler J, Kroisel PM, Felbor U. Identification of pathogenic YY1AP1 splice variants in siblings with Grange syndrome by whole exome sequencing. Am J Med Genet A. 2019;179:295–9.
Di Monaco S, Georges A, Lengele JP, Vikkula M, Persu A. Genomics of fibromuscular dysplasia. Int J Mol Sci. 2018;19.
Acknowledgements
The authors appreciate the participation of all the affected individuals and their families in this study. This work was supported by AMED (grant numbers: JP18ek0109280, JP18dm0107090, JP18ek0109301, JP18ek0109348, and JP18kk020501), JSPS KAKENHI; grant numbers JP17H01539 (N. Matsumoto) and JP16K06254 (A. Takata), the Takeda Science Foundation (N. Miyake and N. Matsumoto), the Yokohama Foundation for Advancement of Medical Science, and the Hayashi Memorial Foundation for Female Natural Scientists.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Saida, K., Kim, C.A., Ceroni, J.R.M. et al. Hemorrhagic stroke and renovascular hypertension with Grange syndrome arising from a novel pathogenic variant in YY1AP1. J Hum Genet 64, 885–890 (2019). https://doi.org/10.1038/s10038-019-0626-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0626-0
This article is cited by
-
The clinical and radiological cerebrovascular abnormalities associated with renovascular hypertension in children: a systematic review
Pediatric Nephrology (2022)
-
Microcephalic osteodysplastic primordial dwarfism type II is associated with global vascular disease
Orphanet Journal of Rare Diseases (2021)
-
Autosomal dominant Alport syndrome due to a COL4A4 mutation with an additional ESPN variant detected by whole-exome analysis
CEN Case Reports (2020)
